Glucose Concentration in Cell Culture Medium Influences the BRCA1-Mediated Regulation of the Lipogenic Action of IGF-I in Breast Cancer Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
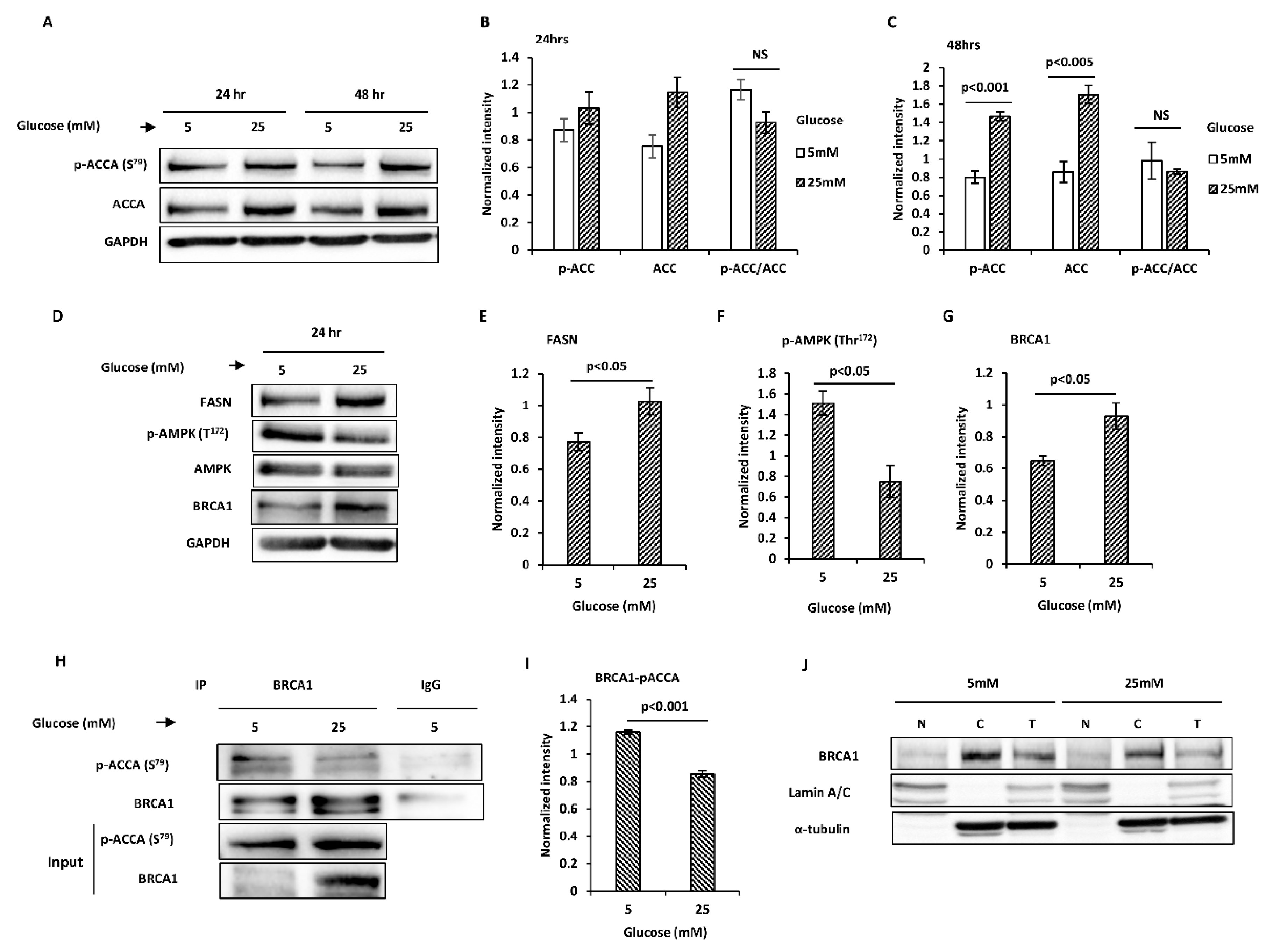
2.1. Glucose Level Regulates Key Proteins Involved in Endogenous Fatty Acid Synthesis in MCF7 Breast Cancer Cells
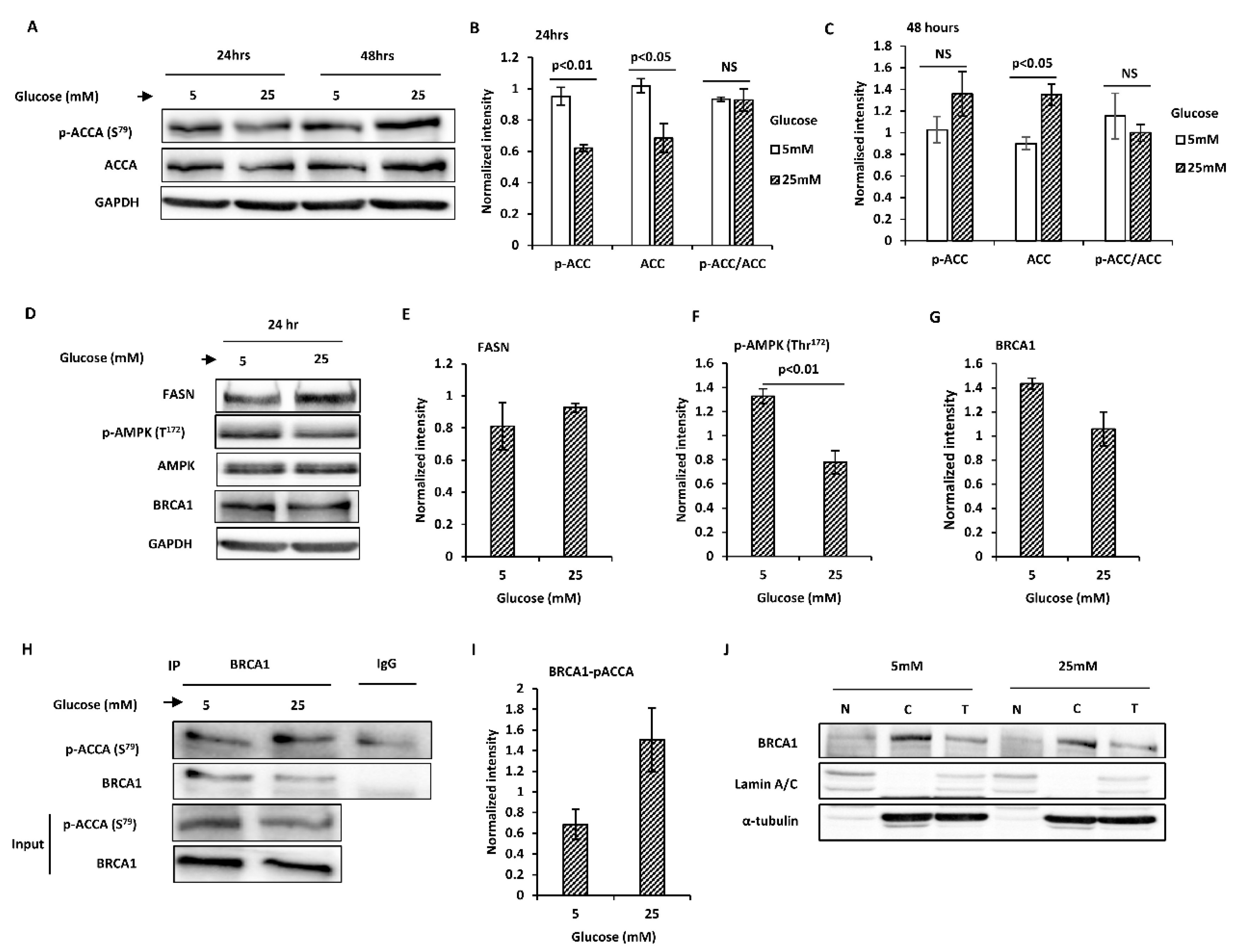
2.2. Glucose Level Regulates Key Proteins Involved in Endogenous Fatty Acid Synthesis in T47D Breast Cancer Cells
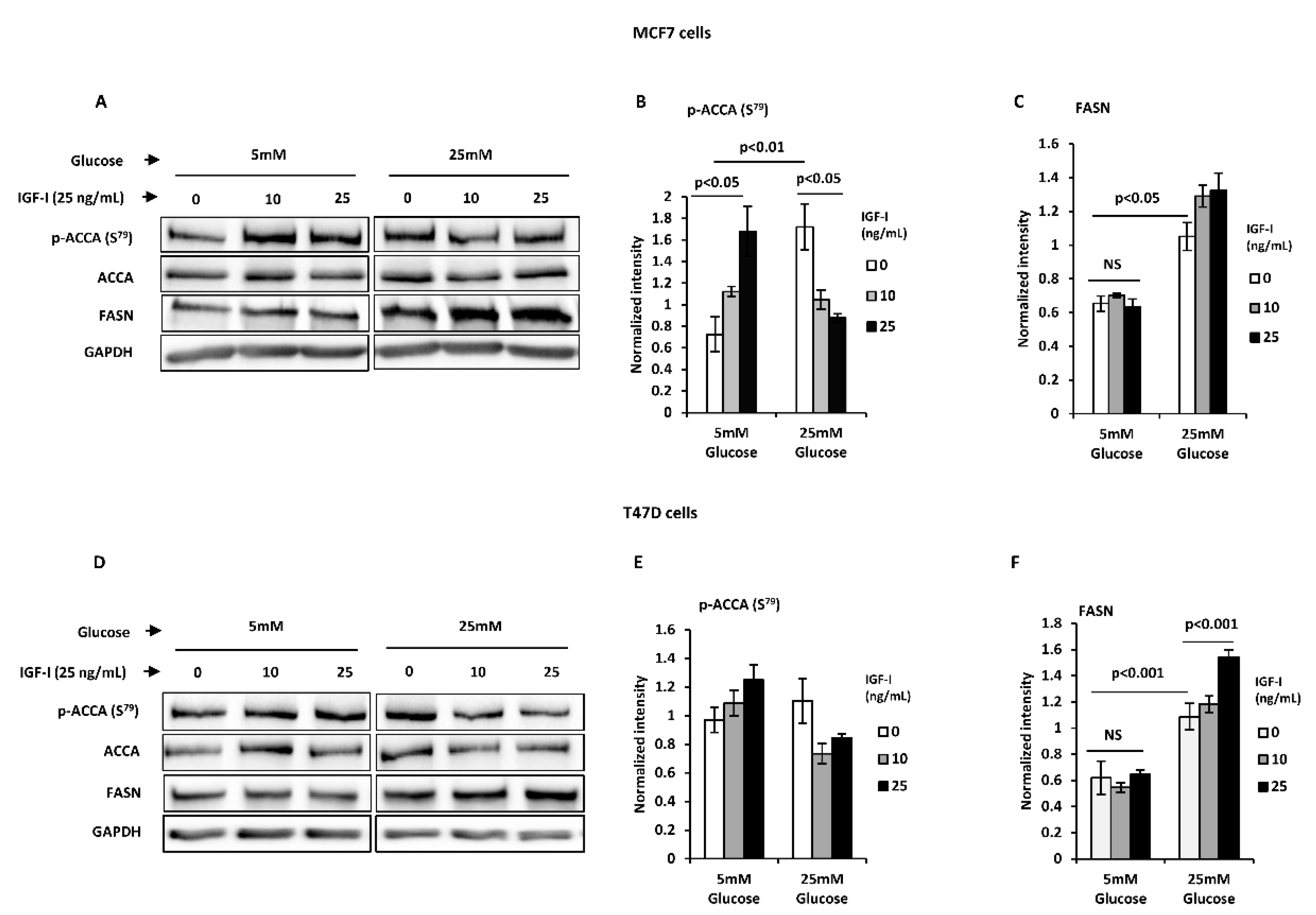
2.3. Normal Glucose Blocks an IGF-I-Induced Lipogenic Phenotype in T47D and MCF7 Breast Cancer Cells
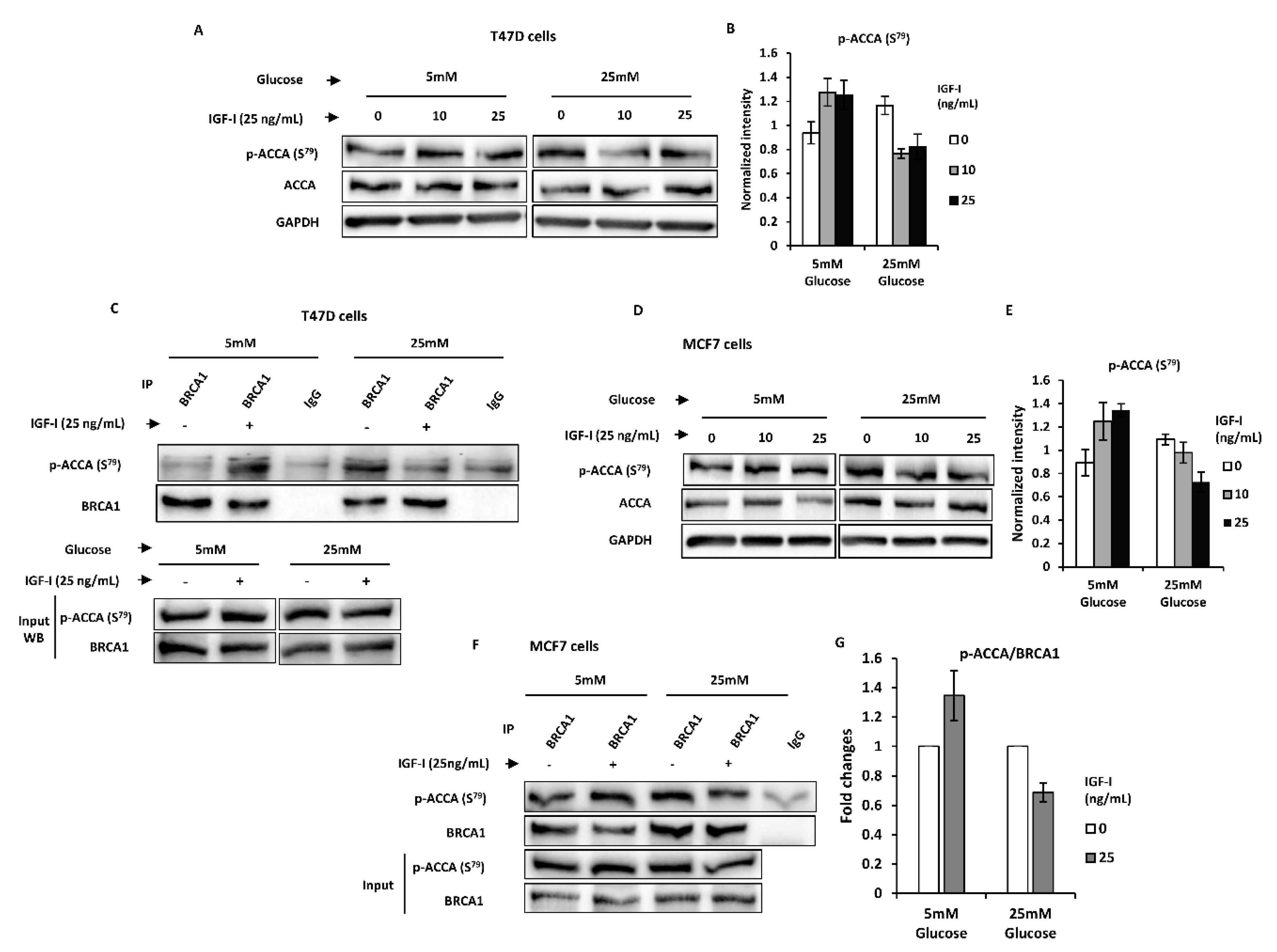
2.4. Normal Glucose Promotes BRCA1 Function of Suppressing an IGF-I-Induced Lipogenic Phenotype in MCF7 and T47D Breast Cancer Cells
2.5. High Glucose Modulates IGF-I-Induced Cell Growth in Breast Cancer Cells, but Not in Benign Mammary Epithelial Cells
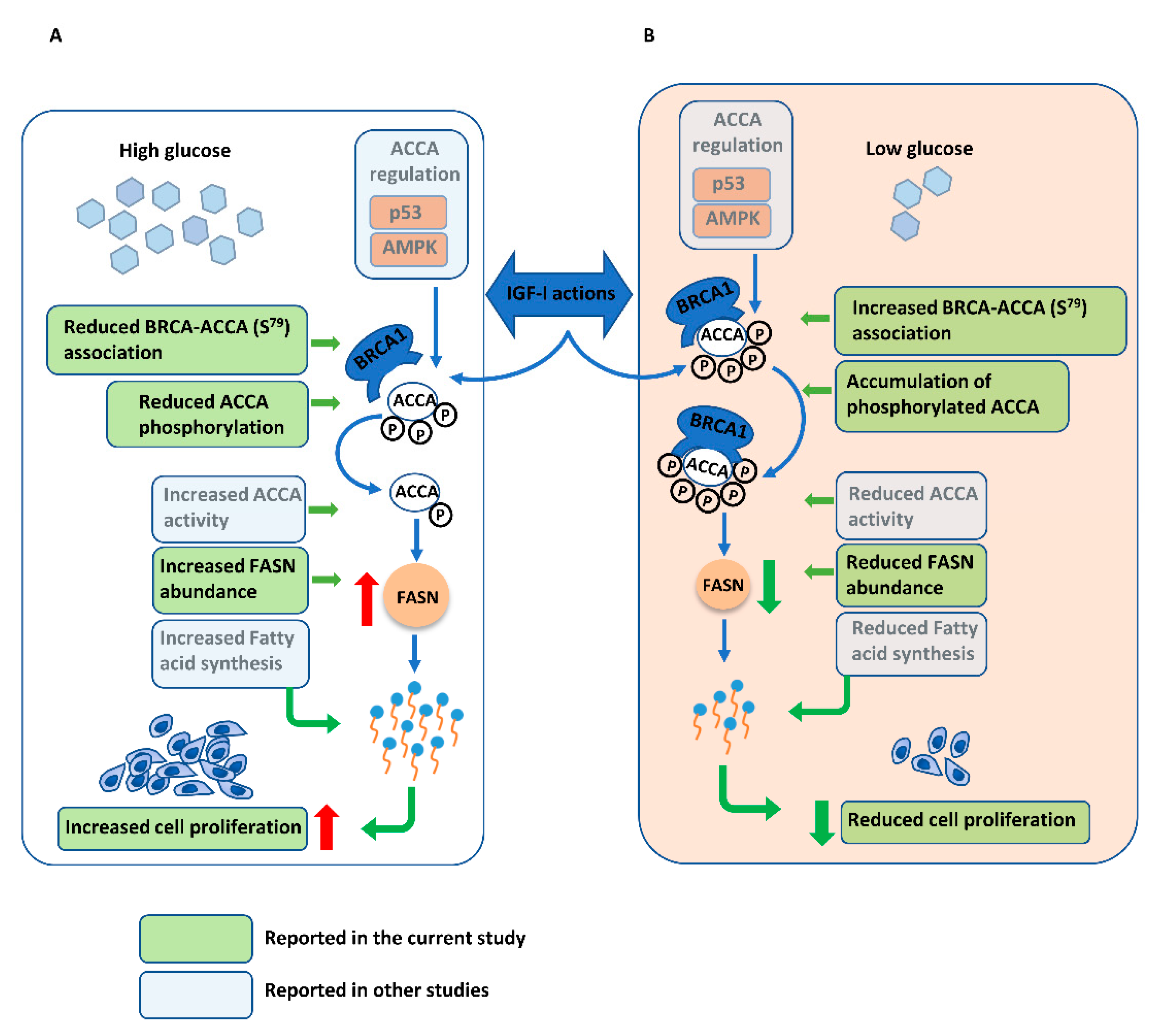
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Western Blotting
4.4. Cell Counting
4.5. Co-Immunoprecipitation
4.6. Subcellular Fractionation
4.7. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACCA | ACCA: Acetyl CoA carboxylase α |
| ACL | ATP citrate lyase |
| AMPK | AMP-activated protein kinase |
| AKT | Protein kinase B |
| ATTC | American type culture collection |
| BRCA1 | Breast cancer 1, early onset |
| ECCAC | European collection of authenticated cell cultures |
| EMT | Epithelia-to-mesenchymal transition |
| ER | Estrogen receptor |
| ERK | Extracellular signal regulated kinase |
| FASN | Fatty acid synthase |
| GLUT | Glucose transporter |
| IGF-I | Insulin-like growth factor I |
| IGF-II | Insulin-like growth factor II |
| IGF-IR | Insulin-like growth factor I receptor |
| MAPK | Mitogen activated protein kinase |
| mTOR | Mammalian target of rapamycin |
| SDS-PAGE | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| STR | Short tandem repeat |
| TBS-T | Tris-buffered saline Tween-20 |
| VEGF | Vascular endothelial growth factor |
References
- Bray, F.; Me, J.F.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Lambie, H.; Miremadi, A.; Pinder, E.S.; Bell, J.; Wencyk, P.; Paish, E.; Macmillan, R.; Ellis, I.O. Prognostic significance of BRCA1 expression in sporadic breast carcinomas. J. Pathol. 2003, 200, 207–213. [Google Scholar] [CrossRef] [PubMed]
- King, M.-C.; Marks, J.H.; Mandell, J.B. Breast and Ovarian Cancer Risks Due to Inherited Mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Macias, V.; Al-Alem, U.; Deaton, R.J.; Kadjaksy-Balla, A.; Gann, P.H.; Rauscher, G.H. BRCA1 protein expression and subcellular localization in primary breast cancer: Automated digital microscopy analysis of tissue microarrays. PLoS ONE 2017, 12, e0184385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, K.; Honda, K.; Inamoto, T.; Shinohara, H.; Yamauchi, A.; Suga, K.; Okuyama, T.; Shimada, T.; Kodama, H.; Noguchi, S.; et al. Reduction of BRCA1 protein expression in Japanese sporadic breast carcinomas and its frequent loss in BRCA1-associated cases. Clin. Cancer Res. 1999, 5, 1249–1261. [Google Scholar] [PubMed]
- Yang, Q.; Sakurai, T.; Mori, I.; Yoshimura, G.; Nakamura, M.; Nakamura, Y.; Suzuma, T.; Tamaki, T.; Umemura, T.; Kakudo, K. Prognostic significance of BRCA1 expression in Japanese sporadic breast carcinomas. Cancer 2001, 92, 54–60. [Google Scholar] [CrossRef]
- Turner, N.C.; Reis-Filho, J.S.; Russell, A.M.; Springall, R.J.; Ryder, K.; Steele, D.; Savage, K.; E Gillett, C.; Schmitt, F.C.; Ashworth, A.; et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene 2006, 26, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakha, E.A.; El-Sheikh, S.E.; Kandil, M.A.; El-Sayed, M.E.; Green, A.R.; Ellis, I.O. Expression of BRCA1 protein in breast cancer and its prognostic significance. Hum. Pathol. 2008, 39, 857–865. [Google Scholar] [CrossRef]
- Bal, A.; Verma, S.; Joshi, K.; Singla, A.; Thakur, R.; Arora, S.; Singh, G. BRCA1-methylated sporadic breast cancers are BRCA-like in showing a basal phenotype and absence of ER expression. Virchows Archiv. 2012, 461, 305–312. [Google Scholar] [CrossRef]
- Holly, J.M.P.; Zeng, L.; Perks, C.M. Epithelial cancers in the post-genomic era: should we reconsider our lifestyle? Cancer Metastasis Rev. 2013, 32, 673–705. [Google Scholar] [CrossRef] [Green Version]
- Tryggvadottir, L.; Sigvaldason, H.; Olafsdottir, G.H.; Jonasson, J.G.; Jonsson, T.; Tulinius, H.; Eyfjörd, J.E. Population-Based Study of Changing Breast Cancer Risk in Icelandic BRCA2 Mutation Carriers, 1920–2000. J. Natl. Cancer Inst. 2006, 98, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Nkondjock, A.; Ghadirian, P. Diet quality and BRCA-associated breast cancer risk. Breast Cancer Res. Treat. 2006, 103, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Narod, A.S.; Foulkes, W.D. BRCA1 and BRCA2: 1994 and beyond. Nat. Rev. Cancer 2004, 4, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Narod, A.S. Modifiers of risk of hereditary breast and ovarian cancer. Nat. Rev. Cancer 2002, 2, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Dizin, E.; Ray, H.; Luquain, C.; Lefai, E.; Foufelle, F.; Billaud, M.; Lenoir, G.M.; Venezia, N.D.; Huang, I.-C.; et al. BRCA1 Affects Lipid Synthesis through Its Interaction with Acetyl-CoA Carboxylase. J. Biol. Chem. 2006, 281, 3172–3181. [Google Scholar] [CrossRef] [Green Version]
- Koobotse, M.; Holly, J.; Perks, C. Elucidating the novel BRCA1 function as a non-genomic metabolic restraint in ER-positive breast cancer cell lines. Oncotarget 2018, 9, 33562–33576. [Google Scholar] [CrossRef]
- Gallagher, E.J.; Leroith, D. The proliferating role of insulin and insulin-like growth factors in cancer. Trends Endocrinol. Metab. 2010, 21, 610–618. [Google Scholar] [CrossRef] [Green Version]
- Werner, H. Tumor suppressors govern insulin-like growth factor signaling pathways: implications in metabolism and cancer. Oncogene 2011, 31, 2703–2714. [Google Scholar] [CrossRef]
- Jackson, K.C.; Gidlund, E.-K.; Norrbom, J.; Valencia, A.; Thomson, D.M.; Schuh, R.A.; Neufer, P.D.; Spangenburg, E.E. BRCA1 is a novel regulator of metabolic function in skeletal muscle. J. Lipid Res. 2014, 55, 668–680. [Google Scholar] [CrossRef] [Green Version]
- Privat, M.; Radosevic-Robin, N.; Aubel, C.; Cayre, A.; Penault-Llorca, F.; Marceau, G.; Sapin, V.; Bignon, Y.-J.; Morvan, D. BRCA1 Induces Major Energetic Metabolism Reprogramming in Breast Cancer Cells. PLoS ONE 2014, 9, e102438. [Google Scholar] [CrossRef] [Green Version]
- Ortega, F.J.; Moreno-Navarrete, J.M.; Mayas, D.; García-Santos, E.; Gómez-Serrano, M.; Rodriguez-Hermosa, J.I.; Ruiz, B.; Ricart, W.; Tinahones, F.J.; Frühbeck, G.; et al. Breast Cancer 1 (BrCa1) May Be behind Decreased Lipogenesis in Adipose Tissue from Obese Subjects. PLoS ONE 2012, 7, e33233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramadan, S.; Arm, J.; Silcock, J.; Santamaria, G.; Buck, J.; Roy, M.; Leong, K.M.; Lau, P.; Clark, D.; Malycha, P.; et al. Lipid and Metabolite Deregulation in the Breast Tissue of Women CarryingBRCA1andBRCA2Genetic Mutations. Radiology 2015, 275, 675–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasconcelos-Dos-Santos, A.; Loponte, H.F.B.R.; Mantuano, N.R.; A Oliveira, I.; De Paula, I.F.; Teixeira, L.K.; De-Freitas-Junior, J.C.M.; Gondim, K.C.; Heise, N.; Mohana-Borges, R.; et al. Hyperglycemia exacerbates colon cancer malignancy through hexosamine biosynthetic pathway. Oncogoly 2017, 6, e306. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, M.R.; D’Esposito, V.; Costa, V.; Liguoro, D.; Collina, F.; Cantile, M.; Prevete, N.; Passaro, C.; Mosca, G.; De Laurentiis, M.; et al. Glucose impairs tamoxifen responsiveness modulating connective tissue growth factor in breast cancer cells. Oncotarget 2017, 8, 109000–109017. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nat. Cell Biol. 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Maamoun, H.; Benameur, T.; Pintus, G.; Munusamy, S.; Agouni, A. Crosstalk Between Oxidative Stress and Endoplasmic Reticulum (ER) Stress in Endothelial Dysfunction and Aberrant Angiogenesis Associated With Diabetes: A Focus on the Protective Roles of Heme Oxygenase (HO)-1. Front. Physiol. 2019, 10, 70. [Google Scholar] [CrossRef] [Green Version]
- Ryu, T.Y.; Park, J.; Scherer, P.E. Hyperglycemia as a Risk Factor for Cancer Progression. Diabetes Metab. J. 2014, 38, 330–336. [Google Scholar] [CrossRef] [Green Version]
- Klement, R.J.; Fink, M.K. Dietary and pharmacological modification of the insulin/IGF-1 system: exploiting the full repertoire against cancer. Oncogenesis 2016, 5, e193. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Cassim, S.; Raymond, V.-A.; Dehbidi-Assadzadeh, L.; Lapierre, P.; Bilodeau, M. Metabolic reprogramming enables hepatocarcinoma cells to efficiently adapt and survive to a nutrient-restricted microenvironment. Cell Cycle 2018, 17, 903–916. [Google Scholar] [CrossRef]
- Cassim, S.; Raymond, V.-A.; Lacoste, B.; Lapierre, P.; Bilodeau, M. Metabolite profiling identifies a signature of tumorigenicity in hepatocellular carcinoma. Oncotarget 2018, 9, 26868–26883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielinska, H.; Holly, J.; Bahl, A.; Perks, C. Inhibition of FASN and ERα signalling during hyperglycaemia-induced matrix-specific EMT promotes breast cancer cell invasion via a caveolin-1-dependent mechanism. Cancer Lett. 2018, 419, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Chan, D.K.; Haugrud, A.B.; Miskimins, W.K. Mechanisms by Which Low Glucose Enhances the Cytotoxicity of Metformin to Cancer Cells Both In Vitro and In Vivo. PLoS ONE 2014, 9, e108444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Qarakhli, A.M.A.; Yusop, N.; Waddington, R.J.; Moseley, R. Effects of high glucose conditions on the expansion and differentiation capabilities of mesenchymal stromal cells derived from rat endosteal niche. BMC Mol. Cell Biol. 2019, 20, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weil, B.R.; Abarbanell, A.M.; Herrmann, J.L.; Wang, Y.; Meldrum, D.R. High glucose concentration in cell culture medium does not acutely affect human mesenchymal stem cell growth factor production or proliferation. Am. J. Physiol. Integr. Comp. Physiol. 2009, 296, R1735–R1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, M.D.; Ballinger, K.R.; Khetani, S.R. Long-term exposure to abnormal glucose levels alters drug metabolism pathways and insulin sensitivity in primary human hepatocytes. Sci. Rep. 2016, 6, 28178. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Yang, E.S.; Jiang, G.; Nowsheen, S.; Wang, H.; Wang, T.; Wang, Y.; Billheimer, D.; Chakravarthy, A.B.; A Brown, M.; et al. p53-Dependent BRCA1 Nuclear Export Controls Cellular Susceptibility to DNA Damage. Cancer Res. 2011, 71, 5546–5557. [Google Scholar] [CrossRef] [Green Version]
- Abu-Elheiga, L.; Brinkley, W.R.; Zhong, L.; Chirala, S.S.; Woldegiorgis, G.; Wakil, S.J. The subcellular localization of acetyl-CoA carboxylase 2. Proc. Natl. Acad. Sci. USA 2000, 97, 1444–1449. [Google Scholar] [CrossRef] [Green Version]
- Magnard, C.; Bachelier, R.; Vincent, A.; Jaquinod, M.; Kieffer, S.; Lenoir, G.M.; Venezia, N.D. BRCA1 interacts with acetyl-CoA carboxylase through its tandem of BRCT domains. Oncogene 2002, 21, 6729–6739. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Biernacka, K.M.; Holly, J.M.P.; Jarrett, C.; Morrison, A.A.; Morgan, A.; Winters, Z.E.; Foulstone, E.J.; Hamilton-Shield, J.P.; Perks, C. Hyperglycaemia confers resistance to chemotherapy on breast cancer cells: the role of fatty acid synthase. Endocr.-Relat. Cancer 2010, 17, 539–551. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.-X.; Peng, X.-H.; Xiong, Y.; Liao, D.-F.; Cao, D.; Li, L. Dual role of insulin-like growth factor-1 in acetyl-CoA carboxylase-alpha activity in human colon cancer cells HCT-8: downregulating its expression and phosphorylation. Mol. Cell. Biochem. 2011, 357, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Hunkeler, M.; Hagmann, A.; Stuttfeld, E.; Chami, M.; Guri, Y.; Stahlberg, H.; Maier, T. Structural basis for regulation of human acetyl-CoA carboxylase. Nat. Cell Biol. 2018, 558, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Brownsey, R.; Boone, A.; Elliott, J.; Kulpa, J.; Lee, W. Regulation of acetyl-CoA carboxylase. Biochem. Soc. Trans. 2006, 34, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-W.; Lin, Y.-H.; Pai, M.-H.; Lo, A.-C.; Lee, Y.-C.; Fang, I.-C.; Lin, J.; Hsieh, R.-K.; Chang, Y.-F.; Chen, C.-L. Association between Phosphorylated AMP-Activated Protein Kinase and Acetyl-CoA Carboxylase Expression and Outcome in Patients with Squamous Cell Carcinoma of the Head and Neck. PLoS ONE 2014, 9, e96183. [Google Scholar] [CrossRef] [Green Version]
- Pelicano, H.; Zhang, W.; Liu, J.; Hammoudi, N.; Dai, J.; Xu, R.-H.; Pusztai, L.; Huang, P. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: role of mTOR pathway and therapeutic potential. Breast Cancer Res. 2014, 16, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Radde, B.N.; Ivanova, M.M.; Mai, H.X.; Salabei, J.K.; Hill, B.G.; Klinge, C.M. Bioenergetic differences between MCF-7 and T47D breast cancer cells and their regulation by oestradiol and tamoxifen. Biochem. J. 2015, 465, 49–61. [Google Scholar] [CrossRef]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.-P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Yu, L.; Chen, W.; Xu, Y.; Wu, M.; Todorova, D.; Tang, Q.; Feng, B.; Jiang, L.; He, J.; et al. Wild-Type p53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer Cell 2019, 35, 191–203.e8. [Google Scholar] [CrossRef] [Green Version]
- Rossignol, R.; Gilkerson, R.; Aggeler, R.; Yamagata, K.; Remington, S.J.; Capaldi, R.A. Energy Substrate Modulates Mitochondrial Structure and Oxidative Capacity in Cancer Cells. Cancer Res. 2004, 64, 985–993. [Google Scholar] [CrossRef] [Green Version]
- Ždralević, M.; Marchiq, I.; De Padua, M.M.C.; Parks, S.K.; Pouysségur, J. Metabolic Plasiticy in Cancers—Distinct Role of Glycolytic Enzymes GPI, LDHs or Membrane Transporters MCTs. Front. Oncol. 2017, 7, 313. [Google Scholar] [CrossRef]
- Swinnen, J.V.; Beckers, A.; Brusselmans, K.; Organe, S.; Segers, J.; Timmermans, L.; Vanderhoydonc, F.; Deboel, L.; Derua, R.; Waelkens, E.; et al. Mimicry of a Cellular Low Energy Status Blocks Tumor Cell Anabolism and Suppresses the Malignant Phenotype. Cancer Res. 2005, 65, 2441–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harriman, G.; Greenwood, J.; Bhat, S.; Huang, X.; Wang, R.; Paul, D.; Tong, L.; Saha, A.K.; Westlin, W.F.; Kapeller, R.; et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc. Natl. Acad. Sci. USA 2016, 113, E1796–E1805. [Google Scholar] [CrossRef] [Green Version]
- Lally, J.S.; Ghoshal, S.; Deperalta, D.K.; Moaven, O.; Wei, L.; Masia, R.; Erstad, D.J.; Fujiwara, N.; Leong, V.; Houde, V.P.; et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 2019, 29, 174–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashima, T.; Seimiya, H.; Tsuruo, T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br. J. Cancer 2009, 100, 1369–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; A Zielinska, H.; Arshad, A.; Hamilton-Shield, J.P.; Bahl, A.; Holly, J.M.P.; Perks, C. Hyperglycaemia-induced chemoresistance in breast cancer cells: role of the estrogen receptor. Endocr.-Relat. Cancer 2015, 23, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perks, C.; Vernon, E.G.; Rosendahl, A.H.; Tonge, D.; Holly, J.M.P. IGF-II and IGFBP-2 differentially regulate PTEN in human breast cancer cells. Oncogene 2007, 26, 5966–5972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chajès, V.; Cambot, M.; Moreau, K.; Lenoir, G.M.; Joulin, V. Acetyl-CoA Carboxylase α Is Essential to Breast Cancer Cell Survival. Cancer Res. 2006, 66, 5287–5294. [Google Scholar] [CrossRef] [Green Version]
- Lamers, M.L.; Almeida, M.E.S.; Vicente-Manzanares, M.; Horwitz, A.F.; Santos, M.F. High Glucose-Mediated Oxidative Stress Impairs Cell Migration. PLoS ONE 2011, 6, e22865. [Google Scholar] [CrossRef] [Green Version]
- Pirkmajer, S.; Chibalin, A.V. Serum starvation: caveat emptor. Am. J. Physiol. Physiol. 2011, 301, C272–C279. [Google Scholar] [CrossRef] [Green Version]
- Puche, J.E.; García-Fernández, M.; Muntané, J.; Rioja, J.; González-Barón, S.; Cortazar, I.C. Low Doses of Insulin-Like Growth Factor-I Induce Mitochondrial Protection in Aging Rats. Endocrinology 2008, 149, 2620–2627. [Google Scholar] [CrossRef] [Green Version]
- Garciía-Fernández, M.; Delgado, G.; Puche, J.E.; González-Barón, S.; Cortázar, I.C. Low Doses of Insulin-Like Growth Factor I Improve Insulin Resistance, Lipid Metabolism, and Oxidative Damage in Aging Rats. Endocrinology 2008, 149, 2433–2442. [Google Scholar] [CrossRef] [Green Version]
- Gupta, C.; Tikoo, K. High glucose and insulin differentially modulates proliferation in MCF-7 and MDA-MB-231 cells. J. Mol. Endocrinol. 2013, 51, 119–129. [Google Scholar] [CrossRef] [Green Version]
- San-Millán, I.; Julian, C.G.; Matarazzo, C.; Martinez, J.; Brooks, G.A. Is Lactate an Oncometabolite? Evidence Supporting a Role for Lactate in the Regulation of Transcriptional Activity of Cancer-Related Genes in MCF7 Breast Cancer Cells. Front. Oncol. 2020, 9, 1536. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, T.; Tardito, S. Cell Culture Medium Formulation and Its Implications in Cancer Metabolism. Trends Cancer 2019, 5, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Asayama, Y. Animal-cell culture media: History, characteristics, and current issues. Reprod. Med. Biol. 2017, 16, 99–117. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.-Y.; Wu, C.-C.; Fang, C.-L.; Chen, W.-Y.; Chen, C.-L. Long-term growth comparison studies of FBS and FBS alternatives in six head and neck cell lines. PLoS ONE 2017, 12, e0178960. [Google Scholar] [CrossRef]
- van der Valk, J.; Brunner, D.; De Smet, K.; Fex Svenningsen, Å.; Honegger, P.; Knudsen, L.E.; Lindl, T.; Noraberg, J.; Price, A.; Scarino, M.L.; et al. Optimization of chemically defined cell culture media – Replacing fetal bovine serum in mammalian in vitro methods. Toxicol. in Vitro 2010, 24, 1053–1063. [Google Scholar] [CrossRef] [Green Version]
- Burrows, C.; Holly, J.M.P.; Laurence, N.J.; Vernon, E.G.; Carter, J.V.; Clark, M.A.; McIntosh, J.; McCaig, C.; Winters, Z.E.; Perks, C. Insulin-Like Growth Factor Binding Protein 3 Has Opposing Actions on Malignant and Nonmalignant Breast Epithelial Cells that Are Each Reversible and Dependent upon Cholesterol-Stabilized Integrin Receptor Complexes. Endocrinology 2006, 147, 3484–3500. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koobotse, M.O.; Schmidt, D.; Holly, J.M.P.; Perks, C.M. Glucose Concentration in Cell Culture Medium Influences the BRCA1-Mediated Regulation of the Lipogenic Action of IGF-I in Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 8674. https://doi.org/10.3390/ijms21228674
Koobotse MO, Schmidt D, Holly JMP, Perks CM. Glucose Concentration in Cell Culture Medium Influences the BRCA1-Mediated Regulation of the Lipogenic Action of IGF-I in Breast Cancer Cells. International Journal of Molecular Sciences. 2020; 21(22):8674. https://doi.org/10.3390/ijms21228674
Chicago/Turabian StyleKoobotse, Moses O., Dayane Schmidt, Jeff M. P. Holly, and Claire M. Perks. 2020. "Glucose Concentration in Cell Culture Medium Influences the BRCA1-Mediated Regulation of the Lipogenic Action of IGF-I in Breast Cancer Cells" International Journal of Molecular Sciences 21, no. 22: 8674. https://doi.org/10.3390/ijms21228674