Design, Synthesis, and In Vitro/In Vivo Anti-Cancer Activities of Novel (20S)-10,11-Methylenedioxy-Camptothecin Heterocyclic Derivatives

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
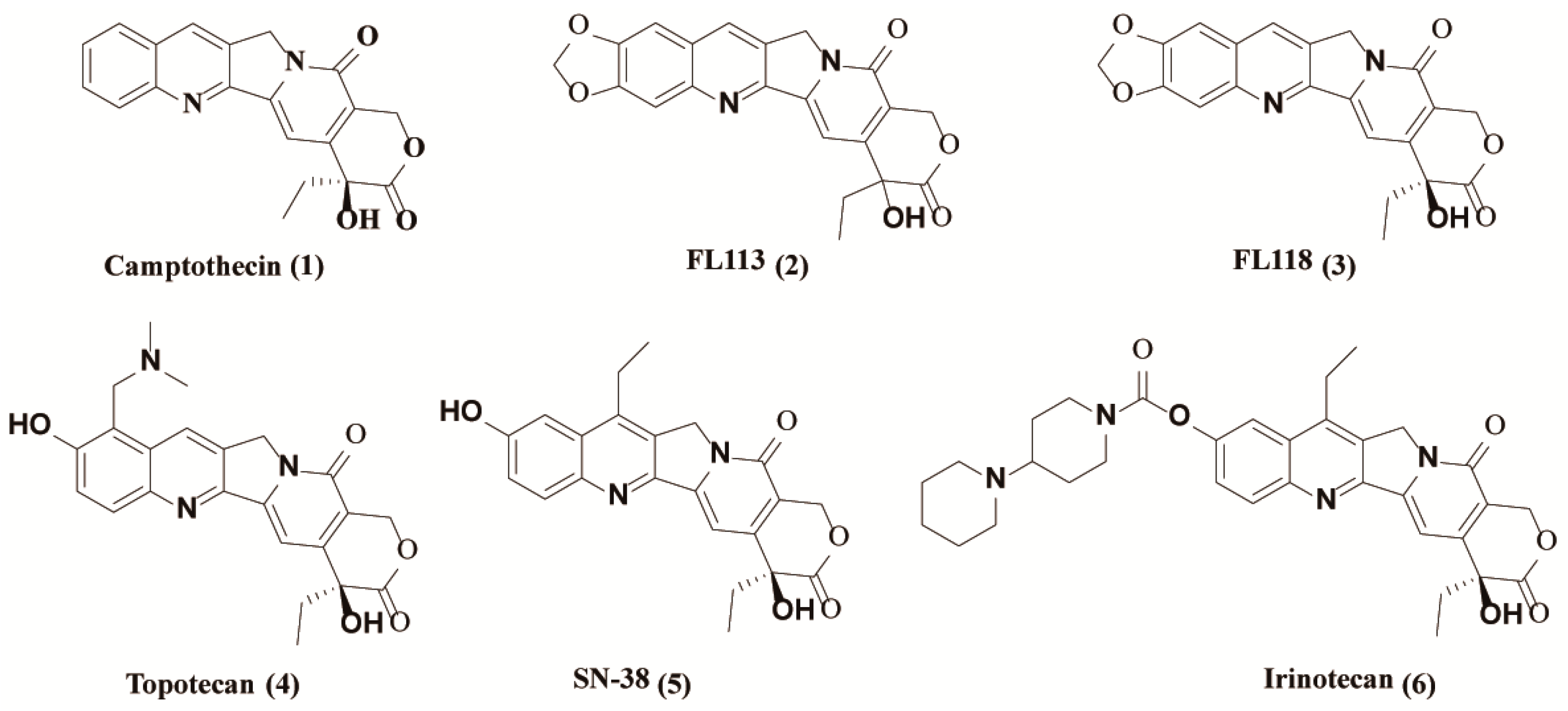
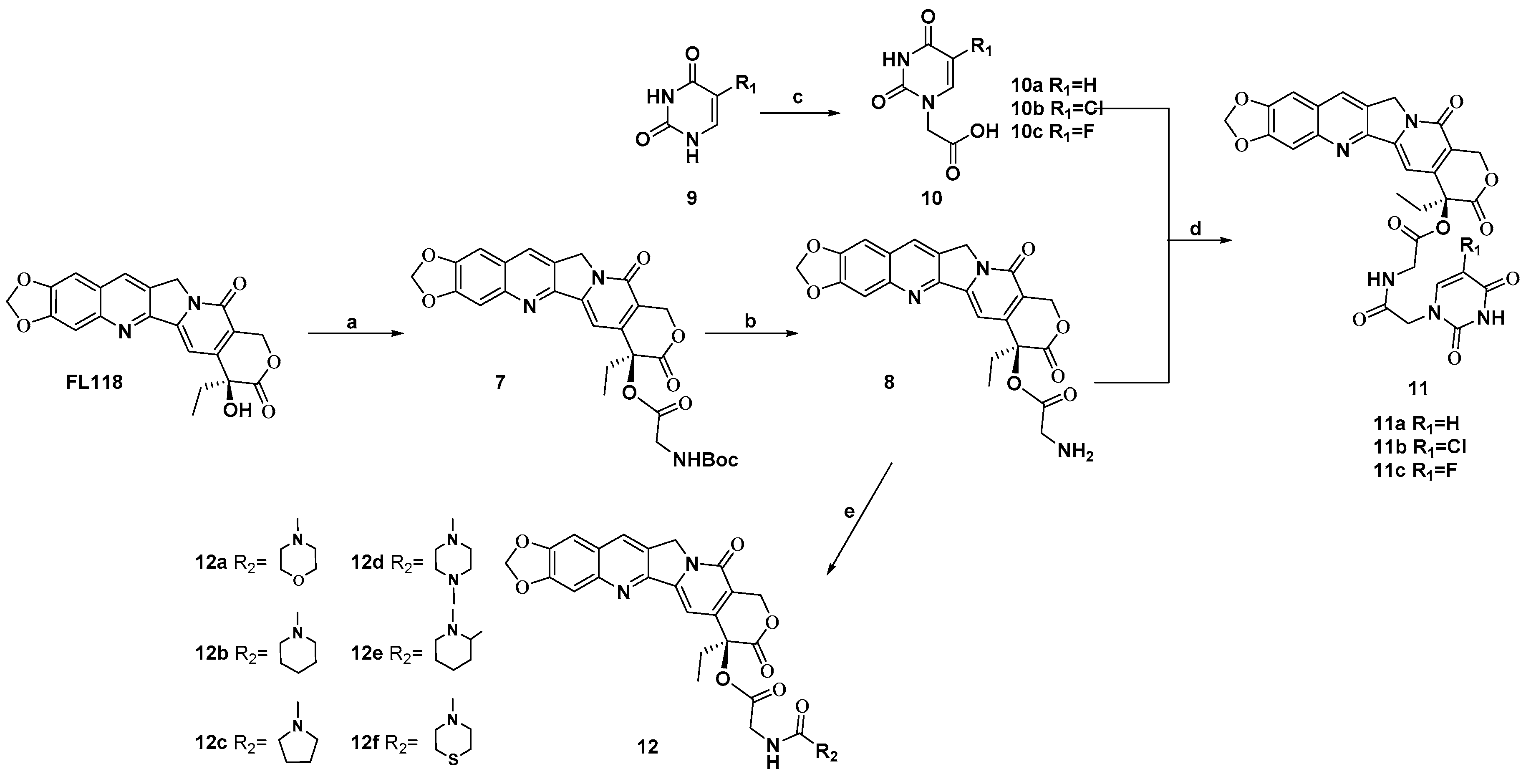
2.1. Chemistry
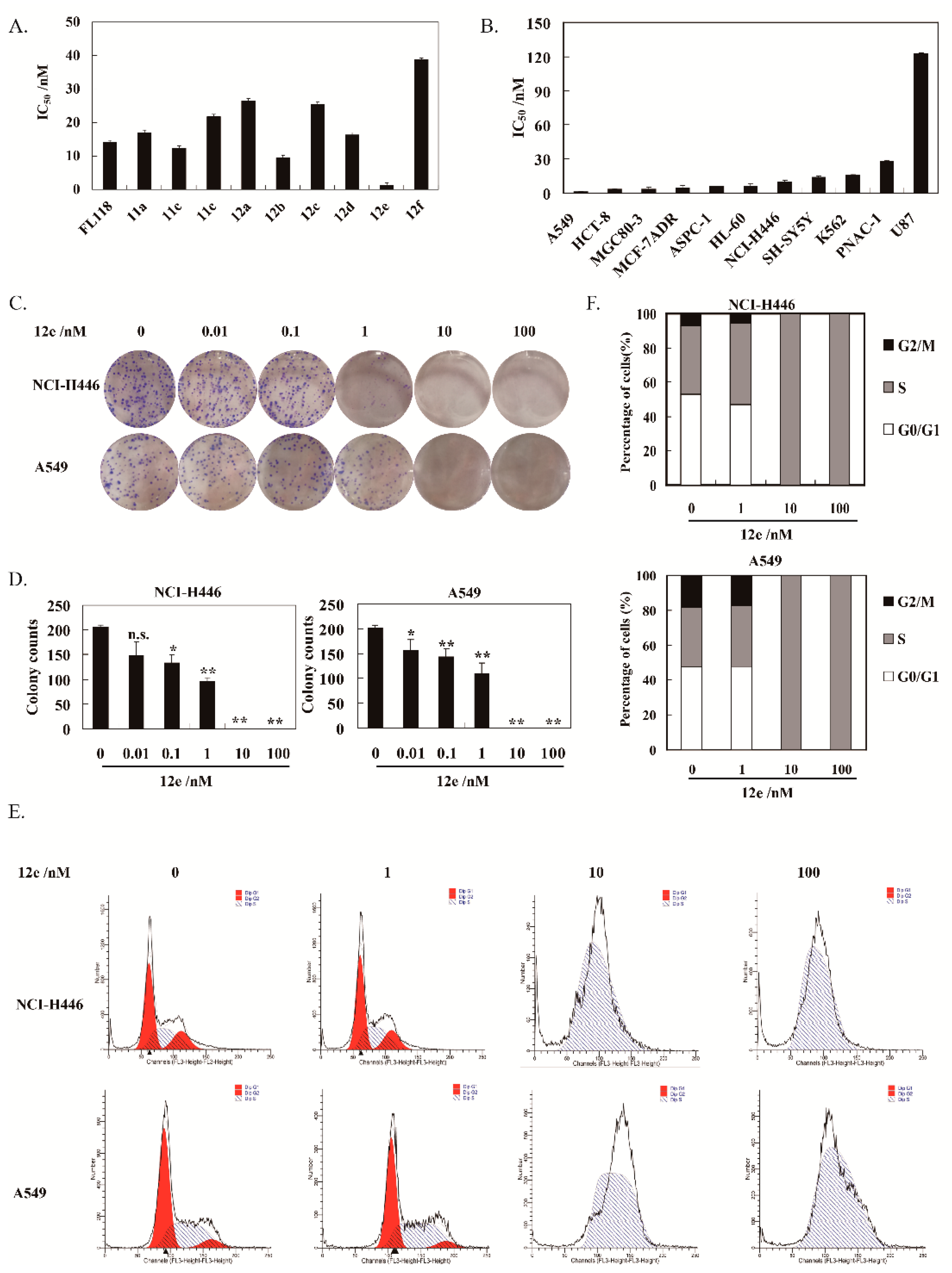
2.2. 12e Inhibits the Proliferation of Various Tumor Cells
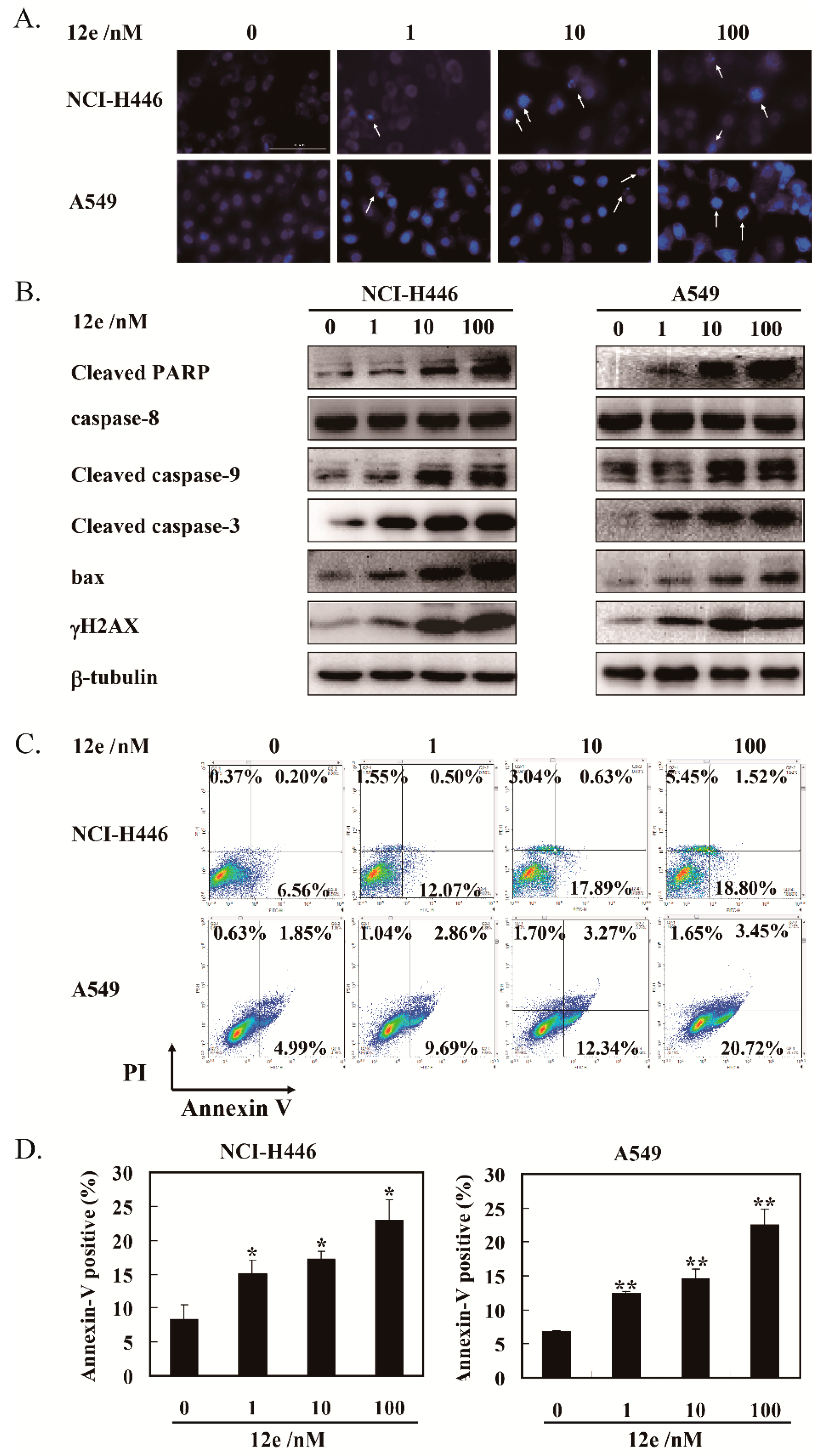
2.3. 12e Induces Cell Apoptosis of A549 Cells and NCI-H446 Cells
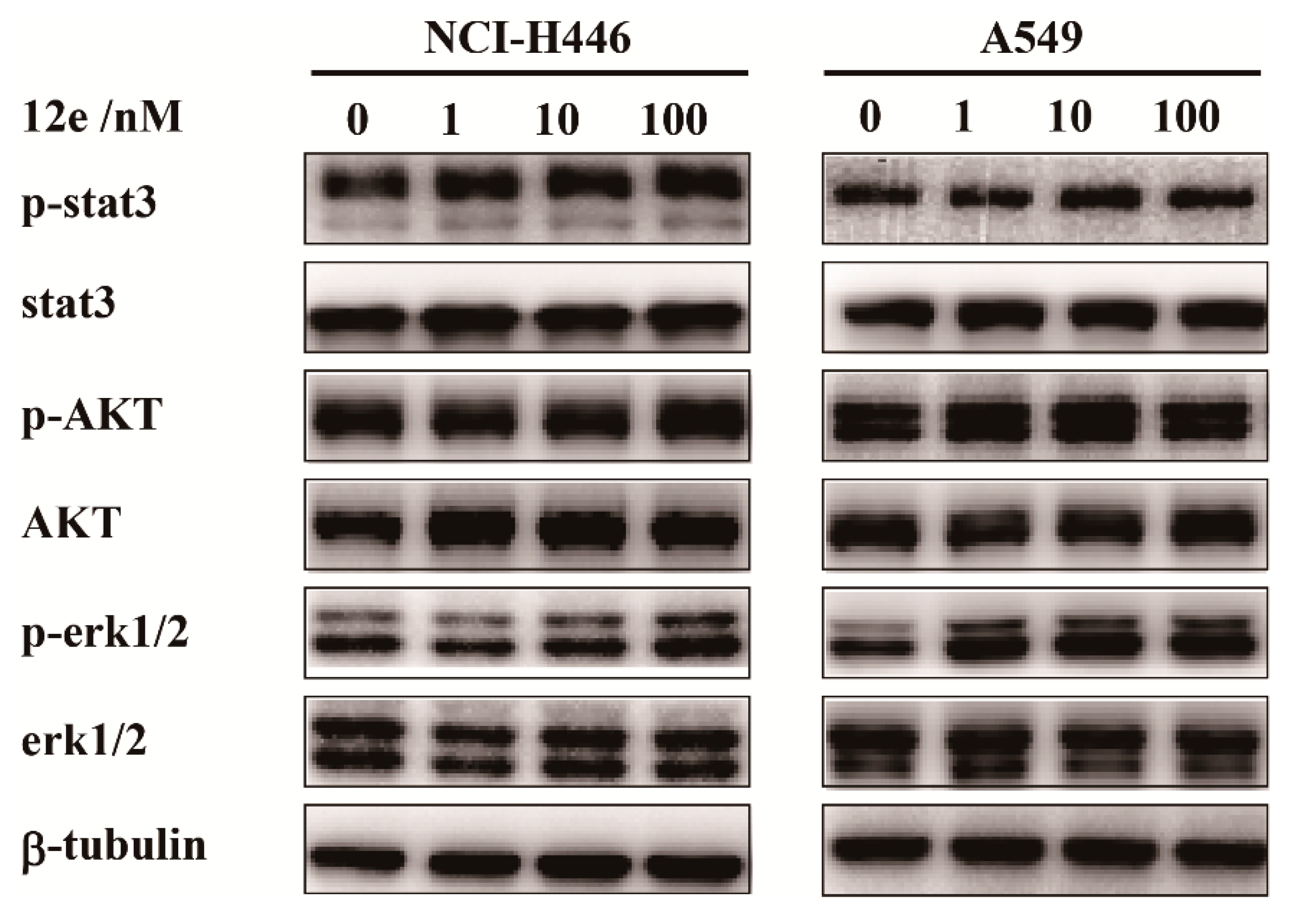
2.4. 12e does not Impair Cell Proliferation Signaling in A549 Cells and NCI-H446 Cells
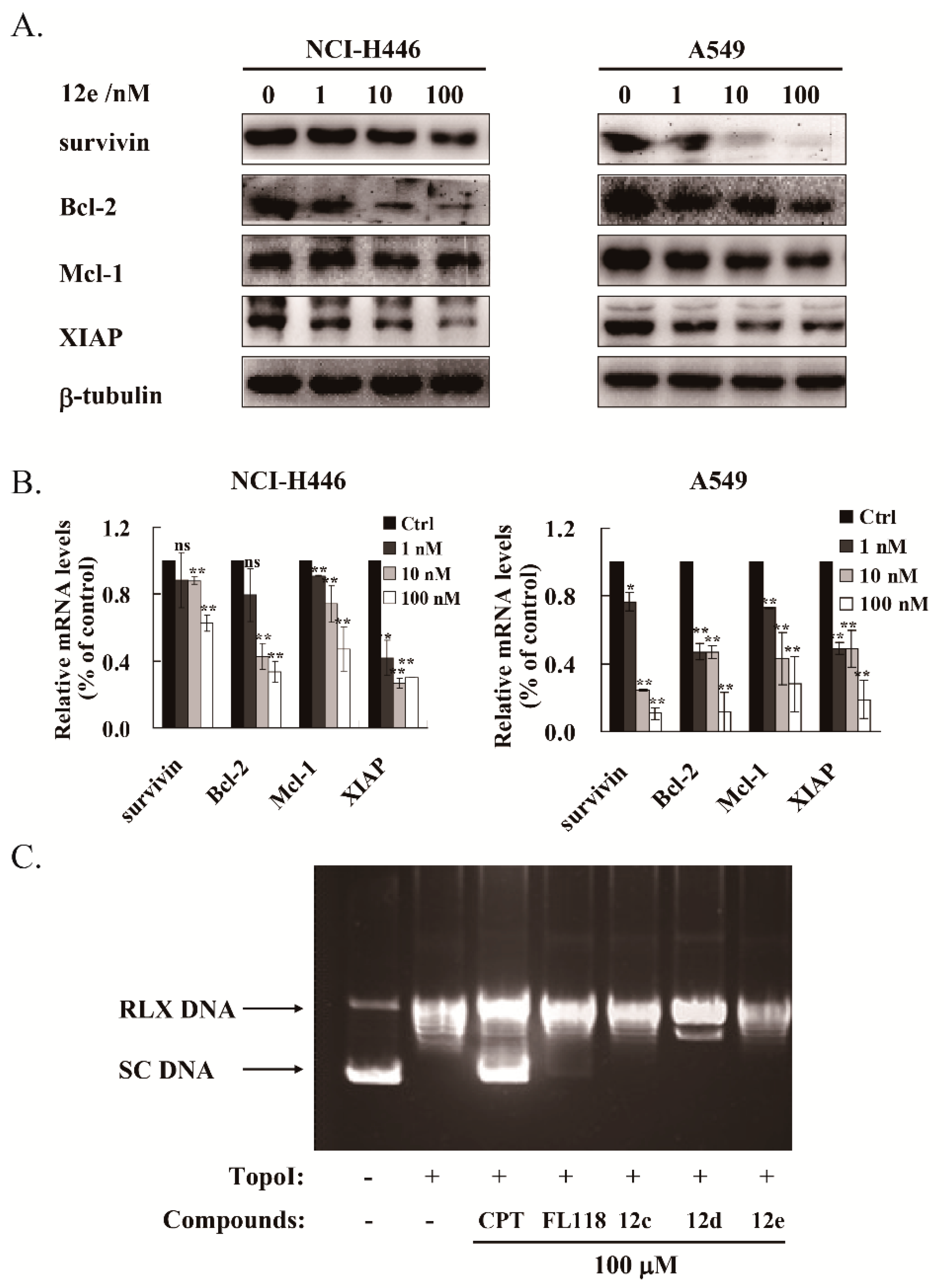
2.5. 12e Reduces the Transcription and Expression of Multiple Anti-Apoptotic Proteins
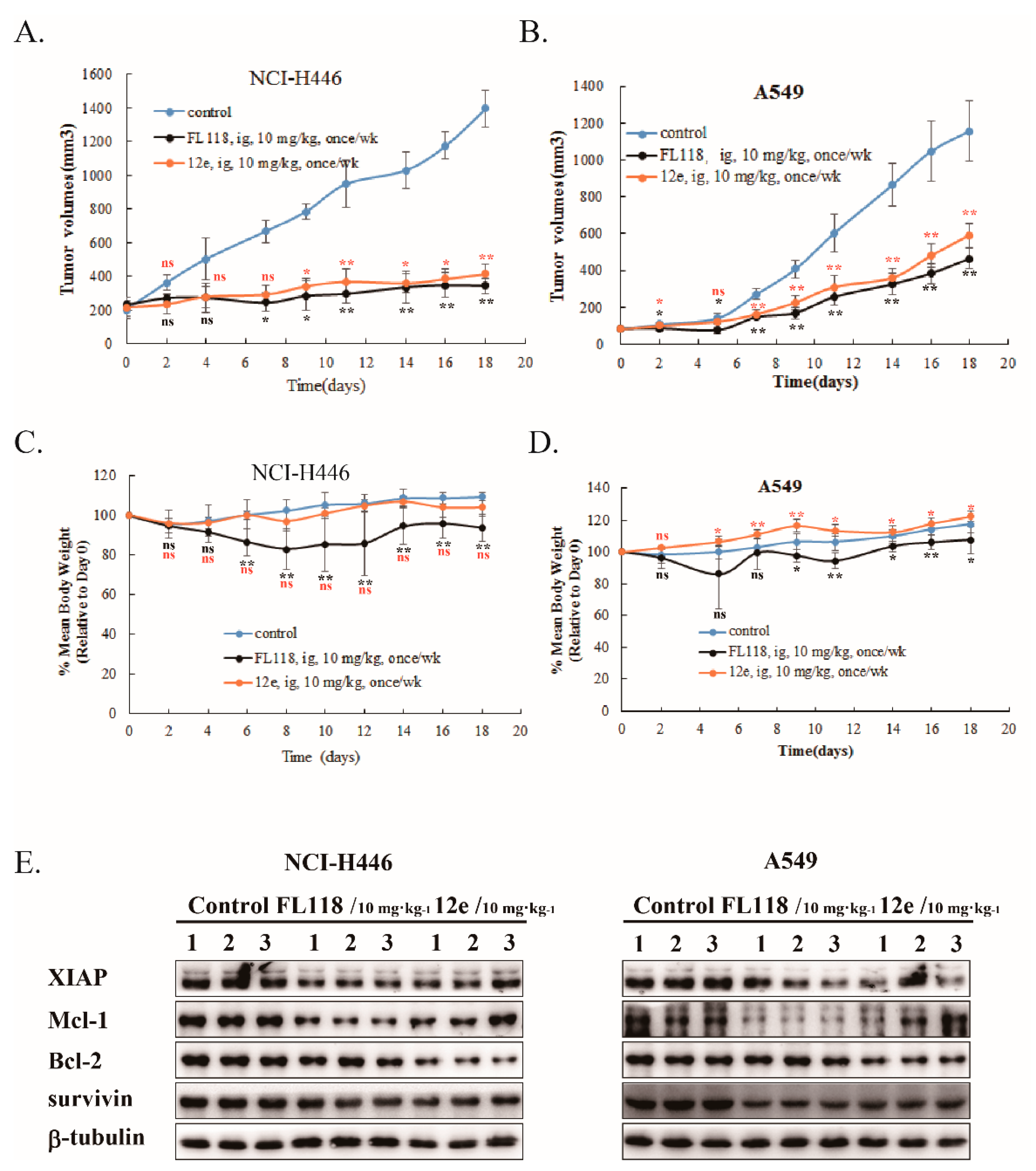
2.6. 12e Suppresses the Growth of Lung Cancer Cells Xenograft In Vivo
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. Materials and Methods
4.1.2. Synthesis of FL118
4.1.3. Synthesis of (S)-7-ethyl-8,11-dioxo-7,8,11,13-tetrahydro-10H-[1,3]dioxolo[4,5-g]pyrano[3′,4′:6,7] indolizino[1,2-b]quinolin-7-yl (tert-butoxycarbonyl)glycinate (7)
4.1.4. Synthesis of (S)-7-ethyl-8,11-dioxo-7,8,11,13-tetrahydro-10H-[1,3]dioxolo[4,5-g]pyrano[3′,4′:6,7] indolizino[1,2-b]quinolin-7-yl glycinate (8)
4.1.5. General Procedure for the Synthesis of Compounds 11a–11c
(S)-7-ethyl-8,11-dioxo-7,8,11,13-tetrahydro-10H-[1,3]dioxolo[4,5-g]pyrano[3′,4′:6,7] indolizino[1,2-b]quinolin-7-yl (2-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetyl)glycinate (11a)
(S)-7-ethyl-8,11-dioxo-7,8,11,13-tetrahydro-10H-[1,3]dioxolo[4,5-g]pyrano[3′,4′:6,7]indolizino[1,2-b]quinolin-7-yl (2-(5-chloro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetyl)glycinate (11b)
(S)-7-ethyl-8,11-dioxo-7,8,11,13-tetrahydro-10H-[1,3]dioxolo[4,5-g]pyrano[3′,4′:6,7]indolizino[1,2-b]quinolin-7-yl (2-(5-fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetyl)glycinate (11c)
4.1.6. General Procedure for the Synthesis of Compounds 12a–12f
(S)-7-ethyl-8,11-dioxo-7,8,11,13-tetrahydro-10H-[1,3]dioxolo[4,5-g]pyrano[3′,4′:6,7]indolizino[1,2-b]quinolin-7-yl (morpholine-4-carbonyl)glycinate (12a)
(S)-7-ethyl-8,11-dioxo-7,8,11,13-tetrahydro-10H-[1,3]dioxolo[4,5-g]pyrano[3′,4′:6,7]indolizino[1,2-b]quinolin-7-yl (piperidine-1-carbonyl)glycinate (12b)
(S)-7-ethyl-8,11-dioxo-7,8,11,13-tetrahydro-10H-[1,3]dioxolo[4,5-g]pyrano[3′,4′:6,7]indolizino[1,2-b]quinolin-7-yl (pyrrolidine-1-carbonyl)glycinate (12c)
(S)-7-ethyl-8,11-dioxo-7,8,11,13-tetrahydro-10H-[1,3]dioxolo[4,5-g]pyrano[3′,4′:6,7]indolizino[1,2-b]quinolin-7-yl (4-methylpiperazine-1-carbonyl)glycinate (12d)
(S)-7-ethyl-8,11-dioxo-7,8,11,13-tetrahydro-10H-[1,3]dioxolo[4,5-g]pyrano[3′,4′:6,7]indolizino[1,2-b]quinolin-7-yl (2-methylpiperidine-1-carbonyl)glycinate (12e)
(S)-7-ethyl-8,11-dioxo-7,8,11,13-tetrahydro-10H-[1,3]dioxolo[4,5-g]pyrano[3′,4′:6,7]indolizino[1,2-b]quinolin-7-yl (thiomorpholine-4-carbonyl)glycinate (12f)
4.2. Biological Evalutation
4.2.1. Cell Culture
4.2.2. Cell Proliferation Assay
4.2.3. Hoechst 33342 Staining
4.2.4. Clonogenic Assay
4.2.5. Annexin V-FITC/PI Double-Staining Assay
4.2.6. Cell Cycle Analysis
4.2.7. Western Blotting
4.2.8. RT-PCR
4.2.9. DNA Relaxation Assay
4.2.10. In Vivo Studies
4.2.11. Data Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Laversanne, M.; Brewster, D.; Mbalawa, C.G.; Kohler, B.; Piñeros, M.; Steliarova-Foucher, E.; Swaminathan, R.; Antoni, S.; et al. Cancer Incidence in Five Continents: Inclusion criteria, highlights from Volume X and the global status of cancer registration. Int. J. Cancer 2015, 137, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, M.M.; Berral-González, A.; Bueno-Fortes, S.; Alonso-López, D.; Rivas, J.D.L. Mining Drug-Target Associations in Cancer: Analysis of Gene Expression and Drug Activity Correlations. Biomolecules 2020, 10, 667. [Google Scholar] [CrossRef] [PubMed]
- Wall, M.E.; Wani, M.C.; Cook, C.E.; Palmer, K.H.; McPhail, A.T.; Sim, G.A. Plant Antitumor Agents. I. The Isolation and Structure of Camptothecin, a Novel Alkaloidal Leukemia and Tumor Inhibitor from Camptotheca acuminata1,2. J. Am. Chem. Soc. 1966, 88, 3888–3890. [Google Scholar] [CrossRef]
- Liew, S.T.; Yang, L.-X. Design, synthesis and development of novel camptothecin drugs. Curr. Pharm. Des. 2008, 14, 1078–1097. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-Y.; Zu, Y.-G.; Shi, R.-Z.; Yao, L.-P. Review Camptothecin: Current Perspectives. Curr. Med. Chem. 2006, 13, 2021–2039. [Google Scholar] [CrossRef] [PubMed]
- Zunino, F.; Dallavalle, S.; Laccabue, D.; Beretta, G.L.; Merlini, L.; Pratesi, G. Current Status and Perspectives in the Development of Camptothecins. Curr. Pharm. Des. 2002, 8, 2505–2520. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Cao, S.; Cheng, Q.; Keefe, J.T.; Rustum, Y.M.; Li, F. A Novel Small Molecule FL118 That Selectively Inhibits Survivin, Mcl-1, XIAP and cIAP2 in a p53-Independent Manner, Shows Superior Antitumor Activity. PLoS ONE 2012, 7, e45571. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ackermann, E.J.; Bennett, C.F.; Rothermel, A.L.; Plescia, J.; Tognin, S.; Villa, A.; Marchisio, P.C.; Altieri, D.C. Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat. Cell Biol. 1999, 1, 461–466. [Google Scholar] [CrossRef]
- Li, F.; Ling, X. Survivin Study: An update of “What is the next wave?”. J. Cell. Physiol. 2006, 208, 476–486. [Google Scholar] [CrossRef]
- Zhao, J.; Ling, X.; Cao, S.; Liu, X.; Wan, S.; Jiang, T.; Li, F. Antitumor Activity of FL118, a Survivin, Mcl-1, XIAP, and cIAP2 Selective Inhibitor, Is Highly Dependent on Its Primary Structure and Steric Configuration. Mol. Pharm. 2014, 11, 457–467. [Google Scholar] [CrossRef]
- Ling, X.; Xu, C.; Fan, C.; Zhong, K.; Li, F.; Wang, X. FL118 Induces p53-Dependent Senescence in Colorectal Cancer Cells by Promoting Degradation of MdmX. Cancer Res. 2014, 74, 7487–7497. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Mai, X.; Liu, F.; Lin, M.; Dong, X.; Xu, Q.; Hao, C.; Zhang, L.; Yu, R.; Jiang, T. Synthesis of novel 10,11-methylenedioxy-camptothecin glycoside derivatives and investigation of their anti-tumor effects in vivo. RSC Adv. 2019, 9, 11142–11150. [Google Scholar] [CrossRef]
- Wani, M.C.; Nicholas, A.W.; Manikumar, G.; Wall, M.E. Plant antitumor agents. 25. Total synthesis and antileukemic activity of ring A substituted camptothecin analogs. Structure-activity correlations. J. Med. Chem. 1987, 30, 1774–1779. [Google Scholar] [CrossRef] [PubMed]
- DiSaia, P.J.; Sinkovics, J.G.; Rutledge, F.N.; Smith, J.P. Cell-mediated immunity to human malignant cells. Am. J. Obstet. Gynecol. 1972, 114, 979–989. [Google Scholar] [CrossRef]
- Adamovics, J.A.; Hutchinson, C.R. Prodrug analogs of the antitumor alkaloid camptothecin. J. Med. Chem. 1979, 22, 310–314. [Google Scholar] [CrossRef]
- Yaegashi, T.; Sawada, S.; Nagata, H.; Furuta, T.; Yokokura, T.; Miyasaka, T. Synthesis and Antitumor Activity of 20(S)-Camptothecin Derivatives. A-Ring-Substituted 7-Ethylcamptothecins and Their E-Rig-Modified Water-Soluble Derivatives. Chem. Pharm. Bull. 1994, 42, 2518–2525. [Google Scholar] [CrossRef]
- Zhao, T.; Lv, H.; Zu, Y.; Qu, Z.; Yao, L.; Su, L.; Liu, C.; Wang, L. Synthesis and antitumor activity of novel 20s-camptothecin analogues. Bioorg. Med. Chem. Lett. 2009, 19, 513–515. [Google Scholar] [CrossRef]
- Wang, M.-J.; Liu, Y.-Q.; Chang, L.-C.; Wang, C.-Y.; Zhao, Y.-L.; Zhao, X.-B.; Qian, K.; Nan, X.; Yang, L.; Yang, X.-M.; et al. Design, Synthesis, Mechanisms of Action, and Toxicity of Novel 20(S)-Sulfonylamidine Derivatives of Camptothecin as Potent Antitumor Agents. J. Med. Chem. 2014, 57, 6008–6018. [Google Scholar] [CrossRef]
- Song, Z.-L.; Wang, M.-J.; Li-Ting, Y.; Wu, D.; Wang, Y.-H.; Yan, L.-T.; Morris-Natschke, S.L.; Liu, Y.-Q.; Zhao, Y.-L.; Wang, C.-Y.; et al. Design, synthesis, cytotoxic activity and molecular docking studies of new 20(S)-sulfonylamidine camptothecin derivatives. Eur. J. Med. Chem. 2016, 115, 109–120. [Google Scholar] [CrossRef]
- Zhao, H.; Lee, C.; Sai, P.; Choe, Y.H.; Boro, M.; Pendri, A.; Guan, S.; Greenwald, R.B. 20-O-acylcamptothecin derivatives: Evidence for lactone stabilization. J. Org. Chem. 2000, 65, 4601–4606. [Google Scholar] [CrossRef]
- Yang, L.-X.; Pan, X.; Wang, H.-J. Novel camptothecin derivatives. Part 1: Oxyalkanoic acid esters of camptothecin and their in vitro and in vivo antitumor activity. Bioorg. Med. Chem. Lett. 2002, 12, 1241–1244. [Google Scholar] [CrossRef]
- De Groot, F.M.H.; Busscher, G.F.; Aben, R.W.M.; Scheeren, H.W. Novel 20-carbonate linked prodrugs of camptothecin and 9-aminocamptothecin designed for activation by tumour-associated plasmin. Bioorg. Med. Chem. Lett. 2002, 12, 2371–2376. [Google Scholar] [CrossRef]
- Lerchen, H.G.; Baumgarten, J.; dem Bruch, K.; Lehmann, T.E.; Sperzel, M.; Kempka, G.; Fiebig, H.H. Synthesis of 20-O-linked 20(S)-camptothecin Glycoconjugates: Impact of the side chain of the ester-linked amino acid on epimerization during the acylation reaction and on hydrolytic stability of the final glycoconjugates. J. Med. Chem. 2001, 44, 4186–4195. [Google Scholar] [CrossRef] [PubMed]
- Lum, C.; Alamgeer, M. Lum Technological and Therapeutic Advances in Advanced Small Cell Lung Cancer. Cancers 2019, 11, 1570. [Google Scholar] [CrossRef]
- Ling, X.; Liu, X.; Zhong, K.; Smith, N.; Prey, J.; Li, F. FL118, a novel camptothecin analogue, overcomes irinotecan and topotecan resistance in human tumor xenograft models. Am. J. Transl. Res. 2015, 7, 1765–1781. [Google Scholar]
- Rabi, T.; Li, F. Multiple mechanisms involved in a low concentration of FL118 enhancement of AMR-MeOAc to induce pancreatic cancer cell apoptosis and growth inhibition. Am. J. Cancer Res. 2018, 8, 2267–2283. [Google Scholar]
- Yang, Z.; Ji, L.; Jiang, G.; Liu, R.; Liu, Z.; Yang, Y.Q.; Ma, H.Z. FL118, a novel camptothecin analogue, suppressed migration and invasion of human breast cancer cells by inhibiting epithelial-mesenchymal transition via the Wnt/β-catenin signaling pathway. Biosci. Trends 2018, 12, 40–46. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Ling, X.; Wu, W.; Fan, C.; Xu, C.; Liao, J.; Rich, L.J.; Huang, R.-Y.; Repasky, E.A.; Wang, X.; Li, F. An ABCG2 non-substrate anticancer agent FL118 targets drug-resistant cancer stem-like cells and overcomes treatment resistance of human pancreatic cancer. J. Exp. Clin. Cancer Res. 2018, 37, 240. [Google Scholar] [CrossRef]
- Bröker, L.E.; Kruyt, F.A.; Giaccone, G. Cell Death Independent of Caspases: A Review. Clin. Cancer Res. 2005, 11, 3155–3162. [Google Scholar] [CrossRef]
- Yu, J.W.; Shi, Y. FLIP and the death effector domain family. Oncogene 2008, 27, 6216–6227. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P. The change of cellular membranes on apoptosis: Fluorescence detection. Exp. Oncol. 2012, 34, 263–268. [Google Scholar] [PubMed]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Little, A.; Smith, P.; Cook, S. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene 2013, 32, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.-L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef]
- Li, F.; Jiang, T.; Li, Q.; Ling, X. Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: Did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am. J. Cancer Res. 2017, 7, 2350–2394. [Google Scholar]
- Chan, K.T.; Meng, F.Y.; Li, Q.; Ho, C.Y.; Lam, T.S.; To, Y.; Lee, W.H.; Li, M.; Chu, K.H.; Toh, M. Cucurbitacin B induces apoptosis and S phase cell cycle arrest in BEL-7402 human hepatocellular carcinoma cells and is effective via oral administration. Cancer Lett. 2010, 294, 118–124. [Google Scholar] [CrossRef]
- Steelman, L.S.; Abrams, S.L.; Whelan, J.E.; Bertrand, F.E.; Ludwig, D.; Bäsecke, J.; Libra, M.; Stivala, F.; Milella, M.; Tafuri, A.; et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia 2008, 22, 686–707. [Google Scholar] [CrossRef]
- Böğürcü-Seidel, N.; Sevimli-Gur, C.; Ozmen, B.; Bedir, E.; Korkmaz, K.S. ALCAPs induce mitochondrial apoptosis and activate DNA damage response by generating ROS and inhibiting topoisomerase I enzyme activity in K562 leukemia cell line. Biochem. Biophys. Res. Commun. 2011, 409, 738–744. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, X.; Wu, G.; Zhang, Y.; Zhang, X.; Yin, R.; Qi, X.; Li, J.; Jiang, T. Design, Synthesis, and In Vitro/In Vivo Anti-Cancer Activities of Novel (20S)-10,11-Methylenedioxy-Camptothecin Heterocyclic Derivatives. Int. J. Mol. Sci. 2020, 21, 8495. https://doi.org/10.3390/ijms21228495
Dai X, Wu G, Zhang Y, Zhang X, Yin R, Qi X, Li J, Jiang T. Design, Synthesis, and In Vitro/In Vivo Anti-Cancer Activities of Novel (20S)-10,11-Methylenedioxy-Camptothecin Heterocyclic Derivatives. International Journal of Molecular Sciences. 2020; 21(22):8495. https://doi.org/10.3390/ijms21228495
Chicago/Turabian StyleDai, Xiufen, Guanzhao Wu, Yixuan Zhang, Xiaomin Zhang, Ruijuan Yin, Xin Qi, Jing Li, and Tao Jiang. 2020. "Design, Synthesis, and In Vitro/In Vivo Anti-Cancer Activities of Novel (20S)-10,11-Methylenedioxy-Camptothecin Heterocyclic Derivatives" International Journal of Molecular Sciences 21, no. 22: 8495. https://doi.org/10.3390/ijms21228495
APA StyleDai, X., Wu, G., Zhang, Y., Zhang, X., Yin, R., Qi, X., Li, J., & Jiang, T. (2020). Design, Synthesis, and In Vitro/In Vivo Anti-Cancer Activities of Novel (20S)-10,11-Methylenedioxy-Camptothecin Heterocyclic Derivatives. International Journal of Molecular Sciences, 21(22), 8495. https://doi.org/10.3390/ijms21228495