The Glucose Metabolic Pathway as A Potential Target for Therapeutics: Crucial Role of Glycosylation in Alzheimer’s Disease

 , , ,
, , ,
Abstract
:1. Introduction
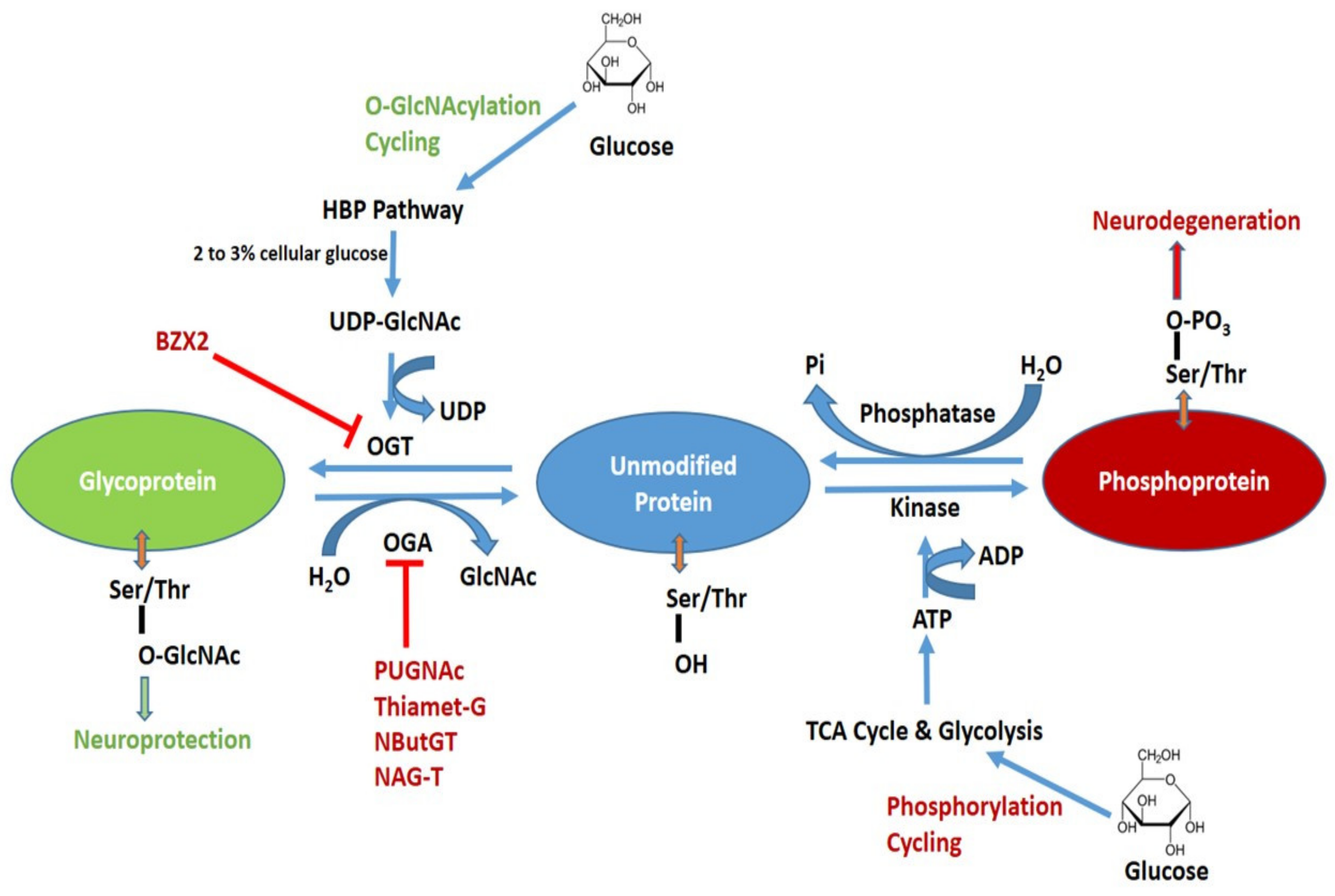
2. Biosynthesis and Modulation of O-GlcNA Cylation
3. Glycosylation in Alzheimer’s Disease
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ADAM | A disintegrin and metalloprotease |
| AICD | APP intracellular domain |
| APP | β-amyloid precursor protein |
| Aph-1 | Anterior pharynx-defective 1 |
| Asn | Asparagine |
| Aβ | β-amyloid |
| BACE1 | β-site APP cleaving enzyme-1 |
| FDG | Fluoro-2-deoxy-D-glucose |
| GalNAc | N-acetylgalactosamine |
| GFAT | Glutamine-fructose-6-phosphate amidotransferase |
| GlcNAc | N-acetylglucosamine |
| GLUT | Glucose transporter |
| GnT-III | β1,4-N-acetylglucosaminyltransferase III |
| HBP | Hexosamine biosynthetic pathway |
| hAPP | human APP |
| LLO | Lipid-linked oligosaccharide |
| MAP | Microtubule-associated protein |
| Mgat3−/− | GnT-III-deficient |
| mOGT | Mitochondrial OGT |
| ncOGT | Nucleocytoplasmic OGT |
| NFTs | Neurofibrillary tangles |
| NICD | Notch intracellular domain |
| OGA | O-GlcNAcase |
| OGA-L | OGA Large |
| OGA-S | OGA short |
| O-GlcNAcylation | O-linked β-N-acetylglucosaminylation |
| OGT | O-GlcNAc transferase |
| OST | Oligosaccharyltransferase |
| Pen-2 | Presenilin enhancer 2 |
| PET | Positron emission tomography |
| PSEN | Presenilin |
| PTMs | Post-translational modifications |
| ROS | Reactive oxygen species |
| Ser | Serine |
| sOGT | short OGT |
| TCA | Tricarboxylic acid |
| Thr | Threonine |
| T2DM | Type II diabetes mellitus |
| UDP-GlcNAc | Uridine 5′-diphospho-N-acetylglucosamine |
| 3 × Tg-AD | Triple transgenic murine model of AD |
References
- Lauc, G.; Pezer, M.; Rudan, I.; Campbell, H. Mechanisms of disease: The human N-glycome. Biochim. Biophys. Acta 2016, 1860, 1574–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Videira, P.A.Q.; Castro-Caldas, M. Linking glycation and glycosylation with inflammation and mitochondrial dysfunction in Parkinson’s disease. Front. Neurosci. 2018, 12, 381. [Google Scholar] [CrossRef] [PubMed]
- Alzheimers Dement. Alzheimer’s disease facts and figures. Alzheimer Dement. 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Liu, K.; Paterson, A.J.; Chin, E.; Kudlow, J.E. Glucose stimulates protein modification by O-linked GlcNAc in pancreatic beta cells: Linkage of O-linked GlcNAc to beta cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 2820–2825. [Google Scholar] [CrossRef] [Green Version]
- McClain, D.A.; Lubas, W.A.; Cooksey, R.C.; Hazel, M.; Parker, G.J.; Love, D.C.; Hanover, J.A. Altered glycan-dependent signaling induces insulin resistance and hyperleptinemia. Proc. Natl. Acad. Sci. USA 2002, 99, 10695–10699. [Google Scholar] [CrossRef] [Green Version]
- Zachara, N.E.; Hart, G.W. Cell signaling, the essential role of O-GlcNAc! Biochim. Biophys. Acta 2006, 1761, 599–617. [Google Scholar] [CrossRef]
- Jaeken, J. Congenital disorders of glycosylation (CDG): It’s all in it! J. Inherit. Metab. Dis. 2003, 26, 99–118. [Google Scholar] [CrossRef]
- Varki, A.; Cummings, R.D.; Aebi, M.; Packer, N.H.; Seeberger, P.H.; Esko, J.D.; Stanley, P.; Hart, G.; Darvill, A.; Kinoshita, T.; et al. Symbol nomenclature for graphical representations of glycans. Glycobiology 2015, 25, 1323–1324. [Google Scholar] [CrossRef] [Green Version]
- Nairn, A.V.; York, W.S.; Harris, K.; Hall, E.M.; Pierce, J.M.; Moremen, K.W. Regulation of glycan structures in animal tissues: Transcript profiling of glycan-related genes. J. Biol. Chem. 2008, 283, 17298–17313. [Google Scholar] [CrossRef] [Green Version]
- Abou-Abbass, H.; Abou-El-Hassan, H.; Bahmad, H.; Zibara, K.; Zebian, A.; Youssef, R.; Ismail, J.; Zhu, R.; Zhou, S.; Dong, X.; et al. Glycosylation and other PTMs alterations in neurodegenerative diseases: Current status and future role in neurotrauma. Electrophoresis 2016, 37, 1549–1561. [Google Scholar] [CrossRef] [PubMed]
- Medina-Cano, D.; Ucuncu, E.; Nguyen, L.S.; Nicouleau, M.; Lipecka, J.; Bizot, J.C.; Thiel, C.; Foulquier, F.; Lefort, N.; Faivre-Sarrailh, C.; et al. High N-glycan multiplicity is critical for neuronal adhesion and sensitizes the developing cerebellum to N-glycosylation defect. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Spiro, R.G. Protein glycosylation: Nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 2002, 12, 43R–56R. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.R.; Hart, G.W. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 1984, 259, 3308–3317. [Google Scholar] [PubMed]
- Haltiwanger, R.S.; Holt, G.D.; Hart, G.W. Enzymatic addition of O-GlcNAc to nuclear and cytoplasmic proteins. Identification of a uridine diphospho-N-acetylglucosamine: Peptide β-N-acetylglucosaminyltransferase. J. Biol. Chem. 1990, 265, 2563–2568. [Google Scholar]
- Nalivaeva, N.N.; Turner, A.J. Post-translational modifications of proteins: Acetylcholinesterase as a model system. Proteomics 2001, 1, 735–747. [Google Scholar] [CrossRef]
- Lefebvre, T.; Guinez, C.; Dehennaut, V.; Beseme-Dekeyser, O.; Morelle, W.; Michalski, J.C. Does O-GlcNAc play a role in neurodegenerative diseases? Expert Rev. Proteom. 2005, 2, 265–275. [Google Scholar] [CrossRef]
- Hart, G.W.; Housley, M.P.; Slawson, C. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 2007, 446, 1017–1022. [Google Scholar] [CrossRef]
- Xu, S.L.; Chalkley, R.J.; Maynard, J.C.; Wang, W.; Ni, W.; Jiang, X.; Shin, K.; Cheng, L.; Savage, D.; Huhmer, A.F.; et al. Proteomic analysis reveals O-GlcNAc modification on proteins with key regulatory functions in Arabidopsis. Proc. Natl. Acad. Sci. USA 2017, 114, E1536–E1543. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface 2018, 15. [Google Scholar] [CrossRef]
- Wang, Z.; Gucek, M.; Hart, G.W. Cross-talk between GlcNAcylation and phosphorylation: Site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc. Natl. Acad. Sci. USA 2008, 105, 13793–13798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatta, E.; Lefebvre, T.; Gaetani, S.; dos Santos, M.; Marrocco, J.; Mir, A.M.; Cassano, T.; Maccari, S.; Nicoletti, F.; Mairesse, J. Evidence for an imbalance between tau O-GlcNAcylation and phosphorylation in the hippocampus of a mouse model of Alzheimer’s disease. Pharmacol. Res. 2016, 105, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Sharma, N.; Barone, E.; Lanzillotta, C.; Castellani, A.; Iavarone, F.; Vincenzoni, F.; Castagnola, M.; Butterfield, D.A.; Gaetani, S.; et al. Proteomic identification of altered protein O-GlcNAcylation in a triple transgenic mouse model of Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3309–3321. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, B.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Brandt, R.; Gong, C.X. Regulation between O-GlcNAcylation and phosphorylation of neurofilament-M and their dysregulation in Alzheimer disease. FASEB J. 2008, 22, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Li, B.; Liu, Y.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Dysregulation of insulin signaling, glucose transporters, O-GlcNAcylation, and phosphorylation of tau and neurofilaments in the brain: Implication for Alzheimer’s disease. Am. J. Pathol. 2009, 175, 2089–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.X.; Wang, J.Z.; Iqbal, K.; Grundke-Iqbal, I. Inhibition of protein phosphatase 2A induces phosphorylation and accumulation of neurofilaments in metabolically active rat brain slices. Neurosci. Lett. 2003, 340, 107–110. [Google Scholar] [CrossRef]
- Joiner, C.M.; Li, H.; Jiang, J.; Walker, S. Structural characterization of the O-GlcNAc cycling enzymes: Insights into substrate recognition and catalytic mechanisms. Curr. Opin. Struct. Biol. 2019, 56, 97–106. [Google Scholar] [CrossRef]
- Gao, Y.; Wells, L.; Comer, F.I.; Parker, G.J.; Hart, G.W. Dynamic O-glycosylation of nuclear and cytosolic proteins: Cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J. Biol. Chem. 2001, 276, 9838–9845. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, J.; Zang, L.; Zhu, H.; Guo, J.; Zhang, J.; Wen, L.; Chen, Y.; Li, Y.; Chen, X.; et al. Production of glycopeptide derivatives for exploring substrate specificity of human OGA toward sugar moiety. Front. Chem. 2018, 6, 646. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, M.B.; Nam, Y.; Jiang, J.; Sliz, P.; Walker, S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 2011, 469, 564–567. [Google Scholar] [CrossRef]
- Okuyama, R.; Marshall, S. UDP-N-acetylglucosaminyl transferase (OGT) in brain tissue: Temperature sensitivity and subcellular distribution of cytosolic and nuclear enzyme. J. Neurochem. 2003, 86, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Wells, L.; Gao, Y.; Mahoney, J.A.; Vosseller, K.; Chen, C.; Rosen, A.; Hart, G.W. Dynamic O-glycosylation of nuclear and cytosolic proteins: Further characterization of the nucleocytoplasmic β-N-acetylglucosaminidase, O-GlcNAcase. J. Biol. Chem. 2002, 277, 1755–1761. [Google Scholar] [CrossRef] [Green Version]
- Comtesse, N.; Maldener, E.; Meese, E. Identification of a nuclear variant of MGEA5, a cytoplasmic hyaluronidase and a β-N-acetylglucosaminidase. Biochem. Biophys. Res. Commun. 2001, 283, 634–640. [Google Scholar] [CrossRef]
- Keembiyehetty, C.N.; Krzeslak, A.; Love, D.C.; Hanover, J.A. A lipid-droplet-targeted O-GlcNAcase isoform is a key regulator of the proteasome. J. Cell Sci. 2011, 124, 2851–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, W.Y.; Chatham, J.C.; Darley-Usmar, V.; McMahon, L.L.; Zhang, J. O-GlcNAcylation and neurodegeneration. Brain Res. Bull. 2017, 133, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Ryan, P.; Xu, M.; Davey, A.K.; Danon, J.J.; Mellick, G.D.; Kassiou, M.; Rudrawar, S. O-GlcNAc modification protects against protein misfolding and aggregation in neurodegenerative disease. ACS Chem. Neurosci. 2019, 10, 2209–2221. [Google Scholar] [CrossRef]
- Olivier-Van Stichelen, S.; Wang, P.; Comly, M.; Love, D.C.; Hanover, J.A. Nutrient-driven O-linked N-acetylglucosamine (O-GlcNAc) cycling impacts neurodevelopmental timing and metabolism. J. Biol. Chem. 2017, 292, 6076–6085. [Google Scholar] [CrossRef] [Green Version]
- Pinho, T.S.; Verde, D.M.; Correia, S.C.; Cardoso, S.M.; Moreira, P.I. O-GlcNAcylation and neuronal energy status: Implications for Alzheimer’s disease. Ageing Res. Rev. 2018, 46, 32–41. [Google Scholar] [CrossRef]
- Clark, R.J.; McDonough, P.M.; Swanson, E.; Trost, S.U.; Suzuki, M.; Fukuda, M.; Dillmann, W.H. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J. Biol. Chem. 2003, 278, 44230–44237. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lu, F.; Wang, J.Z.; Gong, C.X. Concurrent alterations of O-GlcNAcylation and phosphorylation of tau in mouse brains during fasting. Eur. J. Neurosci. 2006, 23, 2078–2086. [Google Scholar] [CrossRef]
- Lim, S.; Haque, M.M.; Nam, G.; Ryoo, N.; Rhim, H.; Kim, Y.K. Monitoring of intracellular tau aggregation regulated by OGA/OGT inhibitors. Int. J. Mol. Sci. 2015, 16, 20212–20224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsen, K.T.; Iverfeldt, K. O-GlcNAcylation increases non-amyloidogenic processing of the amyloid-β precursor protein (APP). Biochem. Biophys. Res. Commun. 2011, 404, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitworth, G.E.; Macauley, M.S.; Stubbs, K.A.; Dennis, R.J.; Taylor, E.J.; Davies, G.J.; Greig, I.R.; Vocadlo, D.J. Analysis of PUGNAc and NAG-thiazoline as transition state analogues for human O-GlcNAcase: Mechanistic and structural insights into inhibitor selectivity and transition state poise. J. Am. Chem. Soc. 2007, 129, 635–644. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Shan, X.; Jones, B.A.; Zhao, G.; Woodward, M.L.; Li, X.; Zhu, Y.; McEachern, E.J.; Silverman, M.A.; Watson, N.V.; et al. Pharmacological inhibition of O-GlcNAcase (OGA) prevents cognitive decline and amyloid plaque formation in bigenic tau/APP mutant mice. Mol. Neurodegener. 2014, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Macauley, M.S.; Shan, X.; Yuzwa, S.A.; Gloster, T.M.; Vocadlo, D.J. Elevation of global O-GlcNAc in rodents using a selective O-GlcNAcase inhibitor does not cause insulin resistance or perturb glucohomeostasis. Chem. Biol. 2010, 17, 949–958. [Google Scholar] [CrossRef] [Green Version]
- Yuzwa, S.A.; Macauley, M.S.; Heinonen, J.E.; Shan, X.; Dennis, R.J.; He, Y.; Whitworth, G.E.; Stubbs, K.A.; McEachern, E.J.; Davies, G.J.; et al. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat. Chem. Biol. 2008, 4, 483–490. [Google Scholar] [CrossRef]
- Paul, S.; Haskali, M.B.; Liow, J.S.; Zoghbi, S.S.; Barth, V.N.; Kolodrubetz, M.C.; Bond, M.R.; Morse, C.L.; Gladding, R.L.; Frankland, M.P.; et al. Evaluation of a PET radioligand to image O-GlcNAcase in brain and periphery of rhesus monkey and knock-out mouse. J. Nucl. Med. 2019, 60, 129–134. [Google Scholar] [CrossRef] [Green Version]
- Bronzuoli, M.R.; Facchinetti, R.; Steardo, L., Jr.; Romano, A.; Stecca, C.; Passarella, S.; Steardo, L.; Cassano, T.; Scuderi, C. palmitoylethanolamide dampens reactive astrogliosis and improves neuronal trophic support in a triple transgenic model of alzheimer’s disease: In vitro and in vivo evidence. Oxidative Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bronzuoli, M.R.; Facchinetti, R.; Valenza, M.; Cassano, T.; Steardo, L.; Scuderi, C. Astrocyte function is affected by aging and not alzheimer’s disease: A preliminary investigation in hippocampi of 3xTg-AD Mice. Front. Pharmacol. 2019, 10, 644. [Google Scholar] [CrossRef] [Green Version]
- Cassano, T.; Pace, L.; Bedse, G.; Lavecchia, A.M.; De Marco, F.; Gaetani, S.; Serviddio, G. Glutamate and mitochondria: Two prominent players in the oxidative stress-induced neurodegeneration. Curr. Alzheimer Res. 2016, 13, 185–197. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef]
- Serviddio, G.; Romano, A.D.; Cassano, T.; Bellanti, F.; Altomare, E.; Vendemiale, G. Principles and therapeutic relevance for targeting mitochondria in aging and neurodegenerative diseases. Curr. Pharm. Des. 2011, 17, 2036–2055. [Google Scholar] [CrossRef] [PubMed]
- Gallelli, C.A.; Calcagnini, S.; Romano, A.; Koczwara, J.B.; de Ceglia, M.; Dante, D.; Villani, R.; Giudetti, A.M.; Cassano, T.; Gaetani, S. Modulation of the oxidative stress and lipid peroxidation by endocannabinoids and their lipid analogues. Antioxidants 2018, 7, 93. [Google Scholar] [CrossRef] [Green Version]
- Trabace, L.; Cassano, T.; Tucci, P.; Steardo, L.; Kendrick, K.M.; Cuomo, V. The effects of nitric oxide on striatal serotoninergic transmission involve multiple targets: An in vivo microdialysis study in the awake rat. Brain Res. 2004, 1008, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L. Glucose metabolism in normal aging and Alzheimer’s disease: Methodological and physiological considerations for PET studies. Clin. Transl. Imaging 2013, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, C.M.; Macklin, L.N.; Cai, Y.; Sharp, A.A.; Yan, X.X.; Reagan, L.P.; Strader, A.D.; Rose, G.M.; Patrylo, P.R. Impaired glucose tolerance and reduced plasma insulin precede decreased AKT phosphorylation and GLUT3 translocation in the hippocampus of old 3 − Tg-AD Mice. J. Alzheimer Dis. 2019, 68, 809–837. [Google Scholar] [CrossRef]
- Macklin, L.; Griffith, C.M.; Cai, Y.; Rose, G.M.; Yan, X.X.; Patrylo, P.R. Glucose tolerance and insulin sensitivity are impaired in APP/PS1 transgenic mice prior to amyloid plaque pathogenesis and cognitive decline. Exp. Gerontol. 2017, 88, 9–18. [Google Scholar] [CrossRef]
- Pardeshi, R.; Bolshette, N.; Gadhave, K.; Ahire, A.; Ahmed, S.; Cassano, T.; Gupta, V.B.; Lahkar, M. Insulin signaling: An opportunistic target to minify the risk of Alzheimer’s disease. Psychoneuroendocrinology 2017, 83, 159–171. [Google Scholar] [CrossRef]
- Bayod, S.; Felice, P.; Andres, P.; Rosa, P.; Camins, A.; Pallas, M.; Canudas, A.M. Downregulation of canonical Wnt signaling in hippocampus of SAMP8 mice. Neurobiol. Aging 2015, 36, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Cisternas, P.; Salazar, P.; Silva-Alvarez, C.; Barros, L.F.; Inestrosa, N.C. Activation of Wnt signaling in cortical neurons enhances glucose utilization through glycolysis. J. Biol. Chem. 2016, 291, 25950–25964. [Google Scholar] [CrossRef] [Green Version]
- Cisternas, P.; Zolezzi, J.M.; Martinez, M.; Torres, V.I.; Wong, G.W.; Inestrosa, N.C. Wnt-induced activation of glucose metabolism mediates the in vivo neuroprotective roles of Wnt signaling in Alzheimer disease. J. Neurochem. 2019, 149, 54–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, B.T.; He, X. Frizzled and LRP5/6 receptors for Wnt/β-catenin signaling. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Neitzel, L.R.; Spencer, Z.T.; Nayak, A.; Cselenyi, C.S.; Benchabane, H.; Youngblood, C.Q.; Zouaoui, A.; Ng, V.; Stephens, L.; Hann, T.; et al. Developmental regulation of Wnt signaling by Nagk and the UDP-GlcNAc salvage pathway. Mech. Dev. 2019, 156, 20–31. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, M.; Sun, J.; Guo, A.; Fernando, R.L.; Chen, Y.; Peng, P.; Zhao, G.; Deng, Y. DPP-4 inhibitor improves learning and memory deficits and AD-like neurodegeneration by modulating the GLP-1 signaling. Neuropharmacology 2019, 157, 107668. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Cassano, T.; Arena, A.; Tramutola, A.; Lavecchia, M.A.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic. Biol. Med. 2016, 91, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Tramutola, A.; Triani, F.; Calcagnini, S.; Di Domenico, F.; Ripoli, C.; Gaetani, S.; Grassi, C.; Butterfield, D.A.; Cassano, T.; et al. Biliverdin reductase-a mediates the beneficial effects of intranasal insulin in Alzheimer disease. Mol. Neurobiol. 2019, 56, 2922–2943. [Google Scholar] [CrossRef] [PubMed]
- Peila, R.; Rodriguez, B.L.; Launer, L.J.; Honolulu-Asia aging, S. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia aging study. Diabetes 2002, 51, 1256–1262. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Tramutola, A.; Lanzillotta, C.; Arena, A.; Blarzino, C.; Cassano, T.; Butterfield, D.A.; Di Domenico, F.; Perluigi, M.; Barone, E. Loss of biliverdin reductase-A favors Tau hyper-phosphorylation in Alzheimer’s disease. Neurobiol. Dis. 2019, 125, 176–189. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008, 582, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Akasaka-Manya, K.; Manya, H.; Sakurai, Y.; Wojczyk, B.S.; Spitalnik, S.L.; Endo, T. Increased bisecting and core-fucosylated N-glycans on mutant human amyloid precursor proteins. Glycoconj. J. 2008, 25, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Tomita, S.; Kirino, Y.; Suzuki, T. Cleavage of Alzheimer’s amyloid precursor protein (APP) by secretases occurs after O-glycosylation of APP in the protein secretory pathway. Identification of intracellular compartments in which APP cleavage occurs without using toxic agents that interfere with protein metabolism. J. Biol. Chem. 1998, 273, 6277–6284. [Google Scholar] [CrossRef] [Green Version]
- McFarlane, I.; Georgopoulou, N.; Coughlan, C.M.; Gillian, A.M.; Breen, K.C. The role of the protein glycosylation state in the control of cellular transport of the amyloid β precursor protein. Neuroscience 1999, 90, 15–25. [Google Scholar] [CrossRef]
- Nakagawa, K.; Kitazume, S.; Oka, R.; Maruyama, K.; Saido, T.C.; Sato, Y.; Endo, T.; Hashimoto, Y. Sialylation enhances the secretion of neurotoxic amyloid-beta peptides. J. Neurochem. 2006, 96, 924–933. [Google Scholar] [CrossRef]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef] [PubMed]
- Kitazume, S.; Tachida, Y.; Kato, M.; Yamaguchi, Y.; Honda, T.; Hashimoto, Y.; Wada, Y.; Saito, T.; Iwata, N.; Saido, T.; et al. Brain endothelial cells produce amyloid {β} from amyloid precursor protein 770 and preferentially secrete the O-glycosylated form. J. Biol. Chem. 2010, 285, 40097–40103. [Google Scholar] [CrossRef] [Green Version]
- Kizuka, Y.; Kitazume, S.; Taniguchi, N. N-glycan and Alzheimer’s disease. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2447–2454. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.S.; Kwon, O.H.; Chung, S. O-GlcNAcylation of amyloid-β precursor protein at threonine 576 residue regulates trafficking and processing. Biochem. Biophys. Res. Commun. 2017, 490, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Cheng, B.; Culwell, A.R.; Esch, F.S.; Lieberburg, I.; Rydel, R.E. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the β-amyloid precursor protein. Neuron 1993, 10, 243–254. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F. α-secretase in Alzheimer’s disease: Molecular identity, regulation and therapeutic potential. J. Neurochem. 2011, 116, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, E.; Prox, J.; Bernreuther, C.; Weber, S.; Schwanbeck, R.; Serneels, L.; Snellinx, A.; Craessaerts, K.; Thathiah, A.; Tesseur, I.; et al. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J. Neurosci. 2010, 30, 4833–4844. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Rossner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escrevente, C.; Morais, V.A.; Keller, S.; Soares, C.M.; Altevogt, P.; Costa, J. Functional role of N-glycosylation from ADAM10 in processing, localization and activity of the enzyme. Biochim. Biophys. Acta 2008, 1780, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Kizuka, Y.; Nakano, M.; Kitazume, S.; Saito, T.; Saido, T.C.; Taniguchi, N. Bisecting GlcNAc modification stabilizes BACE1 protein under oxidative stress conditions. Biochem. J. 2016, 473, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Vanoni, O.; Paganetti, P.; Molinari, M. Consequences of individual N-glycan deletions and of proteasomal inhibition on secretion of active BACE. Mol. Biol. Cell 2008, 19, 4086–4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kizuka, Y.; Kitazume, S.; Fujinawa, R.; Saito, T.; Iwata, N.; Saido, T.C.; Nakano, M.; Yamaguchi, Y.; Hashimoto, Y.; Staufenbiel, M.; et al. An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer’s disease. EMBO Mol. Med. 2015, 7, 175–189. [Google Scholar] [CrossRef]
- Stanley, P. Biological consequences of overexpressing or eliminating N-acetylglucosaminyltransferase-TIII in the mouse. Biochim. Biophys. Acta 2002, 1573, 363–368. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Aragno, M.; Borghi, R.; Autelli, R.; Giliberto, L.; Muraca, G.; Danni, O.; Zhu, X.; Smith, M.A.; et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J. Neurochem. 2008, 104, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Priatel, J.J.; Sarkar, M.; Schachter, H.; Marth, J.D. Isolation, characterization and inactivation of the mouse Mgat3 gene: The bisecting N-acetylglucosamine in asparagine-linked oligosaccharides appears dispensable for viability and reproduction. Glycobiology 1997, 7, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Savonenko, A.V.; Melnikova, T.; Laird, F.M.; Stewart, K.A.; Price, D.L.; Wong, P.C. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc. Natl. Acad. Sci. USA 2008, 105, 5585–5590. [Google Scholar] [CrossRef] [Green Version]
- Willem, M.; Garratt, A.N.; Novak, B.; Citron, M.; Kaufmann, S.; Rittger, A.; DeStrooper, B.; Saftig, P.; Birchmeier, C.; Haass, C. Control of peripheral nerve myelination by the β-secretase BACE1. Science 2006, 314, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Moussa-Pacha, N.M.; Abdin, S.M.; Omar, H.A.; Alniss, H.; Al-Tel, T.H. BACE1 inhibitors: Current status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 2020, 40, 339–384. [Google Scholar] [CrossRef]
- Francis, R.; McGrath, G.; Zhang, J.; Ruddy, D.A.; Sym, M.; Apfeld, J.; Nicoll, M.; Maxwell, M.; Hai, B.; Ellis, M.C.; et al. aph-1 and pen-2 are required for Notch pathway signaling, γ-secretase cleavage of βAPP, and presenilin protein accumulation. Dev. Cell 2002, 3, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Goutte, C.; Tsunozaki, M.; Hale, V.A.; Priess, J.R. APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc. Natl. Acad. Sci. USA 2002, 99, 775–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, H.; Winkler, E.; Edbauer, D.; Prokop, S.; Basset, G.; Yamasaki, A.; Kostka, M.; Haass, C. PEN-2 is an integral component of the γ-secretase complex required for coordinated expression of presenilin and nicastrin. J. Biol. Chem. 2002, 277, 39062–39065. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Nishimura, M.; Arawaka, S.; Levitan, D.; Zhang, L.; Tandon, A.; Song, Y.Q.; Rogaeva, E.; Chen, F.; Kawarai, T.; et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing. Nature 2000, 407, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Nadeau, P.; Song, W.; Donoviel, D.; Yuan, M.; Bernstein, A.; Yankner, B.A. Presenilins are required for γ-secretase cleavage of β-APP and transmembrane cleavage of Notch-1. Nat. Cell Biol. 2000, 2, 463–465. [Google Scholar] [CrossRef]
- De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Guhde, G.; Annaert, W.; Von Figura, K.; Van Leuven, F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998, 391, 387–390. [Google Scholar] [CrossRef]
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. γ-Secretase inhibitors and modulators. Biochim. Biophys. Acta 2013, 1828, 2898–2907. [Google Scholar] [CrossRef] [Green Version]
- Herreman, A.; Van Gassen, G.; Bentahir, M.; Nyabi, O.; Craessaerts, K.; Mueller, U.; Annaert, W.; De Strooper, B. gamma-Secretase activity requires the presenilin-dependent trafficking of nicastrin through the Golgi apparatus but not its complex glycosylation. J. Cell Sci. 2003, 116, 1127–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Nam, D.W.; Park, S.Y.; Song, H.; Hong, H.S.; Boo, J.H.; Jung, E.S.; Kim, Y.; Baek, J.Y.; Kim, K.S.; et al. O-linked β-N-acetylglucosaminidase inhibitor attenuates beta-amyloid plaque and rescues memory impairment. Neurobiol. Aging 2013, 34, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Shirotani, K.; Lu, B.; Gerard, N.P.; Gerard, C.; Hama, E.; Lee, H.J.; Saido, T.C. Metabolic regulation of brain Abeta by neprilysin. Science 2001, 292, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Aβ1-42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef]
- Sato, B.; Katagiri, Y.U.; Iijima, K.; Yamada, H.; Ito, S.; Kawasaki, N.; Okita, H.; Fujimoto, J.; Kiyokawa, N. The human CD10 lacking an N-glycan at Asn(628) is deficient in surface expression and neutral endopeptidase activity. Biochim. Biophys. Acta 2012, 1820, 1715–1723. [Google Scholar] [CrossRef]
- Gao, Y.; Tan, L.; Yu, J.T.; Tan, L. Tau in Alzheimer’s disease: Mechanisms and therapeutic strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef]
- Arnold, C.S.; Johnson, G.V.; Cole, R.N.; Dong, D.L.; Lee, M.; Hart, G.W. The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J. Biol. Chem. 1996, 271, 28741–28744. [Google Scholar] [CrossRef] [Green Version]
- Bellanti, F.; Iannelli, G.; Blonda, M.; Tamborra, R.; Villani, R.; Romano, A.; Calcagnini, S.; Mazzoccoli, G.; Vinciguerra, M.; Gaetani, S.; et al. Alterations of Clock Gene RNA Expression in Brain Regions of a Triple Transgenic Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 59, 615–631. [Google Scholar] [CrossRef] [Green Version]
- Cassano, T.; Magini, A.; Giovagnoli, S.; Polchi, A.; Calcagnini, S.; Pace, L.; Lavecchia, M.A.; Scuderi, C.; Bronzuoli, M.R.; Ruggeri, L.; et al. Early intrathecal infusion of everolimus restores cognitive function and mood in a murine model of Alzheimer’s disease. Exp. Neurol. 2019, 311, 88–105. [Google Scholar] [CrossRef]
- Cassano, T.; Romano, A.; Macheda, T.; Colangeli, R.; Cimmino, C.S.; Petrella, A.; LaFerla, F.M.; Cuomo, V.; Gaetani, S. Olfactory memory is impaired in a triple transgenic model of Alzheimer disease. Behav. Brain Res. 2011, 224, 408–412. [Google Scholar] [CrossRef]
- Cassano, T.; Serviddio, G.; Gaetani, S.; Romano, A.; Dipasquale, P.; Cianci, S.; Bellanti, F.; Laconca, L.; Romano, A.D.; Padalino, I.; et al. Glutamatergic alterations and mitochondrial impairment in a murine model of Alzheimer disease. Neurobiol. Aging 2012, 33, 1121.e1–1121.e12. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Pace, L.; Tempesta, B.; Lavecchia, A.M.; Macheda, T.; Bedse, G.; Petrella, A.; Cifani, C.; Serviddio, G.; Vendemiale, G.; et al. Depressive-like behavior is paired to monoaminergic alteration in a murine model of Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2014, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scuderi, C.; Bronzuoli, M.R.; Facchinetti, R.; Pace, L.; Ferraro, L.; Broad, K.D.; Serviddio, G.; Bellanti, F.; Palombelli, G.; Carpinelli, G.; et al. Ultramicronized palmitoylethanolamide rescues learning and memory impairments in a triple transgenic mouse model of Alzheimer’s disease by exerting anti-inflammatory and neuroprotective effects. Transl. Psychiatry 2018, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Yuzwa, S.A.; Shan, X.; Macauley, M.S.; Clark, T.; Skorobogatko, Y.; Vosseller, K.; Vocadlo, D.J. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 2012, 8, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shan, X.; Safarpour, F.; Erro Go, N.; Li, N.; Shan, A.; Huang, M.C.; Deen, M.; Holicek, V.; Ashmus, R.; et al. Pharmacological inhibition of O-GlcNAcase enhances autophagy in brain through an mTOR-independent pathway. ACS Chem. Neurosci. 2018, 9, 1366–1379. [Google Scholar] [CrossRef]
- Song, Y.; Brady, S.T. Post-translational modifications of tubulin: Pathways to functional diversity of microtubules. Trends Cell Biol. 2015, 25, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Wloga, D.; Gaertig, J. Post-translational modifications of microtubules. J. Cell Sci. 2010, 123, 3447–3455. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Name | Subjects | Effects | References |
|---|---|---|---|
| OGT Inhibitor: | |||
| BZX2 | Tau-BiFC cells | ↑ tau phosphorylation at Ser199 and Ser396 | [41] |
| ↑ tau aggregation | |||
| OGA Inhibitors: | |||
| O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino-N-phenylcarbamate | SH-SY5Y cells | ↑ O-GlcNAcylation, ↑sAPPα, | [42] |
| (PUGNAc) | PC12 cells | ↓ Aβ levels | [43] |
| ↓ Phosphorylation level of tau at Ser-199, Ser-202, Thr-205, Thr-212, Ser-214, Ser-262, and Ser396 | |||
| 1,2-dideoxy-2′-methyl-alpha-d-glucopyranoso[2,1-d]-Delta2′-thiazoline | Human O-GlcNAcase | Highly selective competitive OGA inhibitor | [44] |
| (NAG-thiazoline) | NIH 3T3 cells | ↑ Global GlcNAcylation levels | [21] |
| 1,2-dideoxy-2′-propyl-alpha-D-glucopyranoso-[2,1-D]-delta2′-thiazoline | Sprague-Dawley rats | ↑ O-GlcNAc levels | [45] |
| (NButGT) | C57BL/6J mice | No alteration in glucose tolerance and insulin signaling pathways | [46] |
| No insulin resistance | |||
| 3ar,5r,6s,7r,7ar)-2-(Ethylamino)-5-(Hydroxymethyl)-5,6,7,7a-Tetrahydro-3ah-Pyrano[3,2-D][1,3]thiazole-6,7-Diol | TAPP mice | ↑ Global GlcNAcylation levels | [47] |
| (Thiamet-G) | Improves performance in the Morris water maze (MWM) test | ||
| ↓ Aβ levels | |||
| N-(5-(((2S,4S)-2-methyl-4-(6-fluoropyridin-2-yloxy)piperidin-1-yl)methyl)thiazol-2-yl)acetamide (LSN3316612) | Rhesus monkey | ↑ O-GlcNAc levels | [48] |
| Oga knock-out mice | ↓ Tau phosphorylation | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bukke, V.N.; Villani, R.; Archana, M.; Wawrzyniak, A.; Balawender, K.; Orkisz, S.; Ferraro, L.; Serviddio, G.; Cassano, T. The Glucose Metabolic Pathway as A Potential Target for Therapeutics: Crucial Role of Glycosylation in Alzheimer’s Disease . Int. J. Mol. Sci. 2020, 21, 7739. https://doi.org/10.3390/ijms21207739
Bukke VN, Villani R, Archana M, Wawrzyniak A, Balawender K, Orkisz S, Ferraro L, Serviddio G, Cassano T. The Glucose Metabolic Pathway as A Potential Target for Therapeutics: Crucial Role of Glycosylation in Alzheimer’s Disease . International Journal of Molecular Sciences. 2020; 21(20):7739. https://doi.org/10.3390/ijms21207739
Chicago/Turabian StyleBukke, Vidyasagar Naik, Rosanna Villani, Moola Archana, Agata Wawrzyniak, Krzysztof Balawender, Stanislaw Orkisz, Luca Ferraro, Gaetano Serviddio, and Tommaso Cassano. 2020. "The Glucose Metabolic Pathway as A Potential Target for Therapeutics: Crucial Role of Glycosylation in Alzheimer’s Disease " International Journal of Molecular Sciences 21, no. 20: 7739. https://doi.org/10.3390/ijms21207739
APA StyleBukke, V. N., Villani, R., Archana, M., Wawrzyniak, A., Balawender, K., Orkisz, S., Ferraro, L., Serviddio, G., & Cassano, T. (2020). The Glucose Metabolic Pathway as A Potential Target for Therapeutics: Crucial Role of Glycosylation in Alzheimer’s Disease . International Journal of Molecular Sciences, 21(20), 7739. https://doi.org/10.3390/ijms21207739