The Status of Bile Acids and Farnesoid X Receptor in Brain and Liver of Rats with Thioacetamide-Induced Acute Liver Failure


Abstract
:
1. Introduction
2. Results
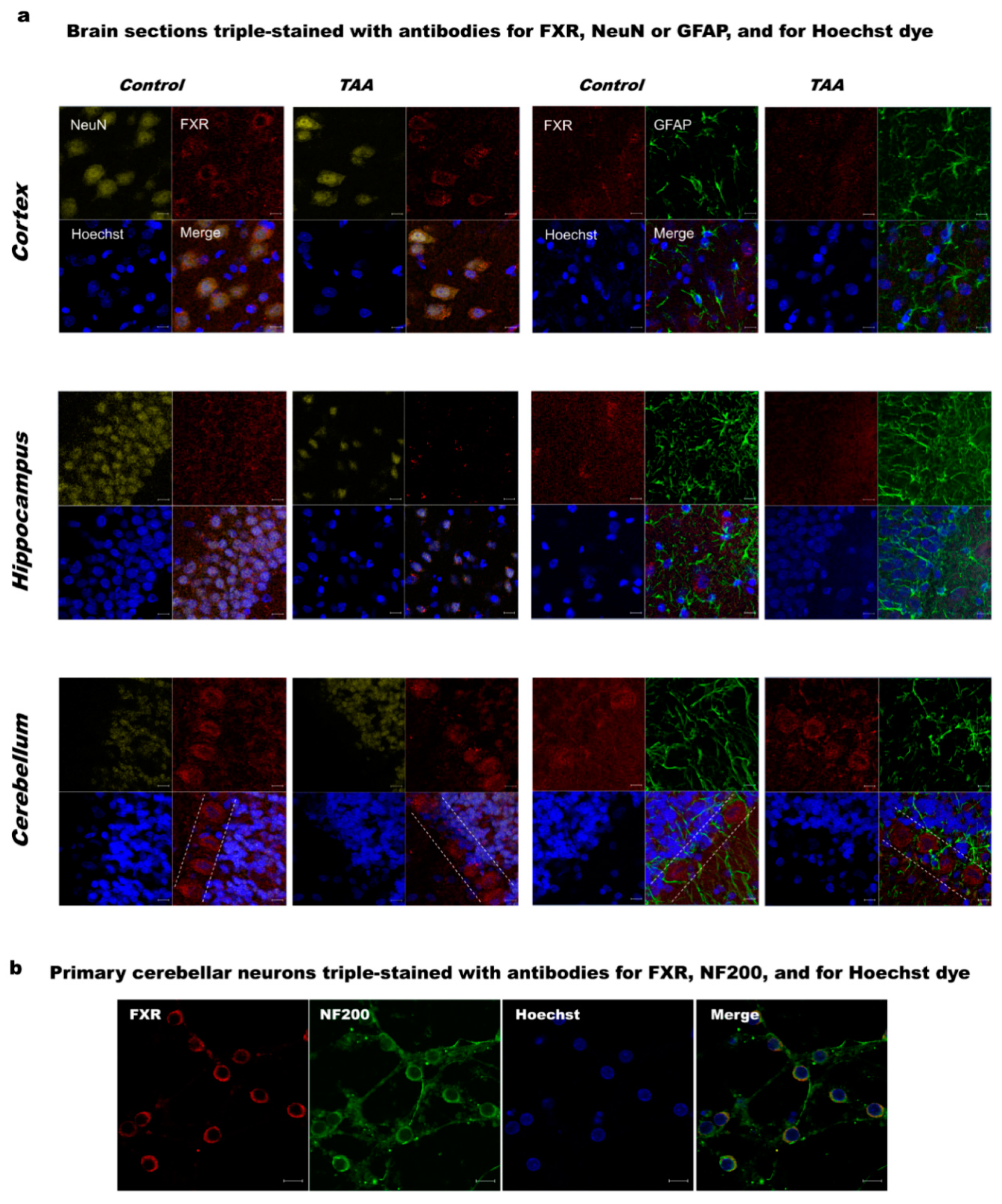
2.1. The Pattern of Immunoreactivity for Farnesoid X Receptor
2.1.1. Brain (Cerebral Cortex, Hippocampus, Cerebellum)
2.1.2. Primary Cultures of Cerebellar Neurons
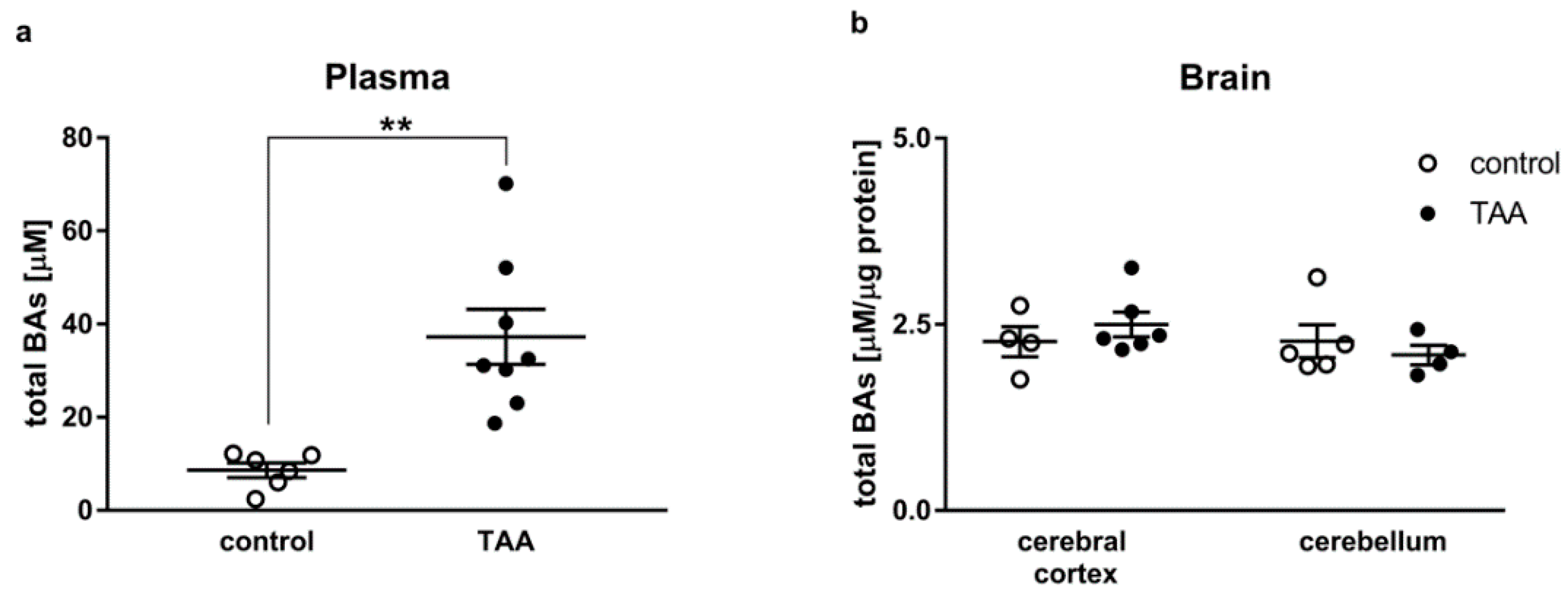
2.2. Total Bile Acids Concentration
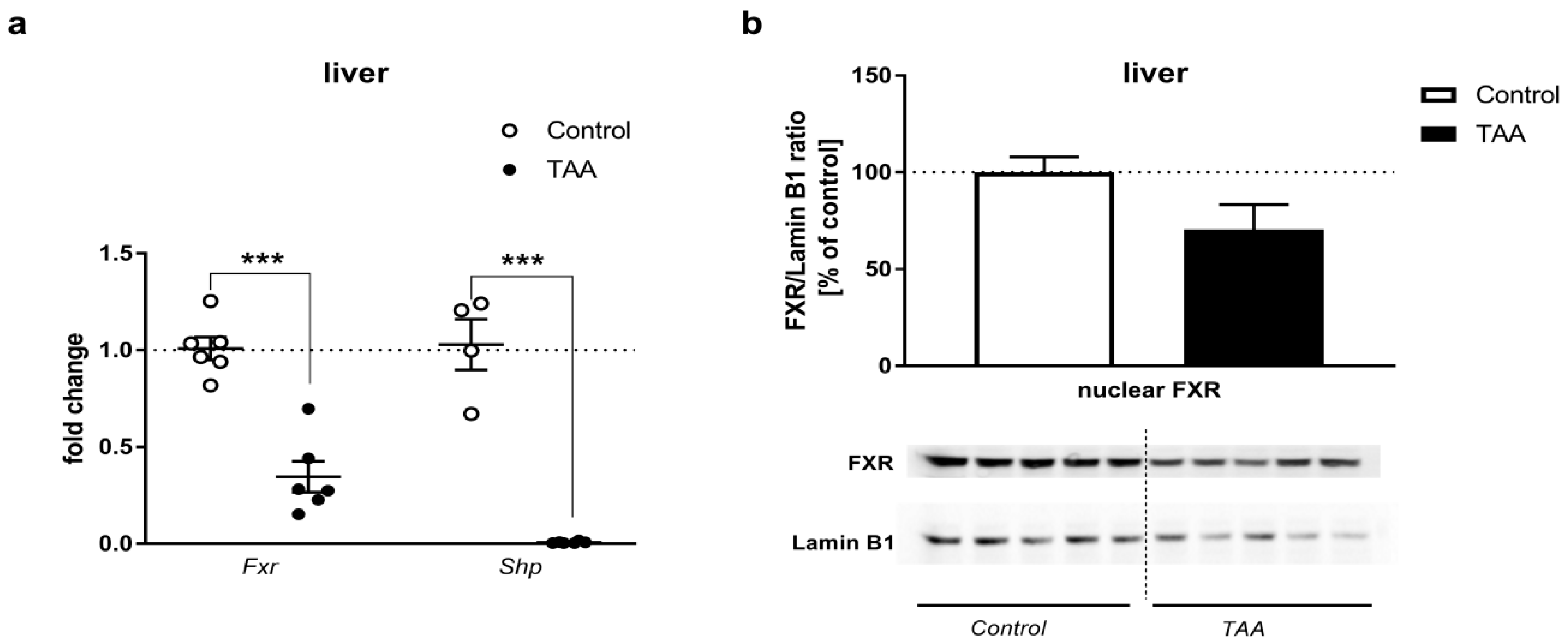
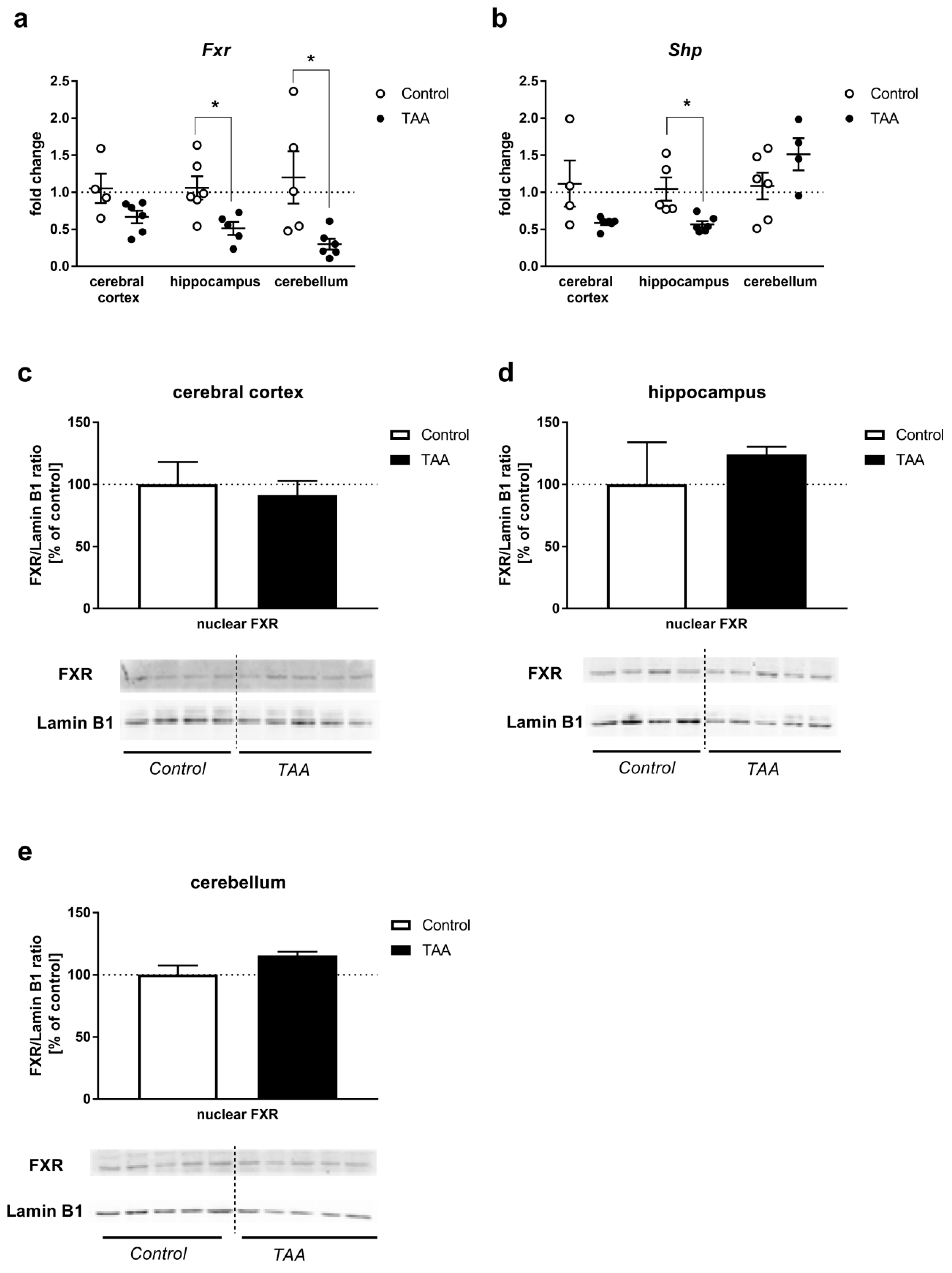
2.3. Real-Time qRT-PCR and Western Blot Analysis of the Expression of FXR and SHP
2.3.1. Liver
2.3.2. Brain
3. Discussion
4. Materials and Methods
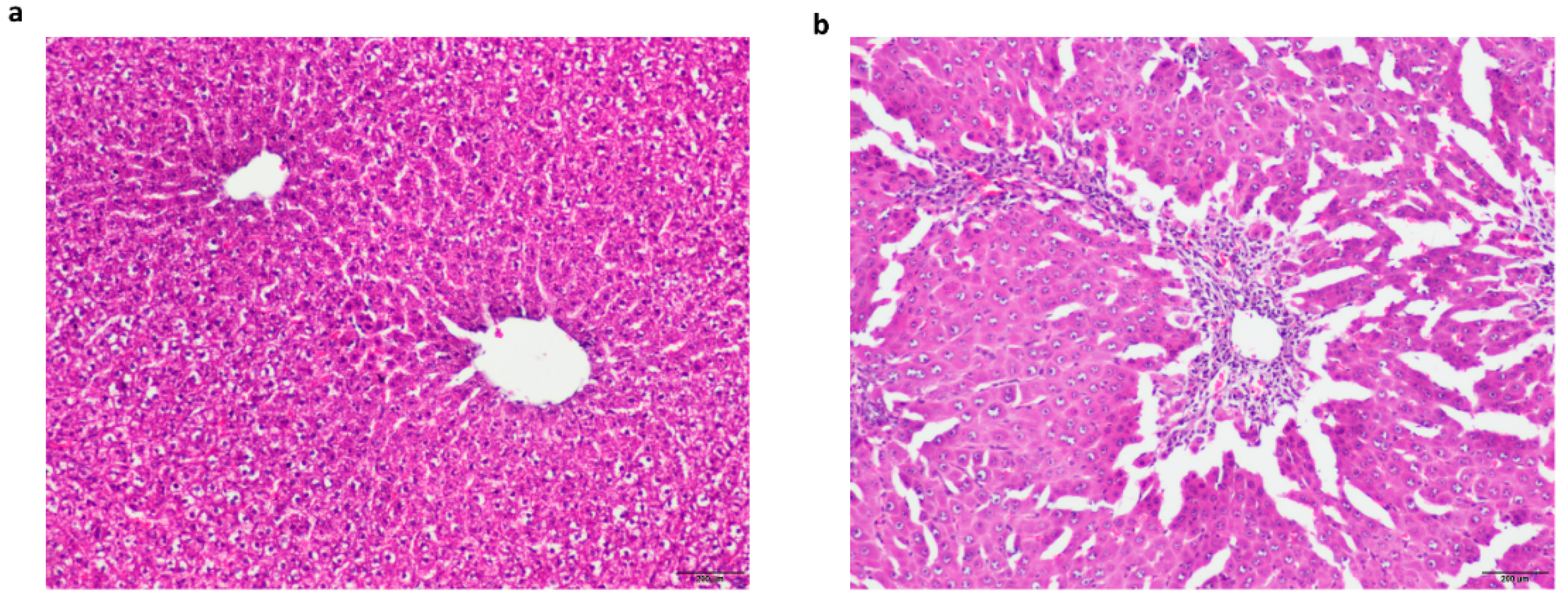
4.1. The Acute Liver Failure Model
4.2. Primary Neuronal Cultures
4.3. Immunolabelling
4.4. Real-Time PCR
4.5. Nuclear Protein Isolation and Western Blot Analysis
4.6. Bile Acid Measurement
4.7. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALF | Acute liver failure |
| BA | Bile acid |
| BBB | Blood–brain barrier |
| FXR | Farnesoid X receptor |
| HE | Hepatic encephalopathy |
| SHP | Small heterodimer partner |
| TAA | Thioacetamide |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | TAA | |
|---|---|---|
| AST (GOT) (U/L) | 153.84 ± 12.4 | 1001.82 ± 141.42 *** |
| ALT (GPT) (U/L) | 60.18 ± 1.92 | 336.38 ± 53.75 *** |
| GGTP (U/L) | 2.88 ± 0.15 | 4.1 ± 0.41 * |
| NH3 (µmol/L) | 55.91 ± 4.52 | 120.85 ± 13.39 ** |
References
- Butterworth, R.F. Pathogenesis of hepatic encephalopathy in cirrhosis: The concept of synergism revisited. Metab. Brain Dis. 2016, 31, 1211–1215. [Google Scholar] [CrossRef]
- Shawcross, D.L.; Wright, G.; Olde Damink, S.W.M.; Jalan, R. Role of ammonia and inflammation in minimal hepatic encephalopathy. Metab. Brain Dis. 2007, 22, 125–138. [Google Scholar] [CrossRef]
- Lima, L.C.D.; Miranda, A.S.; Ferreira, R.N.; Rachid, M.A.; e Silva, A.C.S. Hepatic encephalopathy: Lessons from preclinical studies. World J. Hepatol. 2019, 11, 173–185. [Google Scholar] [CrossRef]
- Bron, B.; Waldram, R.; Silk, D.B.A.; Williams, R. Serum, cerebrospinal fluid, and brain levels of bile acids in patients with fulminant hepatic failure. Gut 1977, 18, 692–696. [Google Scholar] [CrossRef] [Green Version]
- Yin, P.; Wan, D.; Zhao, C.; Chen, J.; Zhao, X.; Wang, W.; Lu, X.; Yang, S.; Gu, J.; Xu, G. A metabonomic study of hepatitis B-induced liver cirrhosis and hepatocellular carcinoma by using RP-LC and HILIC coupled with mass spectrometry. Mol. Biosyst. 2009, 5, 868. [Google Scholar] [CrossRef]
- Wang, X.; Xie, G.; Zhao, A.; Zheng, X.; Huang, F.; Wang, Y.; Yao, C.; Jia, W.; Liu, P. Serum Bile Acids Are Associated with Pathological Progression of Hepatitis B-Induced Cirrhosis. J. Proteome Res. 2016, 15, 1126–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, G.; Wang, X.; Huang, F.; Zhao, A.; Chen, W.; Yan, J.; Zhang, Y.; Lei, S.; Ge, K.; Zheng, X.; et al. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int. J. Cancer 2016, 139, 1764–1775. [Google Scholar] [CrossRef]
- McMillin, M.; Frampton, G.; Quinn, M.; Ashfaq, S.; Santos, M.D.L.; Grant, S.; Demorrow, S. Bile Acid Signaling Is Involved in the Neurological Decline in a Murine Model of Acute Liver Failure. Am. J. Pathol. 2016, 186, 312–323. [Google Scholar] [CrossRef]
- Hadjihambi, A.; Harrison, I.F.; Costas-Rodríguez, M.; Vanhaecke, F.; Arias, N.; Gallego-Durán, R.; Mastitskaya, S.; Hosford, P.S.; Olde Damink, S.W.M.; Davies, N.; et al. Impaired brain glymphatic flow in experimental hepatic encephalopathy. J. Hepatol. 2019, 70, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Wang, X.; Jiang, R.; Zhao, A.; Yan, J.; Zheng, X.; Huang, F.; Liu, X.; Panee, J.; Rajani, C.; et al. Dysregulated bile acid signaling contributes to the neurological impairment in murine models of acute and chronic liver failure. EBioMedicine 2018, 37, 294–306. [Google Scholar] [CrossRef] [Green Version]
- Quinn, M.; Mcmillin, M.; Galindo, C.; Frampton, G.; Yong, H.; Demorrow, S. Bile acids permeabilize the blood brain barrier after bile duct ligation in rats via Rac1-dependent mechanisms. Dig. Liver Dis. 2014, 46, 527–534. [Google Scholar] [CrossRef] [Green Version]
- Mertens, K.L.; Kalsbeek, A.; Soeters, M.R.; Eggink, H.M. Bile Acid Signaling Pathways from the Enterohepatic Circulation to the Central Nervous System. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keitel, V.; Org, B.G.; Bidmon, H.J.; Zemtsova, I.; Spomer, L. The Bile Acid Receptor TGR5 (Gpbar-1) Acts as a Neurosteroid Receptor in Brain. Glia 2010, 58, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, J.; Hu, W.; Wang, C.; Lu, X.; Tong, L.; Wu, F. Identification of functional farnesoid X receptors in brain neurons. FEBS Lett. 2016, 590, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.T.; Makishima, M.; Repa, J.J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular Basis for Feedback Regulation of Bile Acid Synthesis by Nuclear Receptors. Mol. Cell 2000, 6, 507–515. [Google Scholar] [CrossRef]
- Massafra, V.; Milona, A.; Vos, H.R.; Ramos, R.J.J.; Gerrits, J.; Willemsen, E.C.L.; Pittol, J.M.R.; Ijssennagger, N.; Houweling, M.; Prinsen, H.C.M.T.; et al. Farnesoid X Receptor Activation Promotes Hepatic Amino Acid Catabolism and Ammonium Clearance in Mice. Gastroenterology 2017, 152, 1462–1476.e10. [Google Scholar] [CrossRef] [Green Version]
- McMillin, M.; Grant, S.; Frampton, G.; Petrescu, A.D.; Kain, J.; Williams, E.; Haines, R.; Canady, L.; Demorrow, S. FXR-Mediated Cortical Cholesterol Accumulation Contributes to the Pathogenesis of Type A Hepatic Encephalopathy. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 47–63. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Wang, T.; Lan, Y.; Yang, L.; Pan, W.; Zhu, Y. Deletion of mouse FXR gene disturbs multiple neurotransmitter systems and alters neurobehavior. Front. Behav. Neurosci. 2015, 9, 70. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo, R.; Cauli, O.; Gomez-Pinedo, U.; Agusti, A.; Hernandez-Rabaza, V.; Garcia-Verdugo, J.M.; Felipo, V. Hyperammonemia induces neuroinflammation that contributes to cognitive impairment in rats with hepatic encephalopathy. Gastroenterology 2010, 139, 675–684. [Google Scholar] [CrossRef]
- Felipo, V. Hepatic encephalopathy: Effects of liver failure on brain function. Nat. Rev. Neurosci 2013, 14, 851–858. [Google Scholar] [CrossRef]
- Balzano, T.; Forteza, J.; Molina, P.; Giner, J.; Monzó, A.; Sancho-Jiménez, J.; Urios, A.; Montoliu, C.; Felipo, V. The Cerebellum of Patients with Steatohepatitis Shows Lymphocyte Infiltration, Microglial Activation and Loss of Purkinje and Granular Neurons. Sci. Rep. 2018, 8, 3004. [Google Scholar] [CrossRef] [PubMed]
- García-García, R.; Cruz-Gómez, Á.J.; Urios, A.; Mangas-Losada, A.; Forn, C.; Escudero-García, D.; Kosenko, E.; Torregrosa, I.; Tosca, J.; Giner-Durán, R.; et al. Learning and Memory Impairments in Patients with Minimal Hepatic Encephalopathy are Associated with Structural and Functional Connectivity Alterations in Hippocampus. Sci. Rep. 2018, 8, 9664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Yin, L.; Xu, L.; Qi, Y.; Li, H.; Xu, Y.; Han, X.; Liu, K.; Peng, J. Protective effect of dioscin against thioacetamide-induced acute liver injury via FXR/AMPK signaling pathway in vivo. Biomed. Pharmacother. 2018, 97, 481–488. [Google Scholar] [CrossRef]
- Keshk, W.A.; Soliman, N.A.; Ali, D.A.; Elseady, W.S. Mechanistic evaluation of AMPK/SIRT1/FXR signaling axis, inflammation, and redox status in thioacetamide-induced liver cirrhosis: The role of Cichorium intybus linn (chicory)-supplemented diet. J. Food Biochem. 2019, 43, e12938. [Google Scholar] [CrossRef]
- Verbeke, L.; Mannaerts, I.; Schierwagen, R.; Govaere, O.; Klein, S.; Vander Elst, I.; Windmolders, P.; Farre, R.; Wenes, M.; Mazzone, M.; et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci. Rep. 2016, 6, 33453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Wang, C.; Ning, C.; Liu, K.; Wang, X.; Liu, Z.; Sun, H.; Ma, X.; Sun, P.; Meng, Q. Hepatoprotection of auraptene from peels of citrus fruits against thioacetamide-induced hepatic fibrosis in mice by activating farnesoid X receptor. Food Funct. 2018, 9, 2684–2694. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Liu, F.; Cheng, Y.; Xiao, X.-R.; Hu, D.-D.; Tang, Y.-M.; Bao, W.-M.; Yang, J.-H.; Jiang, T.; Hu, J.-P.; et al. Celastrol Protects From Cholestatic Liver Injury Through Modulation of SIRT1-FXR Signaling. Mol. Cell. Proteom. 2019, 18, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterworth, R.F.; Norenberg, M.D.; Felipo, V.; Ferenci, P.; Albrecht, J.; Blei, A.T. Experimental models of hepatic encephalopathy: ISHEN guidelines. Liver Int. 2009, 29, 783–788. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Ruiz-Cordero, R.; Tong, X.Y.; Norenberg, M.D. Brain edema in acute liver failure: Role of neurosteroids. Arch. Biochem. Biophys. 2013, 536, 171–175. [Google Scholar] [CrossRef] [Green Version]
- Grant, S.; McMillin, M.; Frampton, G.; Petrescu, A.D.; Williams, E.; Jaeger, V.; Kain, J.; DeMorrow, S. Direct Comparison of the Thioacetamide and Azoxymethane Models of Type A Hepatic Encephalopathy in Mice. Gene Expr. 2018, 18, 171–185. [Google Scholar] [CrossRef]
- Horvatits, T.; Drolz, A.; Roedl, K.; Rutter, K.; Ferlitsch, A.; Fauler, G.; Trauner, M.; Fuhrmann, V. Serum bile acids as marker for acute decompensation and chronic liver failure in patients with non-cholestatic cirrhosis. Liver Int. 2017, 37, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.H.; Yamamoto, S.; Steers, J.; Sevlever, D.; Lin, W.; Shimojima, N.; Castanedes-Casey, M.; Genco, P.; Golde, T.; Richelson, E.; et al. Matrix metalloproteinase-9 contributes to brain extravasation and edema in fulminant hepatic failure mice. J. Hepatol. 2006, 44, 1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimojima, N.; Eckman, C.B.; McKinney, M.; Sevlever, D.; Yamamoto, S.; Lin, W.; Dickson, D.W.; Nguyen, J.H. Altered expression of zonula occludens-2 precedes increased blood-brain barrier permeability in a murine model of fulminant hepatic failure. J. Invest. Surg. 2008, 21, 101–108. [Google Scholar] [CrossRef] [PubMed]
- McMillin, M.A.; Frampton, G.A.; Seiwell, A.P.; Patel, N.S.; Jacobs, A.N.; DeMorrow, S. TGFβ1 exacerbates blood–brain barrier permeability in a mouse model of hepatic encephalopathy via upregulation of MMP9 and downregulation of claudin-5. Lab. Investig. 2015, 95, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Obara-Michlewska, M.; Ding, F.; Popek, M.; Verkhratsky, A.; Nedergaard, M.; Zielinska, M.; Albrecht, J. Interstitial ion homeostasis and acid-base balance are maintained in oedematous brain of mice with acute toxic liver failure. Neurochem. Int. 2018, 118, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Masago, K.; Kihara, Y.; Yanagida, K.; Hamano, F.; Nakagawa, S.; Niwa, M.; Shimizu, T. Lysophosphatidic acid receptor, LPA6, regulates endothelial blood-brain barrier function: Implication for hepatic encephalopathy. Biochem. Biophys. Res. Commun. 2018, 501, 1048–1054. [Google Scholar] [CrossRef]
- Albrecht, J.; Hilgier, W.; Januszewski, S.; Kapuściński, A.; Quack, G. Increase of the brain uptake index for L-ornithine in rats with hepatic encephalopathy. Neuroreport 1994, 5, 671–673. [Google Scholar] [CrossRef]
- Albrecht, J.; Hilgier, W.; Januszewski, S.; Quack, G. Contrasting effects of thioacetamide-induced liver damage on the brain uptake indices of ornithine, arginine and lysine: Modulation by treatment with ornithine aspartate. Metab. Brain Dis. 1996, 11, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Szumanska, G.; Albrecht, J. Lectin histochemistry of the rat brain following thioacetamide-induced hepatic failure. Mol. Chem. Neuropathol. 1997, 32, 163–177. [Google Scholar] [CrossRef]
- Jin, S.; Wang, X.-T.; Liu, L.; Yao, D.; Liu, C.; Zhang, M.; Guo, H.-F.; Liu, X.-D. P-glycoprotein and multidrug resistance-associated protein 2 are oppositely altered in brain of rats with thioacetamide-induced acute liver failure. Liver Int. 2013, 33, 274–282. [Google Scholar] [CrossRef]
- Balasubramaniyan, N.; Ananthanarayanan, M.; Suchy, F.J. Direct methylation of FXR by Set7/9, a lysine methyltransferase, regulates the expression of FXR target genes. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G937–G947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.T.; Tosun, M.; Jeong, H.-H.; Karakas, C.; Semerci, F.; Liu, Z.; Maletić-Savatić, M. Metabolomics of mammalian brain reveals regional differences. BMC Syst. Biol. 2018, 12, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripodi, V.; Contin, M.; Fernández, M.A.; Lemberg, A. Bile acids content in brain of common duct ligated rats. Ann. Hepatol 2012, 11, 930–934. [Google Scholar] [CrossRef]
- Fiorucci, S.; Rizzo, G.; Donini, A.; Distrutti, E.; Santucci, L. Targeting farnesoid X receptor for liver and metabolic disorders. Trends Mol. Med. 2007, 13, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, L.; Farre, R.; Trebicka, J.; Komuta, M.; Roskams, T.; Klein, S.; Elst, I.V.; Windmolders, P.; Vanuytsel, T.; Nevens, F.; et al. Obeticholic Acid, a Farnesoid X Receptor Agonist, Improves Portal Hypertension by Two Distinct Pathways in Cirrhotic Rats. Hepatology 2013, 59, 2286–2298. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Trauner, M.; Jansen, P.; Poupon, R. New paradigms in the treatment of hepatic cholestasis: From UDCA to FXR, PXR and beyond. J. Hepatol. 2015, 62, S25–S37. [Google Scholar] [CrossRef] [Green Version]
- Schousboe, A.; Drejer, J.; Hansen, G.H.; Meier, E. Cultured Neurons as Model Systems for Biochemical and Pharmacological Studies on Receptors for Neurotransmitter Amino Acids. Dev. Neurosci. 1985, 7, 252–262. [Google Scholar] [CrossRef]
- Zielińska, M.; Milewski, K.; Skowrońska, M.; Gajos, A.; Ziemińska, E.; Beręsewicz, A.; Albrecht, J. Induction of inducible nitric oxide synthase expression in ammonia-exposed cultured astrocytes is coupled to increased arginine transport by upregulated y(+)LAT2 transporter. J. Neurochem. 2015, 135, 1272–1281. [Google Scholar] [CrossRef]
- Skowrońska, K.; Obara-Michlewska, M.; Czarnecka, A.; Dąbrowska, K.; Zielińska, M.; Albrecht, J. Persistent Overexposure to N-Methyl-d-Aspartate (NMDA) Calcium-Dependently Downregulates Glutamine Synthetase, Aquaporin 4, and Kir4.1 Channel in Mouse Cortical Astrocytes. Neurotox. Res. 2019, 35, 271–280. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Czarnecka, A.; Milewski, K.; Jaźwiec, R.; Zielińska, M. Intracerebral Administration of S-Adenosylhomocysteine or S-Adenosylmethionine Attenuates the Increases in the Cortical Extracellular Levels of Dimethylarginines Without Affecting cGMP Level in Rats with Acute Liver Failure. Neurotox. Res. 2017, 31, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czarnecka, A.M.; Milewski, K.; Albrecht, J.; Zielińska, M. The Status of Bile Acids and Farnesoid X Receptor in Brain and Liver of Rats with Thioacetamide-Induced Acute Liver Failure. Int. J. Mol. Sci. 2020, 21, 7750. https://doi.org/10.3390/ijms21207750
Czarnecka AM, Milewski K, Albrecht J, Zielińska M. The Status of Bile Acids and Farnesoid X Receptor in Brain and Liver of Rats with Thioacetamide-Induced Acute Liver Failure. International Journal of Molecular Sciences. 2020; 21(20):7750. https://doi.org/10.3390/ijms21207750
Chicago/Turabian StyleCzarnecka, Anna Maria, Krzysztof Milewski, Jan Albrecht, and Magdalena Zielińska. 2020. "The Status of Bile Acids and Farnesoid X Receptor in Brain and Liver of Rats with Thioacetamide-Induced Acute Liver Failure" International Journal of Molecular Sciences 21, no. 20: 7750. https://doi.org/10.3390/ijms21207750