Clonal Hematopoiesis, Cardiovascular Diseases and Hematopoietic Stem Cells

Abstract
:1. Introduction
2. Clonal Hematopoiesis
3. DNMT3A
4. TET2
5. ASXL1
6. JAK2
7. HSPC Biology and Clonal Hematopoiesis
8. Potential Therapeutic Interventions for CH-Related Conditions
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Apoe | apolipoprotein E |
| ARCH | age-related clonal hematopoiesis |
| ASXL1 | additional sex combs-like 1 |
| CH | clonal hematopoiesis |
| CHIP | clonal hematopoiesis of indeterminate potential |
| CVD | cardiovascular diseases |
| DNMT3A | DNA methyltransferase 3 alpha |
| GNAS | GNAS complex locus |
| HSC | hematopoietic stem cells |
| HSPC | hematopoietic stem/progenitor cells |
| IFN-γ | interferon gamma |
| IL-1β | interleukin 1 beta |
| IL-6 | interleukin 6 |
| JAK2 | Janus kinase 2 |
| LDLR | low density lipoprotein receptor |
| NF-kB | nuclear factor kappa B |
| NLRP3 | NLR family pyrin domain containing 3 |
| PPM1D | protein phosphatase, Mg2+/Mn2+ dependent 1D |
| PV | polycythemia vera |
| ROS | reactive oxygen species |
| SIRT1 | sirtuin 1 |
| Stat5 | signal transducer and activator of transcription 5 |
| TET2 | ten-eleven translocation 2 |
| TNF-α | tumor necrosis factor alpha |
| TP53 | tumor protein p53 |
References
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.; Leopoulou, M.; Theofilis, P.; Antonopoulos, A.S.; Siasos, G.; Latsios, G.; Mystakidi, V.C.; Antoniades, C.; Tousoulis, D. A link between inflammation and thrombosis in atherosclerotic cardiovascular diseases: Clinical and therapeutic implications. Atherosclerosis 2020, 309, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Kobold, S. Inflammation: A common contributor to cancer, aging, and cardiovascular diseases-expanding the concept of cardio-oncology. Cardiovasc. Res. 2019, 115, 824–829. [Google Scholar] [CrossRef]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Blokzijl, F.; de Ligt, J.; Jager, M.; Sasselli, V.; Roerink, S.; Sasaki, N.; Huch, M.; Boymans, S.; Kuijk, E.; Prins, P.; et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016, 538, 260–264. [Google Scholar] [CrossRef]
- Yizhak, K.; Aguet, F.; Kim, J.; Hess, J.M.; Kübler, K.; Grimsby, J.; Frazer, R.; Zhang, H.; Haradhvala, N.J.; Rosebrock, D.; et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 2019, 364, eaaw0726:1–eaaw0726:10. [Google Scholar] [CrossRef]
- Moore, M.A.S. Overview of hemopoiesis and hemopoietic reconstruction. In Cell Therapy: Stem Cell Transplantation, Gene Therapy and Cellular Immunotherapy; Morstyn, G., Sheridan, S., Eds.; Cambridge University Press: New York, NY, USA, 1996; pp. 3–17. ISBN 0-521-47315-2. [Google Scholar]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Fey, M.F.; Liechti-Gallati, S.; von Rohr, A.; Borisch, B.; Theilkäs, L.; Schneider, V.; Oestreicher, M.; Nagel, S.; Ziemiecki, A.; Tobler, A. Clonality and X-inactivation patterns in hematopoietic cell populations detected by the highly informative M27 beta DNA probe. Blood 1994, 83, 931–938. [Google Scholar] [CrossRef] [Green Version]
- Busque, L.; Mio, R.; Mattioli, J.; Brais, E.; Blais, N.; Lalonde, Y.; Maragh, M.; Gilliland, D.G. Nonrandom X-inactivation patterns in normal females: Lyonization ratios vary with age. Blood 1996, 88, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Kustikova, O.S.; Geiger, H.; Li, Z.; Brugman, M.H.; Chambers, S.M.; Shaw, C.A.; Pike-Overzet, K.; de Ridder, D.; Staal, F.J.; von Keudell, G.; et al. Retroviral vector insertion sites associated with dominant hematopoietic clones mark “stemness” pathways. Blood 2007, 109, 1897–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.L.; Challen, G.A.; Birmann, B.M.; Druley, T.E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016, 7, 12484:1–12484:7. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedernhofer, L.J.; Gurkar, A.U.; Wang, Y.; Vijg, J.; Hoeijmakers, J.H.J.; Robbins, P.D. Nuclear Genomic Instability and Aging. Annu. Rev. Biochem. 2018, 87, 295–322. [Google Scholar] [CrossRef]
- Moriwaki, S.; Ray, S.; Tarone, R.E.; Kraemer, K.H.; Grossman, L. The effect of donor age on the processing of UV-damaged DNA by cultured human cells: Reduced DNA repair capacity and increased DNA mutability. Mutat. Res. 1996, 364, 117–123. [Google Scholar] [CrossRef]
- Goukassian, D.; Gad, F.; Yaar, M.; Eller, M.S.; Nehal, U.S.; Gilchrest, B.A. Mechanisms and implications of the age-associated decrease in DNA repair capacity. FASEB J. 2000, 14, 1325–1334. [Google Scholar] [CrossRef]
- Intano, G.W.; Cho, E.J.; McMahan, C.A.; Walter, C.A. Age-related base excision repair activity in mouse brain and liver nuclear extracts. J. Gerontol. A Biol. Sci. Med. Sci. 2003, 58, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Bourgoin, V.; Mollica, L.; Dubé, M.P.; Busque, L. Lineage restriction analyses in CHIP indicate myeloid bias for TET2 and multipotent stem cell origin for DNMT3A. Blood 2018, 132, 277–280. [Google Scholar] [CrossRef]
- Arends, C.M.; Galan-Sousa, J.; Hoyer, K.; Chan, W.; Jäger, M.; Yoshida, K.; Seemann, R.; Noerenberg, D.; Waldhueter, N.; Fleischer-Notter, H.; et al. Hematopoietic lineage distribution and evolutionary dynamics of clonal hematopoiesis. Leukemia 2018, 32, 1908–1919. [Google Scholar] [CrossRef]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lépine, G.; Mollica, L.; Szuber, N.; Dubé, M.P.; Busque, L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017, 130, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Desai, P.; Mencia-Trinchant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023. [Google Scholar] [CrossRef]
- Acuna-Hidalgo, R.; Sengul, H.; Steehouwer, M.; van de Vorst, M.; Vermeulen, S.H.; Kiemeney, L.A.L.M.; Veltman, J.A.; Gilissen, C.; Hoischen, A. Ultra-sensitive Sequencing Identifies High Prevalence of Clonal Hematopoiesis-Associated Mutations throughout Adult Life. Am. J. Hum. Genet. 2017, 101, 50–64. [Google Scholar] [CrossRef] [Green Version]
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Mendelson Cohen, N.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Structure and function of mammalian DNA methyltransferases. ChemBioChem. 2011, 12, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Dorsheimer, L.; Assmus, B.; Rasper, T.; Ortmann, C.A.; Ecke, A.; Abou-El-Ardat, K.; Schmid, T.; Brüne, B.; Wagner, S.; Serve, H.; et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019, 4, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Mas-Peiro, S.; Hoffmann, J.; Fichtlscherer, S.; Dorsheimer, L.; Rieger, M.A.; Dimmeler, S.; Vasa-Nicotera, M.; Zeiher, A.M. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur. Heart J. 2020, 41, 933–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bick, A.G.; Pirruccello, J.P.; Griffin, G.K.; Gupta, N.; Gabriel, S.; Saleheen, D.; Libby, P.; Kathiresan, S.; Natarajan, P. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation 2020, 141, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, M.; Park, H.J.; Celik, H.; Ostrander, E.L.; Reyes, J.M.; Guzman, A.; Rodriguez, B.; Lei, Y.; Lee, Y.; Ding, L.; et al. Loss of Dnmt3a Immortalizes Hematopoietic Stem Cells In Vivo. Cell Rep. 2018, 23, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sano, S.; Oshima, K.; Wang, Y.; Katanasaka, Y.; Sano, M.; Walsh, K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ. Res. 2018, 123, 335–341. [Google Scholar] [CrossRef]
- Leoni, C.; Montagner, S.; Rinaldi, A.; Bertoni, F.; Polletti, S.; Balestrieri, C.; Monticelli, S. Dnmt3a restrains mast cell inflammatory responses. Proc. Natl. Acad. Sci. USA 2017, 114, E1490–E1499. [Google Scholar] [CrossRef] [Green Version]
- Gamper, C.J.; Agoston, A.T.; Nelson, W.G.; Powell, J.D. Identification of DNA methyltransferase 3a as a T cell receptor-induced regulator of Th1 and Th2 differentiation. J. Immunol. 2009, 183, 2267–2276. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Zhou, B.; Zhang, Y.; Nguyen, E.T.; Du, J.; Glosson, N.L.; Kaplan, M.H. DNA methyltransferase 3a limits the expression of interleukin-13 in T helper 2 cells and allergic airway inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, 541–546. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef] [Green Version]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langemeijer, S.M.; Kuiper, R.P.; Berends, M.; Knops, R.; Aslanyan, M.G.; Massop, M.; Stevens-Linders, E.; van Hoogen, P.; van Kessel, A.G.; Raymakers, R.A.; et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat. Genet. 2009, 41, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Cremer, S.; Kirschbau, K.; Berkowitsch, A.; John, D.; Kiefer, K.; Dorsheimer, L.; Wagner, J.; Rasper, T.; Abou-El-Ardat, K.; Assmus, B.; et al. Multiple Somatic Mutations for Clonal Hematopoiesis Are Associated With Increased Mortality in Patients With Chronic Heart Failure. Circ. Genom. Precis. Med. 2020, 13, e003003. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Thompson, E.C.; Hastie, R.; Tsangaratou, A.; Rajewsky, K.; Koralov, S.B.; Rao, A. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 14566–14571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Quivoron, C.; Couronné, L.; Della Valle, V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; Do Cruzeiro, M.; Delhommeau, F.; Arnulf, B.; Stern, M.H.; et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 2011, 20, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Cai, X.; Cai, C.L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.C.; Xu, M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Lee, J.; Chrysanthou, S.; Zhao, Y.; Josephs, K.; Sato, H.; Teruya-Feldstein, J.; Zheng, D.; Dawlaty, M.M.; Ito, K. Non-catalytic Roles of Tet2 Are Essential to Regulate Hematopoietic Stem and Progenitor Cell Homeostasis. Cell Rep. 2019, 28, 2480–2490. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [Green Version]
- Sano, S.; Oshima, K.; Wang, Y.; MacLauchlan, S.; Katanasaka, Y.; Sano, M.; Zuriaga, M.A.; Yoshiyama, M.; Goukassian, D.; Cooper, M.A.; et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J. Am. Coll. Cardiol. 2018, 71, 875–886. [Google Scholar] [CrossRef]
- Wang, Y.; Sano, S.; Yura, Y.; Ke, Z.; Sano, M.; Oshima, K.; Ogawa, H.; Horitani, K.; Min, K.D.; Miura-Yura, E.; et al. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. J.C.I. Insight 2020, 5, e135204:1–e135204:16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Leventhal, M.J.; Bao, E.L.; Nasser, J.; Zekavat, S.M.; Szeto, M.D.; Laurie, C.; et al. Inherited Causes of Clonal Hematopoiesis of Indeterminate Potential in TOPMed Whole Genomes. bioRxiv 2014. preprint. [Google Scholar] [CrossRef] [Green Version]
- Itzykson, R.; Fenaux, P. Epigenetics of myelodysplastic syndromes. Leukemia 2014, 28, 497–506. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Gao, J.; Adli, M.; Dey, A.; Trimarchi, T.; Chung, Y.R.; Kuscu, C.; Hricik, T.; Ndiaye-Lobry, D.; Lafave, L.M.; et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 2013, 210, 2641–2659. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, Z.; He, Y.; Pan, F.; Chen, S.; Rhodes, S.; Nguyen, L.; Yuan, J.; Jiang, L.; Yang, X.; et al. Loss of Asxl1 leads to myelodysplastic syndrome-like disease in mice. Blood 2014, 123, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramani, A.; Larjo, A.; Bassein, J.A.; Chang, X.; Hastie, R.B.; Togher, S.M.; Lähdesmäki, H.; Rao, A. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat. Commun. 2015, 6, 7307:1–7307:15. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Kurtenbach, S.; Guo, Y.; Lohse, I.; Durante, M.A.; Li, J.; Li, Z.; Al-Ali, H.; Li, L.; Chen, Z.; et al. Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood 2018, 131, 328–341. [Google Scholar] [CrossRef]
- Nagase, R.; Inoue, D.; Pastore, A.; Fujino, T.; Hou, H.A.; Yamasaki, N.; Goyama, S.; Saika, M.; Kanai, A.; Sera, Y.; et al. Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J. Exp. Med. 2018, 215, 1729–1747. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernig, G.; Mercher, T.; Okabe, R.; Levine, R.L.; Lee, B.H.; Gilliland, D.G. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood 2006, 107, 4274–4281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacout, C.; Pisani, D.F.; Tulliez, M.; Gachelin, F.M.; Vainchenker, W.; Villeval, J.L. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood 2006, 108, 1652–1660. [Google Scholar] [CrossRef] [PubMed]
- Tiedt, R.; Hao-Shen, H.; Sobas, M.A.; Looser, R.; Dirnhofer, S.; Schwaller, J.; Skoda, R.C. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood 2008, 111, 3931–3940. [Google Scholar] [CrossRef] [PubMed]
- Akada, H.; Yan, D.; Zou, H.; Fiering, S.; Hutchison, R.E.; Mohi, M.G. Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood 2010, 115, 3589–3597. [Google Scholar] [CrossRef] [Green Version]
- Marty, C.; Lacout, C.; Martin, A.; Hasan, S.; Jacquot, S.; Birling, M.C.; Vainchenker, W.; Villeval, J.L. Myeloproliferative neoplasm induced by constitutive expression of JAK2V617F in knock-in mice. Blood 2010, 116, 783–787. [Google Scholar] [CrossRef]
- Yan, D.; Hutchison, R.E.; Mohi, G. Critical requirement for Stat5 in a mouse model of polycythemia vera. Blood 2012, 119, 3539–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spivak, J.L.; Merchant, A.; Williams, D.M.; Rogers, O.; Zhao, W.; Duffield, A.; Resar, L.S.; Moliterno, A.R.; Zhao, Z.J. Thrombopoietin is required for full phenotype expression in a JAK2V617F transgenic mouse model of polycythemia vera. PLoS ONE 2020, 15, e0232801:1–e0232801:17. [Google Scholar] [CrossRef]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaan8292:1–eaan8292:10. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.J.; Peloso, G.M.; Yu, H.; Butterworth, A.S.; Wang, X.; Mahajan, A.; Saleheen, D.; Emdin, C.; Alam, D.; Alves, A.C.; et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat. Genet. 2017, 49, 1758–1766. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Liu, W.; Fidler, T.; Wang, Y.; Tang, Y.; Woods, B.; Welch, C.; Cai, B.; Silvestre-Roig, C.; Ai, D.; et al. Macrophage Inflammation, Erythrophagocytosis, and Accelerated Atherosclerosis in Jak2V617F Mice. Circ. Res. 2018, 123, e35–e47. [Google Scholar] [CrossRef] [PubMed]
- Oguro, H. The Roles of Cholesterol and Its Metabolites in Normal and Malignant Hematopoiesis. Front Endocrinol. 2019, 10, 204:1–204:15. [Google Scholar] [CrossRef] [PubMed]
- Morgan, P.K.; Fang, L.; Lancaster, G.I.; Murphy, A.J. Hematopoiesis is regulated by cholesterol efflux pathways and lipid rafts: Connections with cardiovascular diseases. J. Lipid Res. 2020, 61, 667–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, P.; Courties, G.; Wei, Y.; Leuschner, F.; Gorbatov, R.; Robbins, C.S.; Iwamoto, Y.; Thompson, B.; Carlson, A.L.; Heidt, T.; et al. Myocardial infarction accelerates atherosclerosis. Nature 2012, 487, 325–329. [Google Scholar] [CrossRef] [Green Version]
- Perdigones, N.; Perin, J.C.; Schiano, I.; Nicholas, P.; Biegel, J.A.; Mason, P.J.; Babushok, D.V.; Bessler, M. Clonal hematopoiesis in patients with dyskeratosis congenita. Am. J. Hematol. 2016, 91, 1227–1233. [Google Scholar] [CrossRef] [Green Version]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef] [Green Version]
- Parmar, K.; Mauch, P.; Vergilio, J.A.; Sackstein, R.; Down, J.D. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc. Natl. Acad. Sci. USA 2007, 104, 5431–5436. [Google Scholar] [CrossRef] [Green Version]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Vener, C.; Novembrino, C.; Catena, F.B.; Fracchiolla, N.S.; Gianelli, U.; Savi, F.; Radaelli, F.; Fermo, E.; Cortelezzi, A.; Lonati, S.; et al. Oxidative stress is increased in primary and post-polycythemia vera myelofibrosis. Exp. Hematol. 2010, 38, 1058–1065. [Google Scholar] [CrossRef]
- Musolino, C.; Allegra, A.; Saija, A.; Alonci, A.; Russo, S.; Spatari, G.; Penna, G.; Gerace, D.; Cristani, M.; David, A.; et al. Changes in advanced oxidation protein products, advanced glycation end products, and s-nitrosylated proteins, in patients affected by polycythemia vera and essential thrombocythemia. Clin. Biochem. 2012, 45, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Juntilla, M.M.; Patil, V.D.; Calamito, M.; Joshi, R.P.; Birnbaum, M.J.; Koretzky, G.A. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood 2010, 115, 4030–4038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, E.K.; Izukawa, T.; Young, S.; Rosen, G.; Jamali, M.; Zhang, L.; Johnson, D.; Bain, E.; Hilland, J.; Ferrone, C.K.; et al. Comorbid and inflammatory characteristics of genetic subtypes of clonal hematopoiesis. Blood Adv. 2019, 3, 2482–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abegunde, S.O.; Buckstein, R.; Wells, R.A.; Rauh, M.J. An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Exp. Hematol. 2018, 59, 60–65. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Belyavsky, A.V. Niches of Hematopoietic Stem Cells in Bone Marrow. Mol Biol 2019, 53, 1012–1019. [Google Scholar] [CrossRef]
- Janel, A.; Dubois-Galopin, F.; Bourgne, C.; Berger, J.; Tarte, K.; Boiret-Dupré, N.; Boisgard, S.; Verrelle, P.; Déchelotte, P.; Tournilhac, O.; et al. The chronic lymphocytic leukemia clone disrupts the bone marrow microenvironment. Stem Cells Dev. 2014, 23, 2972–2982. [Google Scholar] [CrossRef]
- Vanegas, N.P.; Vernot, J.P. Loss of quiescence and self-renewal capacity of hematopoietic stem cell in an in vitro leukemic niche. Exp. Hematol. Oncol. 2017, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Verovskaya, E.; Calero-Nieto, F.; Reynaud, D.; Zhang, S.Y.; Herault, A.; Bakker, S.; Pietras, E.; Svendsen, A.F.; Wang, X.; Kinston, S.; et al. Inflammatory changes in the bone marrow microenvironment drive both niche and blood system remodeling during aging. Exp. Hematol. 2018, 64, S43–S44. [Google Scholar] [CrossRef]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Zhu, H.; Huang, J.; Zhu, Y.; Hong, M.; Zhu, H.; Zhang, J.; Li, S.; Yang, L.; Lian, Y.; et al. The synergy of Vitamin C with decitabine activates TET2 in leukemic cells and significantly improves overall survival in elderly patients with acute myeloid leukemia. Leuk. Res. 2018, 66, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Sun, J.; He, X.; Zhu, Y.; Ding, Z.; Dong, H.; Feng, Y.; Du, J.; Wang, H.; Wu, X.; Zhang, L.; et al. SIRT1 Activation Disrupts Maintenance of Myelodysplastic Syndrome Stem and Progenitor Cells by Restoring TET2 Function. Cell Stem Cell 2018, 23, 355–369. [Google Scholar] [CrossRef] [Green Version]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Miranda, M.X.; van Tits, L.J.; Lohmann, C.; Arsiwala, T.; Winnik, S.; Tailleux, A.; Stein, S.; Gomes, A.P.; Suri, V.; Ellis, J.L.; et al. The Sirt1 activator SRT3025 provides atheroprotection in Apoe-/-mice by reducing hepatic Pcsk9 secretion and enhancing Ldlr expression. Eur. Heart J. 2015, 36, 51–59. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, W.; Wang, W.; Fidler, T.; Woods, B.; Levine, R.L.; Tall, A.R.; Wang, N. Inhibition of JAK2 Suppresses Myelopoiesis and Atherosclerosis in Apoe-/-Mice. Cardiovasc. Drugs Ther. 2020, 34, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Jia, J.; Yu, Z.; Duanmu, Z.; He, H.; Chen, S.; Qu, C. Inhibition of JAK2/STAT3/SOCS3 signaling attenuates atherosclerosis in rabbit. BMC Cardiovasc. Disord. 2020, 20, 133:1–133:9. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, B.; Gupta, N.; Schnoeder, T.M.; Oelschlegel, A.M.; Shahzad, K.; Goldschmidt, J.; Philipsen, L.; Weinert, S.; Ghosh, A.; Saalfeld, F.C.; et al. JAK2-V617F promotes venous thrombosis through β1/β2 integrin activation. J. Clin. Investig. 2018, 128, 4359–4371. [Google Scholar] [CrossRef]
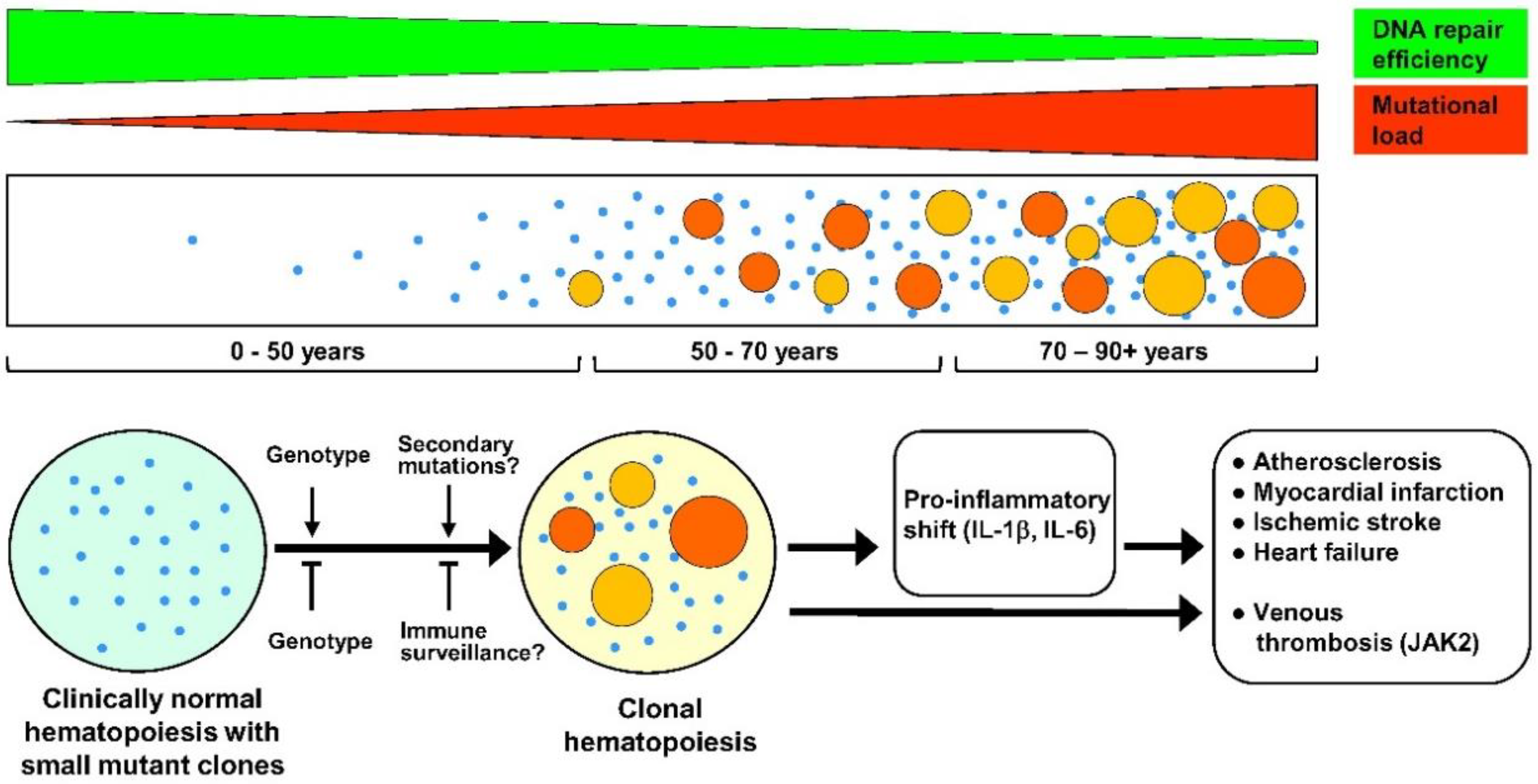

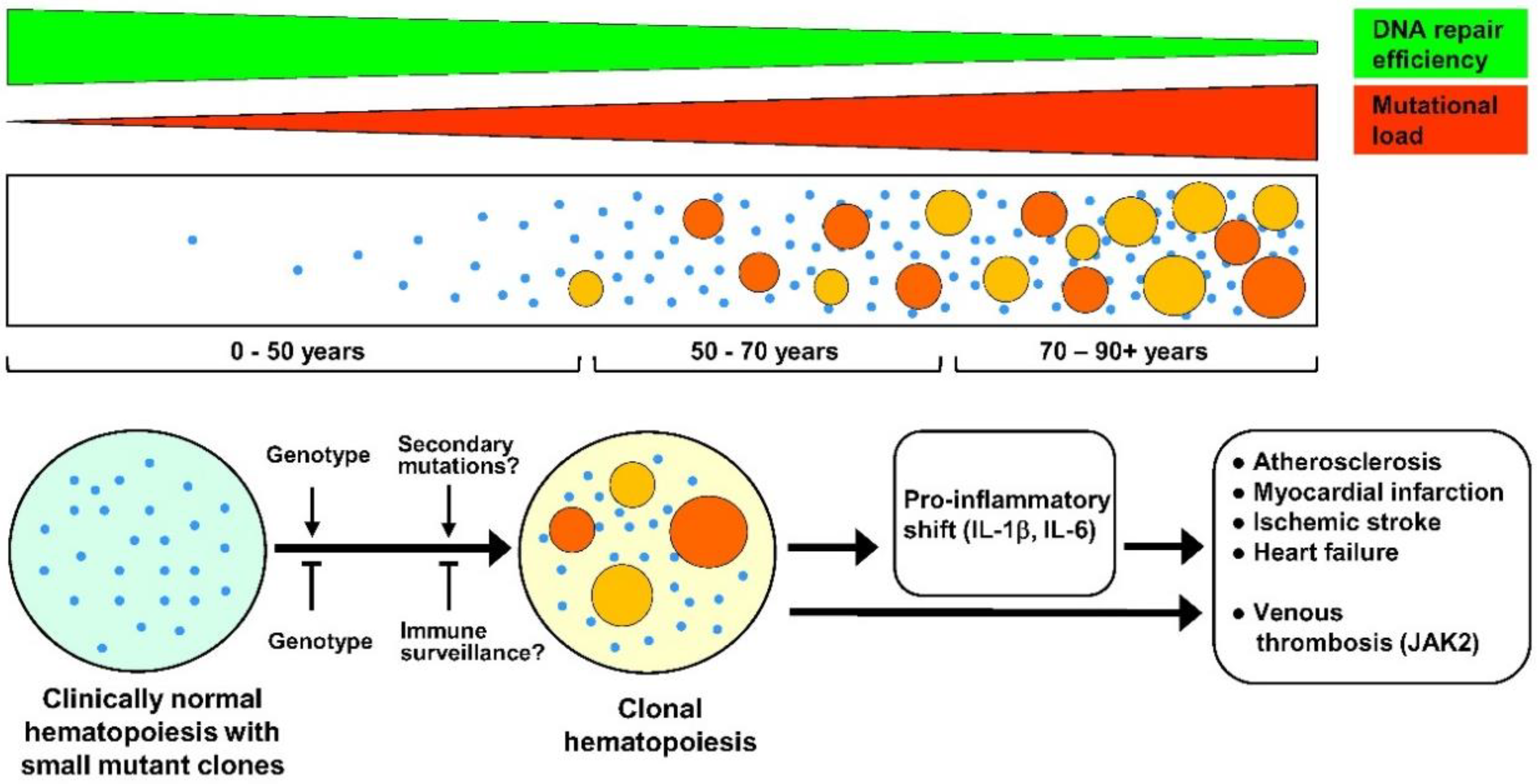{kind=link}
| Gene | Clinical Effects of Gene Mutations | Effects of Gene Deficiency/Mutations in Experimental Models |
|---|---|---|
| DNMT3A (DNA methyltransferase 3 alpha) | Increased risks of cardiovascular diseases, in particular: coronary heart disease, ischemic stroke, early onset myocardial infarction, coronary artery calcification. Increased risks of adverse outcomes and mortality in patients with chronic heart failure. | Upregulation of multipotency genes, strong enhancement of hematopoietic stem cell (HSC) self-renewal and expansion. Pro-inflammatory shift, cardiac hypertrophy and fibrosis, diminished cardiac function. |
| TET2 (ten-eleven translocation 2) | Increased risks of cardiovascular diseases, in particular: coronary heart disease, ischemic stroke, early onset myocardial infarction, coronary artery calcification. Increased risks of adverse outcomes and mortality in patients with chronic heart failure. | Enhancement of hematopoietic stem cell self-renewal, enlargement of the HSC compartment, myeloid shift with eventual myeloproliferation. Accelerated atherosclerosis and significant increase in atherosclerotic plaque size, proatherogenic interleukin 1β secretion in macrophages. Worsening of cardiac remodeling and function in experimental models of heart failure. Cardiac dysfunction due to hypertrophy and fibrosis, enhanced inflammatory signature in heart macrophages. |
| ASXL1 (additional sex combs-like 1) | Increased risks of coronary heart disease and ischemic stroke. | C-terminally truncated forms produce enlarged hematopoietic stem cell pool and increased susceptibility to leukemic transformation. No data as yet on effects on atherosclerosis and cardiovascular diseases. |
| JAK2 (Janus kinase 2) | Myeloproliferative diseases, polycythemia vera. Enhanced production of erythrocytes and thrombocytes. Increased risks of venous thrombosis. Increased risks of coronary heart disease. | Development of polycythemia vera-like pathology in mice, increased propensity for neutrophil extracellular trap formation. Increased atherosclerosis with early lesion formation and increased complexity in advanced state. Enhanced production of proinflammatory cytokines and chemokines, in particular interleukin 6 and interleukin 1β. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kandarakov, O.; Belyavsky, A. Clonal Hematopoiesis, Cardiovascular Diseases and Hematopoietic Stem Cells. Int. J. Mol. Sci. 2020, 21, 7902. https://doi.org/10.3390/ijms21217902
Kandarakov O, Belyavsky A. Clonal Hematopoiesis, Cardiovascular Diseases and Hematopoietic Stem Cells. International Journal of Molecular Sciences. 2020; 21(21):7902. https://doi.org/10.3390/ijms21217902
Chicago/Turabian StyleKandarakov, Oleg, and Alexander Belyavsky. 2020. "Clonal Hematopoiesis, Cardiovascular Diseases and Hematopoietic Stem Cells" International Journal of Molecular Sciences 21, no. 21: 7902. https://doi.org/10.3390/ijms21217902
APA StyleKandarakov, O., & Belyavsky, A. (2020). Clonal Hematopoiesis, Cardiovascular Diseases and Hematopoietic Stem Cells. International Journal of Molecular Sciences, 21(21), 7902. https://doi.org/10.3390/ijms21217902