Medicinal Chemistry Targeting Mitochondria: From New Vehicles and Pharmacophore Groups to Old Drugs with Mitochondrial Activity

Abstract

1. Introduction
2. Mitochondria as Pharmacological Targets
3. The Stem Cell Problem in Solid Tumors
4. Energy Metabolism in Hypoxic Tumor Cells
5. Polyphenols as Mitochondrial-Disrupting Agents Targeting Tumor and Cancer Stem Cells

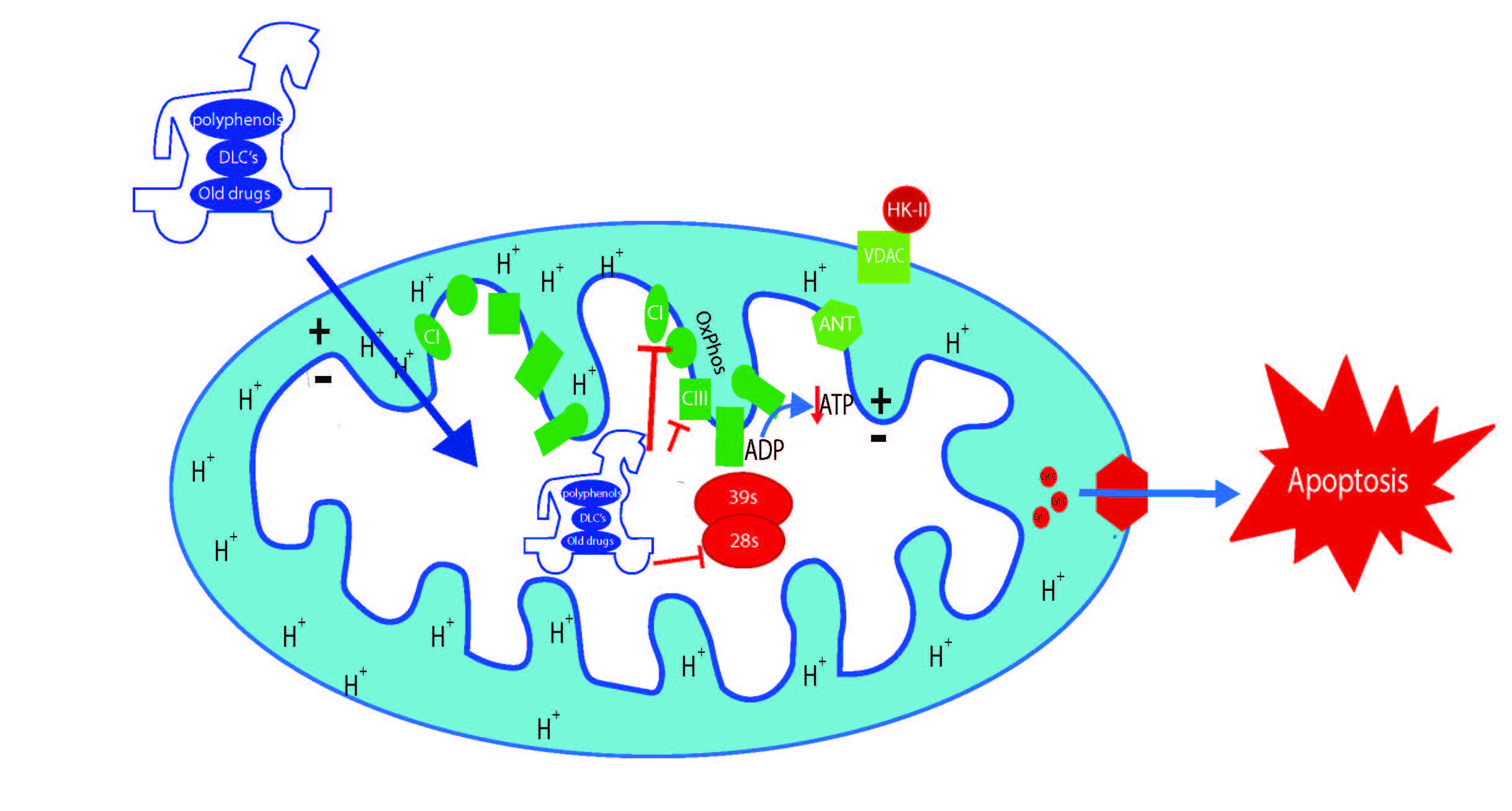{kind=link}
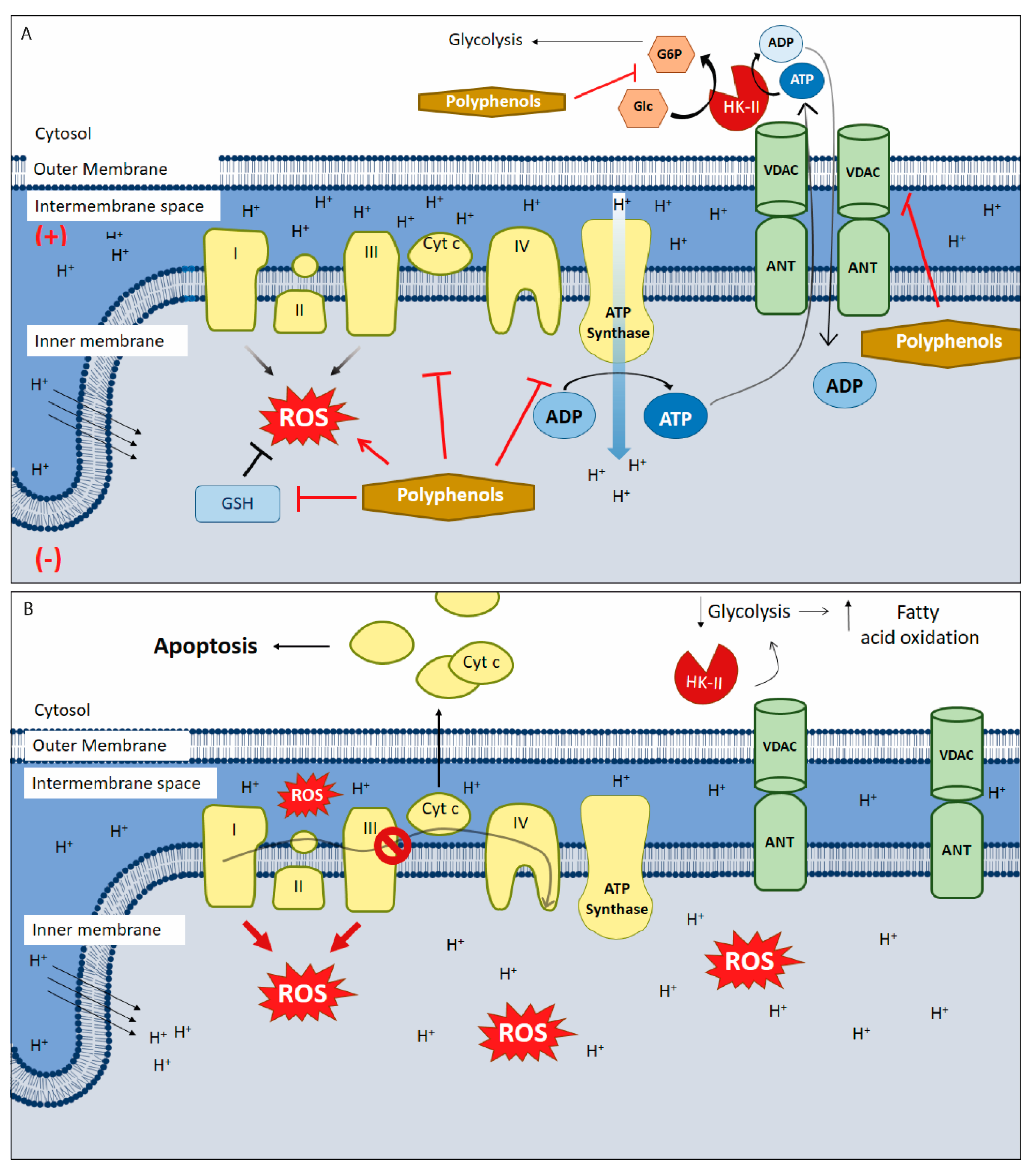{kind=link}
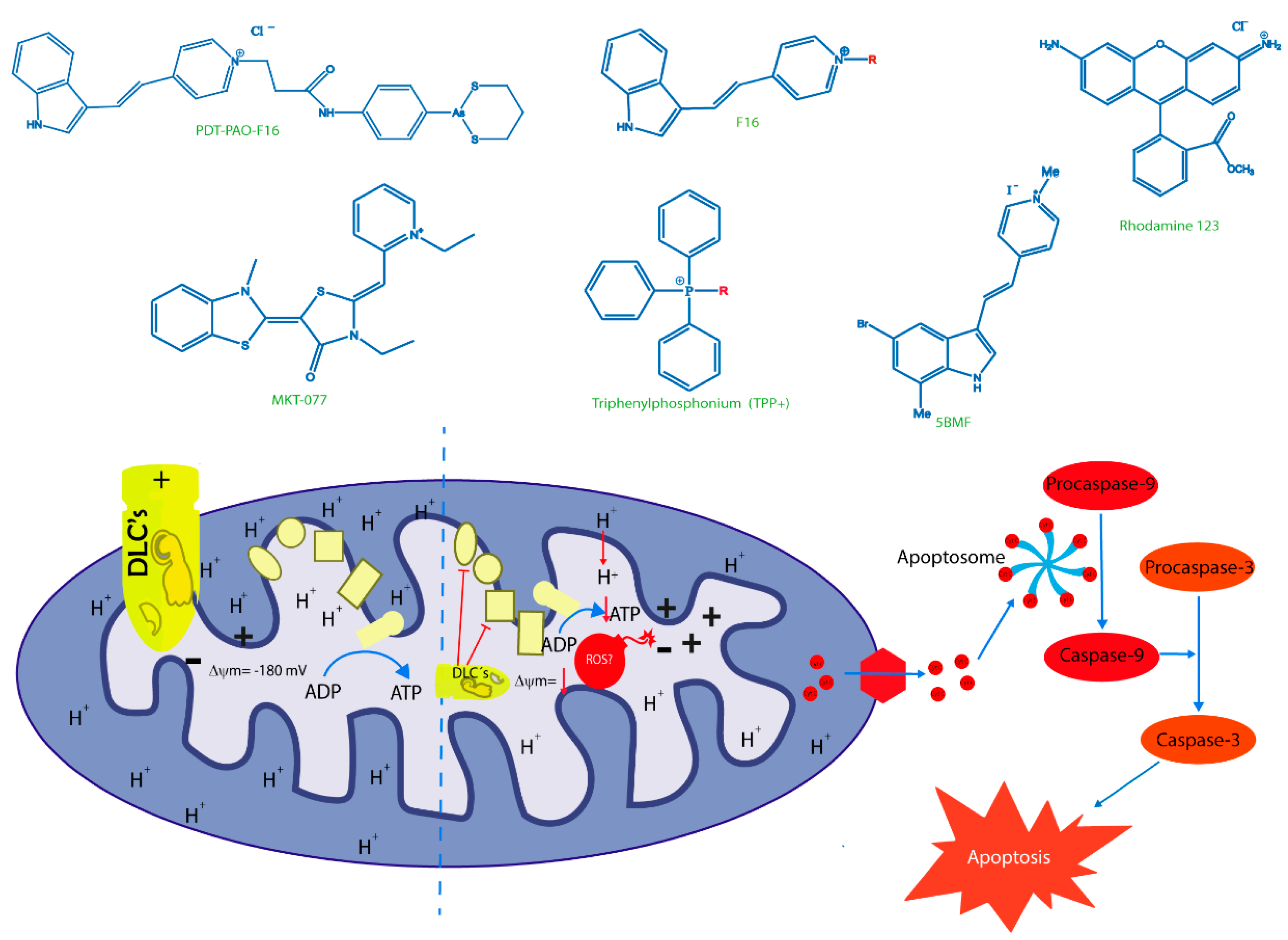{kind=link}
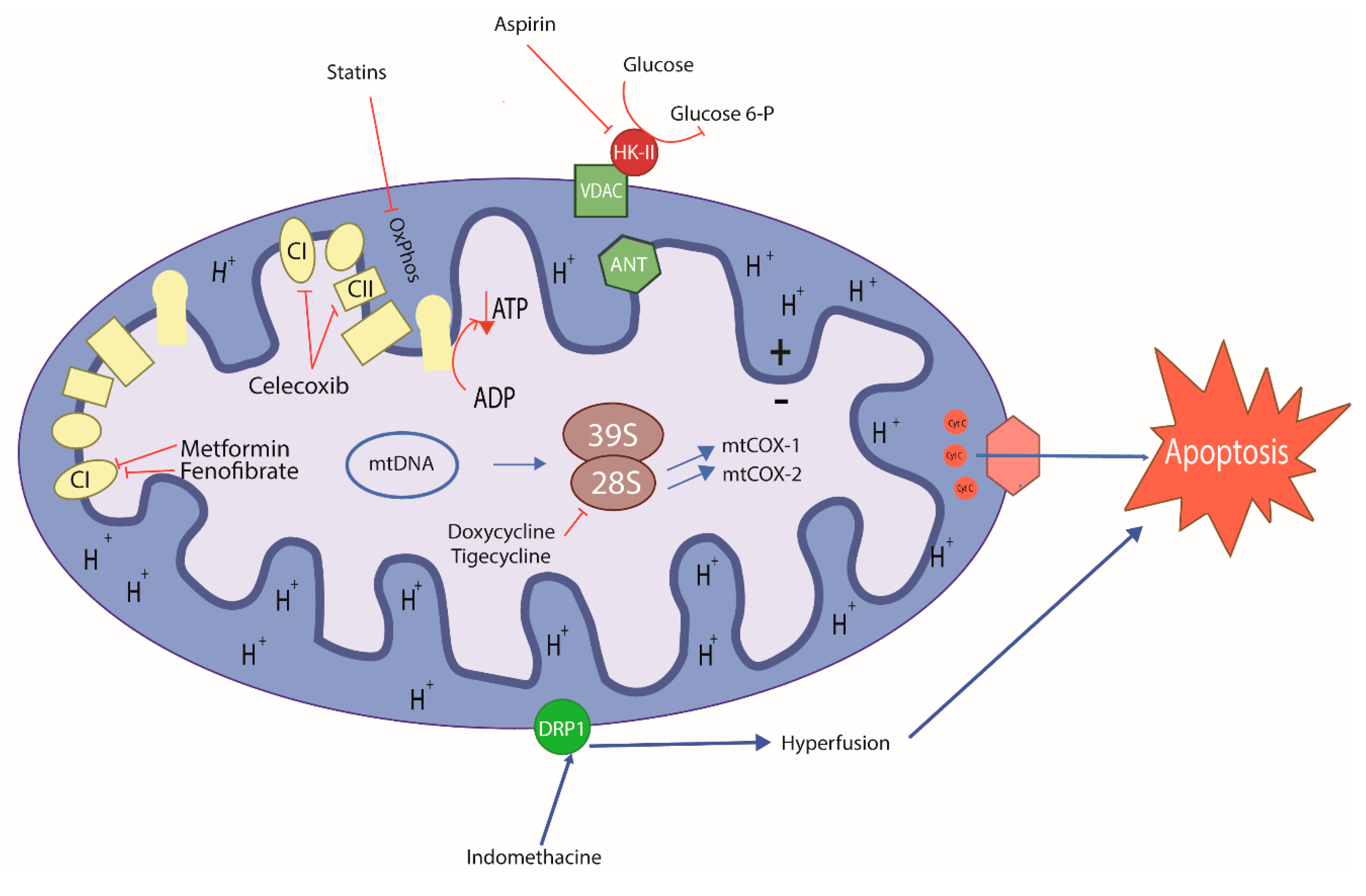{kind=link}
| Polyphenol | Chemical Structure | Model | Target | Consequences | Ref. |
|---|---|---|---|---|---|
| Luteolin (flavonoid) |  | SW1990 xenograft Model | ↓ bcl-2 | Mitochondrial Permeabilization Cellular death ↓Tumor Growth | [47] |
| HT-29 | ↑ GSH ↑ caspase 3 and 9 | ↓ ∆ψm ↓Proliferation Apoptosis | [48] | ||
| SKM-1 Rat cancerous hepatocytes | ↑ ROS ↑ caspase 3 and 9 | ↓ ∆ψm Apoptosis | [52,53] | ||
| Heperidin (flavonoid) |  | Mouse lung cancer | ↑ GSH, SOD | ↓Tumor Incidence, ↓PCNA | [54] |
| SGC-7901, MGC-803 and HGC-27 Gastric xenograft model | ↑ ROS; ↓bcl-2 ↑ Caspase 3 and 9 | ↓Proliferation ↓ ∆ψm Apoptosis ↓Tumor Growth | [55] | ||
| Curcumin (flavonoid) |  | p53-deficient H1299 | ↓bcl-2; ↑ Bax | ↓Proliferation Necrosis | [56] |
| A549, SPC-A1 | ↑ ROS | ↓ ∆ψm ↓Proliferation Apoptosis | [57] | ||
| HCT116, HT29 | ↓ HK-II | HK-II mitochondrial dissociation Apoptosis | [58] | ||
| Ellagic acid (Hydroxybenzoates) |  | TSGH8301 SH-SY5Y | ↑ Caspase 3 and 9 | ↓ ∆ψm ↓Proliferation Apoptosis | [59,60] |
| B-lymphocytes from CLL patients | ↑ ROS | Apoptosis | [53,61] | ||
| HOP62 and H1975 Mouse lung cancer | ↓ OXPHOS | ↓ATP ↓Tumor Growth | [60] | ||
| Resveratrol (Silbene) |  | H446 TRAMP cells | ↓bcl-2 | ↓ ∆ψm ↓ Cell Viability | [62,63] |
| HeLa and MDA-MB-231 | ↑ ROS; ↓ GSH ↓ OXPHOS | ↓Proliferation ↑ Mitophagy | [64] | ||
| Epigallocatechin Gallate (EGCG) (Catechin) |  | Hep2 | ↓bcl-2; ↑ Bax | ↓ ∆ψm | [65] |
| MIA PaCa-2 and SMMC7721 | ↑ ROS | Apoptosis | [66,67] | ||
| SCC-25 | ↑ ROS | Cytotoxicity | [68] | ||
| REN | ↓ OXPHOS (I, III Complex) | ↓Proliferation Apoptosis | [69] |
6. New Trojan Horses for Targeting Tumor Cell Mitochondria
7. Old Drugs Targeting Tumor and Cancer Stem Cell Mitochondria
| Name | Pharmacophore | Mitochondrial Target | Year | References |
|---|---|---|---|---|
| CoQ10 |  | OXPHOS | 2000 | [107] |
| α-Tocopherol |  | OXPHOS | 2004 | [108] |
| Gallic Acid |  | ΔΨm, uncoupling effect | 20142017 | [23,109] |
| Doxorubicin |  | ROS generation | 2014 | [110] |
| F16 |  | ΔΨm | 2014 | [95] |
| Chlorambucil |  | mtDNA | 2013 | [111] |
| DNP (2,4-dinitrophenol) |  | ΔΨm | 2006 | [112] |
| Lonidamide |  | OXPHOS (complex I) | 2019 | [113] |
| Metformin |  | OXPHOS (complex I) | 2016 | [114,115] |
| Paraquat |  | ROS generation | 2020 | [115] |
| Artemisinin |  | ΔΨm | 2017 | [116] |
| Curcumin |  | ROS generation, ΔΨm, AKT inhibition and STAT3 phosphorylation | 2014 | [117,118] |
| Benzoic acid derivatives |  | OXPHOS uncoupling effect | 2016 | [26] |
8. Concluding Remarks and Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Vidali, S.; Aminzadeh, S.; Lambert, B.; Rutherford, T.; Sperl, W.; Kofler, B.; Feichtinger, R.G. Mitochondria: The ketogenic diet—A metabolism-based therapy. Int. J. Biochem. Cell Biol. 2015, 63, 55–59. [Google Scholar] [CrossRef]
- De Berardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2. [Google Scholar] [CrossRef]
- Han, Y.; Cho, U.; Kim, S.; Park, I.S.; Cho, J.H.; Dhanasekaran, D.N.; Song, Y.S. Tumour microenvironment on mitochondrial dynamics and chemoresistance in cancer. Free Radic. Res. 2018, 52, 1271–1287. [Google Scholar] [CrossRef]
- Begg, K.; Tavassoli, M. Inside the hypoxic tumour: Reprogramming of the DDR and radioresistance. Cell Death Discov. 2020, 6. [Google Scholar] [CrossRef]
- Carnero, A.; Lleonart, M. The hypoxic microenvironment: A determinant of cancer stem cell evolution. Inside Cell 2016, 1, 96–105. [Google Scholar] [CrossRef]
- Pastò, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and mitochondrial respiration: A neglected connection. Cancer Res. 2015, 75, 3685–3686. [Google Scholar] [CrossRef]
- Warburg, O. On Respiratory Impairment in Cancer Cells. Science 1956, 124, 267–272. [Google Scholar]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy Samuel. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef]
- Cai, F.F.; Kohler, C.; Zhang, B.; Chen, W.J.; Barekati, Z.; Garritsen, H.S.P.; Lenner, P.; Toniolo, P.; Zhang, J.J.; Zhong, X.Y. Mutations of mitochondrial DNA as potential biomarkers in breast cancer. Anticancer Res. 2011, 31, 4267–4271. [Google Scholar]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef]
- Gammon, L.; Mackenzie, I.C. Roles of hypoxia, stem cells and epithelial-mesenchymal transition in the spread and treatment resistance of head and neck cancer. J. Oral Pathol. Med. 2016, 45, 77–82. [Google Scholar] [CrossRef]
- Magrì, A.; Reina, S.; De Pinto, V. VDAC1 as pharmacological target in cancer and neurodegeneration: Focus on its role in apoptosis. Front. Chem. 2018, 6, 108. [Google Scholar] [CrossRef]
- Shoshan-barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. Molecular Aspects of Medicine VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Liu, C.; Wang, X.; Zhang, Y. The Roles of HK2 on Tumorigenesis of Cervical Cancer. Technol. Cancer Res. Treat. 2019, 18, 1533033819871306. [Google Scholar] [CrossRef]
- Ralph, S.J.; Neuzil, J. Mitochondria as targets for cancer therapy. Mol. Nutr. Food Res. 2009, 53, 9–28. [Google Scholar] [CrossRef]
- Samuel, B.; Lampidis, T.; McIsaac, R.; Bo Chen, L. Anticarcinoma activity in vivo of rhodamine 123, a Mitochondrial-Specific Dye. Science 1983, 222, 169–172. [Google Scholar]
- Murphy, M.P. Biochimica et Biophysica Acta Targeting lipophilic cations to mitochondria. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Rohlena, J.; Dong, L.-F.; Ralph, S.J.; Neuzil, J. Anticancer Drugs Targeting the Mitochondrial Electron Transport Chain. Antioxid. Redox Signal. 2011, 15, 2951–2974. [Google Scholar] [CrossRef] [PubMed]
- Jara, J.A.; Castro-Castillo, V.; Saavedra-Olavarría, J.; Peredo, L.; Pavanni, M.; Jaña, F.; Letelier, M.E.; Parra, E.; Becker, M.I.; Morello, A.; et al. Antiproliferative and uncoupling effects of delocalized, lipophilic, cationic gallic acid derivatives on cancer cell lines. Validation in vivo in singenic mice. J. Med. Chem. 2014, 57, 2440–2454. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Acuña, C.; Fuentes-Retamal, S.; Guzmán-Rivera, D.; Peredo-Silva, L.; Madrid-Rojas, M.; Rebolledo, S.; Castro-Castillo, V.; Pavani, M.; Catalán, M.; Maya, J.D.; et al. Destabilization of mitochondrial functions as a target against breast cancer progression: Role of TPP+-linked-polyhydroxybenzoates. Toxicol. Appl. Pharmacol. 2016, 309, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Panina, S.B.; Baran, N.; Brasil da Costa, F.H.; Konopleva, M.; Kirienko, N.V. A mechanism for increased sensitivity of acute myeloid leukemia to mitotoxic drugs. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Xiao, Y.; Fu, B.; Qin, Z. TPP-based mitocans: A potent strategy for anticancer drug design. RSC Med. Chem. 2020, 11, 858–875. [Google Scholar] [CrossRef]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Thomlinson, R.H.; Gray, L.H. The Histological Structure of Some Human Lung Cancers and the Possible Implications for Radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K.; Kharazinejad, E.; Majidpoor, J. Hypoxia in solid tumors: A key promoter of cancer stem cell (CSC) resistance. J. Cancer Res. Clin. Oncol. 2020, 146, 19–31. [Google Scholar] [CrossRef]
- Hirayama, A.; Kami, K.; Sugimoto, M.; Sugawara, M.; Toki, N.; Onozuka, H.; Kinoshita, T.; Saito, N.; Ochiai, A.; Tomita, M.; et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009, 69, 4918–4925. [Google Scholar] [CrossRef]
- Zhang, B.; Xie, Z.; Li, B. The clinicopathologic impacts and prognostic significance of GLUT1 expression in patients with lung cancer: A meta-analysis. Gene 2019, 689, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Otto Warburg, B.; Wind, F.; Negelein, N. The metabolism of tumors in the body. J. Gen. Physiol. 1923, 309, 397–519. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; De Milito, A.; Demiroglu-Zergeroglu, A.; Gullbo, J.; D’Arcy, P.; Linder, S. Eradicating Quiescent Tumor Cells by Targeting Mitochondrial Bioenergetics. Trends Cancer 2016, 2, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.; Pandey, S. Exploiting mitochondrial vulnerabilities to trigger apoptosis selectively in cancer cells. Cancers 2019, 11, 916. [Google Scholar] [CrossRef]
- Durazzo, A.; Lucarini, M.; Souto, E.B.; Cicala, C.; Caiazzo, E.; Izzo, A.A.; Novellino, E.; Santini, A. Polyphenols: A concise overview on the chemistry, occurrence, and human health. Phyther. Res. 2019, 33, 2221–2243. [Google Scholar] [CrossRef]
- Potì, F.; Santi, D.; Spaggiari, G.; Zimetti, F.; Zanotti, I. Polyphenol health effects on cardiovascular and neurodegenerative disorders: A review and meta-analysis. Int. J. Mol. Sci. 2019, 20, 351. [Google Scholar] [CrossRef]
- Gu, H.-F.; Mao, X.-Y.; Du, M. Prevention of breast cancer by dietary polyphenols—Role of cancer stem cells Hao-Feng. Crit. Rev. Food Sci. Nutr. 2020, 60, 810–825. [Google Scholar] [CrossRef]
- Mileo, A.M.; Miccadei, S. Polyphenols as Modulator of Oxidative Stress in Cancer Disease: New Therapeutic Strategies. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef]
- Ki, W.L.; Hyong, J.L. The roles of polyphenols in cancer chemoprevention. BioFactors 2006, 26, 105–121. [Google Scholar] [CrossRef]
- Asensi, M.; Ortega, A.; Mena, S.; Feddi, F.; Estrela, J.M. Natural polyphenols in cancer therapy. Crit. Rev. Clin. Lab. Sci. 2011, 48, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.L.; Tedesco, I.; Spagnuolo, C.; Russo, M. Antioxidant polyphenols in cancer treatment: Friend, foe or foil? Semin. Cancer Biol. 2017, 46, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Taparia, S.S.; Khanna, A. Procyanidin-rich extract of natural cocoa powder causes ROS-mediated caspase-3 dependent apoptosis and reduction of pro-MMP-2 in epithelial ovarian carcinoma cell lines. Biomed. Pharmacol. 2016, 83, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Khan, H.; Fratantonio, D.; Hasan, M.M.; Sharifi, S.; Fathi, N.; Ullah, H.; Rastrelli, L. Apoptosis induced by luteolin in breast cancer: Mechanistic and therapeutic perspectives. Phytomedicine 2019, 59. [Google Scholar] [CrossRef]
- Cook, M.T. Mechanism of metastasis suppression by luteolin in breast cancer. Breast Cancer Targets Ther. 2018, 10, 89–100. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Chen, L.; Li, H. The dietary compound luteolin inhibits pancreatic cancer growth by targeting BCL-2. Food Funct. 2018, 9, 3018–3027. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Ryu, Y.S.; Hyun, Y.J.; Park, J.E.; Shilnikova, K.; Zhen, A.X.; Kang, H.K.; Koh, Y.S.; Jeong, Y.J.; et al. Luteolin induces apoptotic cell death via antioxidant activity in human colon cancer cells. Int. J. Oncol. 2017, 51, 1169–1178. [Google Scholar] [CrossRef]
- Pandurangan, A.K.; Ananda Sadagopan, S.K.; Dharmalingam, P.; Ganapasam, S. Luteolin, a bioflavonoid inhibits Azoxymethane-induced colorectal cancer through activation of Nrf2 signaling. Toxicol. Mech. Methods 2014, 24, 13–20. [Google Scholar] [CrossRef]
- Pandurangan, A.K.; Esa, N.M. Luteolin, a bioflavonoid inhibits colorectal cancer through modulation of multiple signaling pathways: A review. Asian Pac. J. Cancer Prev. 2014, 15, 5501–5508. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, H.; Jia, Y.; Pan, H.; Ding, H. Luteolin induces apoptosis by ROS/ER stress and mitochondrial dysfunction in gliomablastoma. Cancer Chemother. Pharmacol. 2017, 79, 1031–1041. [Google Scholar] [CrossRef]
- Dong, W.; Lin, Y.; Cao, Y.; Liu, Y.; Xie, X.; Gu, W. Luteolin induce myelodisplatic syndrome-derived cell apoptosis via p53-dependent mitochondrial signaling pathway mediated by reactive oxygen species. Int. J. Mol. Med. 2018, 42, 1106–1115. [Google Scholar] [PubMed]
- Seydi, E.; Salimi, A.; Rasekh, H.R.; Mohsenifar, Z.; Pourahmad, J. Selective Cytotoxicity of Luteolin and Kaempferol on Cancerous Hepatocytes Obtained from Rat Model of Hepatocellular Carcinoma: Involvement of ROS-Mediated Mitochondrial Targeting. Nutr. Cancer 2018, 70, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Kamaraj, S.; Ramakrishnan, G.; Anandakumar, P.; Jagan, S.; Devaki, T. Antioxidant and anticancer efficacy of hesperidin in benzo(a)pyrene induced lung carcinogenesis in mice. Investig. New Drugs 2009, 27, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, D.; Vikash; Song, J.; Wang, J.; Yi, J.; Dong, W. Hesperetin Induces the Apoptosis of Gastric Cancer Cells via Activating Mitochondrial Pathway by Increasing Reactive Oxygen Species. Dig. Dis. Sci. 2015, 60, 2985–2995. [Google Scholar] [CrossRef] [PubMed]
- Feie, L.; Xi, C.; Bing, X.; Zhou, H. Curcumin induce p53-independent necrosis in H1299 cells via a mitochondria-associated pathway. Mol. Med. Rep. 2015, 12, 7806–7814. [Google Scholar]
- Wang, C.; Song, X.; Shang, M.; Zou, W.; Zhang, M.; Wei, H.; Shao, H. Curcumin exerts cytotoxicity dependent on reactive oxygen species accumulation in non-small-cell lung cancer cells. Future Oncol. 2019, 15, 1243–1253. [Google Scholar] [CrossRef]
- Wang, K.; Fan, H.; Chen, Q.; Ma, G.; Zhu, M.; Zhang, X.; Zhang, Y.; Yu, J. Curcumin inhibits aerobic glycolysis and induces mitochondrial-mediated apoptosis through hexokinase II in human colorectal cancer cells in vitro. Anticancer Drugs 2015, 26, 15–24. [Google Scholar] [CrossRef]
- Ho, C.-C.; Huang, A.-C.; Yu, C.-S.; Lien, J.-C.; Wu, S.-H.; Huang, Y.-P.; Huang, H.-Y.; Kuo, J.-H.; Liao, W.-Y.; Yang, J.-S.; et al. Ellagic Acid Induces Apoptosis in TSGH8301 Human Bladder Cancer Cells Through the Endoplasmic Reticulum Stress- and Mitochondria-Dependent Signaling Pathways. Environ. Toxicol. 2014, 29, 1262–1274. [Google Scholar] [CrossRef]
- Alfredsson, C.F.; Ding, M.; Liang, Q.L.; Sundström, B.E.; Nånberg, E. Ellagic acid induces a dose- and time-dependent depolarization of mitochondria and activation of caspase-9 and -3 in human neuroblastoma cells. Biomed. Pharmacol. 2014, 68, 129–135. [Google Scholar] [CrossRef]
- Salimi, A.; Roudkenar, M.H.; Sadeghi, L.; Mohseni, A.; Seydi, E.; Pirahmadi, N.; Pourahmad, J. Ellagic acid, a polyphenolic compound, selectively induces ROS-mediated apoptosis in cancerous B-lynphocytes of CLL patients by directly targeting mitochondria. Redox Biol. 2015, 6, 461–471. [Google Scholar]
- Li, W.; Shi, Y.; Wang, R.; Pan, L.; Ma, L.; Jin, F. Resveratrol promotes the sensitivity of small-cell lung cancer H446 cells to cisplatin by regulating intrinsic apoptosis. Int. J. Oncol. 2018, 53, 2123–2130. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.J.; Eroglu, E.; Stokes, J.A.; Scissum-Gunn, K.; Saldanha, S.N.; Singh, U.P.; Manne, U.; Ponnazhagan, S.; Mishra, M.K. Resveratrol induces mitochondria-mediated, caspaseindependent apoptosis in murine prostate cancer cells. Oncotarget 2017, 8, 20895–20908. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Enríquez, S.; Pacheco-Velázquez, S.C.; Marín-Hernández, Á.; Gallardo-Pérez, J.C.; Robledo-Cadena, D.X.; Hernández-Reséndiz, I.; García-García, J.D.; Belmont-Díaz, J.; López-Marure, R.; Hernández-Esquivel, L.; et al. Resveratrol inhibits cancer cell proliferation by impairing oxidative phosphorylation and inducing oxidative stress. Toxicol. Appl. Pharmacol. 2019, 370, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jeong, Y.J.; Lee, S.W.; Kim, D.; Oh, S.J.; Lim, H.S.; Oh, H.K.; Kim, S.H.; Kim, W.J.; Jung, J.Y. EGCG induces apoptosis in human laryngeal epidermoid carcinoma Hep2 cells via mitochondria with the release of apoptosis-inducing factor and endonuclease G. Cancer Lett. 2010, 290, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Qanungo, S.; Das, M.; Haldar, S.; Basu, A. Epigallocatechin-3-gallate induces mitochondrial membrane depolarization and caspase-dependent apoptosis in pancreatic cancer cells. Carcinogenesis 2005, 26, 958–967. [Google Scholar] [CrossRef]
- Li, W.; Nie, S.; Yu, Q.; Xie, M.Y. (-)-Epigallocatechin-3-Gallate Induces Apoptosis of Human Hepatoma Cells By Mitochondrial Pathways Related To Reactive Oxygen Species. J. Agric. Food Chem. 2009, 57, 6685–6691. [Google Scholar] [CrossRef]
- Sun, Z.; Yu, G.; Huang, C.; Bu, L.; Liu, J. Hypoxia induces TFE3 expression in head and neck squamous cell carcinoma. Oncotarget 2016, 7. [Google Scholar] [CrossRef][Green Version]
- Valenti, D.; De Bari, L.; Manente, G.A.; Rossi, L.; Mutti, L.; Moro, L.; Vacca, R.A. Negative modulation of mitochondrial oxidative phosphorylation by epigallocatechin-3 gallate leads to growth arrest and apoptosis in human malignant pleural mesothelioma cells. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 2085–2096. [Google Scholar] [CrossRef]
- Chen, A.; Jiang, P.; Zeb, F.; Wu, X.; Xu, C.; Chen, L.; Feng, Q. EGCG regulates CTR1 expression through its pro-oxidative property in non-small-cell lung cancer cells. J. Cell. Physiol. 2020, 1–12. [Google Scholar] [CrossRef]
- Sang, S.; Lee, M.J.; Hou, Z.; Ho, C.T.; Yang, C.S. Stability of tea polyphenol (-)-epigallocatechin-3-gallate and formation of dimers and epimers under common experimental conditions. J. Agric. Food Chem. 2005, 53, 9478–9484. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, S.; Fichna, J.; Lewandowska, U. Polyphenols as mitochondria-targeted anticancer drugs. Cancer Lett. 2015, 366, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Moini, H.; Arroyo, A.; Vaya, J.; Packer, L. Bioflavonoid effects on the mitochondrial respiratory electron transport chain and cytochrome c redox state. Redox Rep. 1999, 4, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Tewari, D.; Ahmed, T.; Chirasani, V.R.; Singh, P.K.; Maji, S.K.; Senapati, S.; Bera, A.K. Modulation of the mitochondrial voltage dependent anion channel (VDAC) by curcumin. Biochim. Biophys. Acta Biomembr. 2015, 1848, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Li, M.; Liu, W.-B.; Zhou, Z.S.; Zhang, R.; Li, J.L.; Zhou, K.C. Epigallocatechin gallate inhibits human tongue carcinoma cells via HK2-mediated glycolysis. Oncol. Rep. 2015, 33, 1533–1539. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, J.; Dai, W.; Zhang, Q.; Feng, J.; Wu, L.; Liu, T.; Yu, Q.; Xu, S.; Wang, W.; et al. Genistein suppresses aerobic glycolysis and induces hepatocellular carcinoma cell death. Br. J. Cancer 2017, 117, 1518–1528. [Google Scholar] [CrossRef]
- Wu, H.; Pan, L.; Gao, C.; Xu, H.; Li, Y.; Zhang, L.; Ma, L.; Sun, X.; Qin, H. Quercetin Inhibits the Proliferation of Glycolysis-Addicted HCC Cells by Reducing Hexokinase 2 and Akt-mTOR Pathway. Molecules 2019, 24, 1993. [Google Scholar] [CrossRef]
- Li, W.; Ma, X.; Li, N.; Liu, H.; Dong, Q.; Zhang, J.; Yang, C.; Liu, Y.; Liang, Q.; Zhang, S.; et al. Resveratrol inhibits Hexokinases II mediated glycolysis in non-small cell lung cancer via targeting Akt signaling pathway. Exp. Cell Res. 2016, 349, 320–327. [Google Scholar] [CrossRef]
- García-Aranda, M.; Pérez-Ruiz, E.; Redondo, M. Bcl-2 inhibition to overcome resistance to chemo-and immunotherapy. Int. J. Mol. Sci. 2018, 19, 3950. [Google Scholar] [CrossRef]
- Verma, S.; Singh, A.; Kumari, A.; Tyagi, C.; Goyal, S.; Jamal, S.; Grover, A. Natural polyphenolic inhibitors against the antiapoptotic BCL-2. J. Recept. Signal Transduct. 2017, 37, 391–400. [Google Scholar] [CrossRef]
- Wang, H.; Luo, Y.; Qiao, T.; Wu, Z.; Huang, Z. Luteolin sensitizes the antitumor effect of cisplatin in drug-resistant ovarian cancer via induction of apoptosis and inhibition of cell migration and invasion. J. Ovarian Res. 2018, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, Y.; Chen, K.; Shi, P.; Li, Y.; Deng, M.; Jiang, Z.; Wang, X.; Li, P.; Xu, B. The pan-Bcl2 Inhibitor AT101 Activates the Intrinsic Apoptotic Pathway and Causes DNA Damage in Acute Myeloid Leukemia Stem-Like Cells. Target. Oncol. 2017, 12, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Fong, D.; Chan, M.M. Dietary Phytochemicals Target Cancer Stem Cells for Cancer Chemoprevention. In Mitochondria as Targets for Phytochemicals in Cancer Prevention and Therapy; Springer: Berlin, Germany, 2013; pp. 85–125. ISBN 9781461493266. [Google Scholar]
- García-Heredia, J.M.; Carnero, A. Role of Mitochondria in Cancer Stem Cell Resistance. Cells 2020, 9, 1693. [Google Scholar] [CrossRef]
- Jara, J.A.; Rojas, D.; Castro-Castillo, V.; Fuentes-Retamal, S.; Sandoval-Acuña, C.; Parra, E.; Pavani, M.; Maya, J.D.; Ferreira, J.; Catalán, M. Novel benzoate-lipophilic cations selectively induce cell death in human colorectal cancer cell lines. Toxicol. In Vitro 2020, 65, 104814. [Google Scholar] [CrossRef]
- Lichtshteint, D.; Kabackt, H.R.; Blumet, A.J. Use of a Lipophilic Cation for Determination of Membrane Potential in Neuroblastoma-Glioma Hybrid Cell Suspension. Proc. Natl. Acad. Sci. USA 1979, 76, 650–654. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 425–434. [Google Scholar] [CrossRef]
- Bagkos, G.; Koufopoulos, K.; Piperi, C. A new model for mitochondrial membrane potential production and storage. Med. Hypotheses 2014, 83, 175–181. [Google Scholar] [CrossRef]
- Zielonka, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2018, 117, 10043–10120. [Google Scholar] [CrossRef]
- Koya, K.; Li, Y.; Wang, H.; Ukai, T.; Tatsuta, N.; Kawakami, M.; Shishido, T.; Chen, L.B. MKT-077, a Novel Rhodacyanine Dye in Clinical Trials, Exhibits Anticarcinoma Activity in Preclinical Studies Based on Selective Mitochondrial Accumulation. Cancer Res. 1996, 56, 538–544. [Google Scholar]
- Propper, D.J.; Braybrooke, J.P.; Taylor, D.J.; Lodi, R.; Styles, P.; Cramer, J.A.; Collins, W.C.J.; Levitt, N.C.; Talbot, D.C.; Ganesan, T.S.; et al. Phase I trial of the selective mitochondrial toxin MKT 077 in chemo-resistant solid tumours. Ann. Oncol. 1999, 10, 923–927. [Google Scholar] [CrossRef]
- Jones, L.W.; Narayan, K.S.; Shapiro, C.E.; Sweatman, T.W. Rhodamine-123: Therapy for Hormone Refractory Prostate Cancer, A Phase I Clinical Trial. J. Chemother. 2005, 17, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; Berardi, M.J.; Scorrano, L.; Korsmeyer, S.J.; Leder, P. A novel mitochondriotoxic small molecule that selectively inhibits tumor cell growth. Cancer Cell 2002, 2, 29–42. [Google Scholar] [CrossRef]
- Wang, J.; He, H.; Xiang, C.; Fan, X.; Yang, L.; Yuan, L.; Jiang, F.; Liu, Y. Uncoupling Effect of F16 Is Responsible for Its Mitochondrial Toxicity and Anticancer Activity. Toxicol. Sci. 2018, 161, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Cao, Q.; Wang, X.; Cheng, Z. Design, synthesis and biological evaluation of mitochondria targeting theranostic agents. Chem. Commun. 2014, 50, 8919–8922. [Google Scholar] [CrossRef]
- Rathinavelu, A.; Alhazzani, K.; Kanagasabai, T. Anti-cancer effects of F16: A novel vascular endothelial growth factor receptor—Specific inhibitor. Tumor Biol. 2017, 1–12. [Google Scholar] [CrossRef]
- Qian, K.; Chen, H.; Qu, C.; Qi, J.; Du, B.; Ko, T.; Xiang, Z.; Kandawa-schulz, M.; Wang, Y.; Cheng, Z. Mitochondria-targeted delocalized lipophilic cation complexed with human serum albumin for tumor cell imaging and treatment. Nanomed. Nanotechnol. Biol. Med. 2020, 23, 102087. [Google Scholar] [CrossRef]
- Liu, Y.J.; Fan, X.Y.; Zhang, D.D.; Xia, Y.Z.; Hu, Y.J.; Jiang, F.L.; Zhou, F.L.; Liu, Y. Dual Inhibition of Pyruvate Dehydrogenase Complex and Respiratory Chain Complex Induces Apoptosis by a Mitochondria-Targeted Fluorescent Organic Arsenical in vitro and in vivo. ChemMedChem 2020, 15, 552–558. [Google Scholar] [CrossRef]
- Parton, R.L.; Lenhard, J.R.; Gilman, P.B., Jr. Correlation of Anti-Cancer Activity of Dyes with Redox Potentials. U.S. Patent 10,983,369, 11 May 2006. [Google Scholar]
- Yang, N.; Gilman, P.; Mirzayans, R.; Sun, X.; Touret, N. Characterization of the Apoptotic Response Induced by the Cyanine Dye D112: A Potentially Selective Anti-Cancer Compound. PLoS ONE 2015, 10, e0125381. [Google Scholar] [CrossRef]
- Yang, N.; Weinfeld, M.; Lemieux, H.; Montpetit, B.; Goping, I.S. Photo-activation of the delocalized lipophilic cation D112 potentiates cancer selective ROS production and apoptosis. Cell Death Dis. 2017, 8, e2587. [Google Scholar] [CrossRef]
- Sica, V.; Bravo-San Pedro, J.M.; Stoll, G.; Kroemer, G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int. J. Cancer 2020, 146, 10–17. [Google Scholar] [CrossRef]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Zimorski, V.; Ku, C.; Martin, W.F.; Gould, S.B. Endosymbiotic theory for organelle origins. Curr. Opin. Microbiol. 2014, 22, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Esposti, M.D.; Chouaia, B.; Comandatore, F.; Crotti, E.; Sassera, D.; Lievens, P.M.J.; Daffonchio, D.; Bandi, C. Evolution of mitochondria reconstructed from the energy metabolism of living bacteria. PLoS ONE 2014, 9, e96566. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.J.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999, 716, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, A.; Kotamraju, S.; Kalivendi, S.V.; Matsunaga, T.; Shang, T.; Keszler, A.; Joseph, J.; Kalyanaraman, B. Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. J. Biol. Chem. 2004, 279, 37575–37587. [Google Scholar] [CrossRef]
- Peredo-silva, L.; Fuentes-retamal, S.; Sandoval-acuña, C.; Pavani, M.; Maya, J.D.; Castro-castillo, V.; Madrid-rojas, M.; Rebolledo, S.; Kemmerling, U.; Parra, E.; et al. Derivatives of alkyl gallate triphenylphosphonium exhibit antitumor activity in a syngeneic murine model of mammary adenocarcinoma. Toxicol. Appl. Pharmacol. 2017, 329, 334–346. [Google Scholar] [CrossRef]
- Han, M.; Vakili, M.R.; Soleymani Abyaneh, H.; Molavi, O.; Lai, R.; Lavasanifar, A. Mitochondrial delivery of doxorubicin via triphenylphosphine modification for overcoming drug resistance in MDA-MB-435/DOX cells. Mol. Pharm. 2014, 11, 2640–2649. [Google Scholar] [CrossRef]
- Millard, M.; Gallagher, J.D.; Olenyuk, B.Z.; Neamati, N. A selective mitochondrial-targeted chlorambucil with remarkable cytotoxicity in breast and pancreatic cancers. J. Med. Chem. 2013, 56, 9170–9179. [Google Scholar] [CrossRef]
- Blaikie, F.H.; Brown, S.E.; Samuelsson, L.M.; Brand, M.D.; Smith, R.A.J.; Murphy, M.P. Targeting dinitrophenol to mitochondria: Limitations to the development of a self-limiting mitochondrial protonophore. Biosci. Rep. 2006, 26, 231–243. [Google Scholar] [CrossRef]
- Cheng, G.; Zhang, Q.; Pan, J.; Lee, Y.; Ouari, O.; Hardy, M.; Zielonka, M.; Myers, C.R.; Zielonka, J.; Weh, K.; et al. Targeting lonidamine to mitochondria mitigates lung tumorigenesis and brain metastasis. Nat. Commun. 2019, 10, 2205. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zielonka, J.; Ouari, O.; Lopez, M.; McAllister, D.; Boyle, K.; Barrios, C.S.; Weber, J.J.; Johnson, B.D.; Hardy, M.; et al. Mitochondria-targeted analogues of metformin exhibit enhanced antiproliferative and radiosensitizing effects in pancreatic cancer cells. Cancer Res. 2016, 76, 3904–3915. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.R.; Zielonka, J.; Kalyanaraman, B.; Hartley, R.C.; Murphy, M.P.; Avadhani, N.G. Mitochondria-targeted paraquat and metformin mediate ROS production to induce multiple pathways of retrograde signaling: A dose-dependent phenomenon. Redox Biol. 2020, 36, 101606. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Cao, Y.; Zhu, P.; Zhou, B. A mitochondria-targeting artemisinin derivative with sharply increased antitumor but depressed anti-yeast and anti-malaria activities. Sci. Rep. 2017, 7, 45665. [Google Scholar] [CrossRef] [PubMed]
- Reddy, C.A.; Somepalli, V.; Golakoti, T.; Kanugula, A.K.R.; Karnewar, S.; Rajendiran, K.; Vasagiri, N.; Prabhakar, S.; Kuppusamy, P.; Kotamraju, S.; et al. Mitochondrial-targeted curcuminoids: A strategy to enhance bioavailability and anticancer efficacy of curcumin. PLoS ONE 2014, 9, e89351. [Google Scholar] [CrossRef] [PubMed]
- Hasan, W.; Kori, R.K.; Thakre, K.; Yadav, R.S.; Jat, D. Synthesis, characterization and efficacy of mitochondrial targeted delivery of TPP-curcumin in rotenone-induced toxicity. DARU J. Pharm. Sci. 2019, 27, 557–570. [Google Scholar] [CrossRef]
- Protasoni, M.; Kroon, A.M.; Taanman, J.W. Mitochondria as oncotarget: A comparison between the tetracycline analogs doxycycline and COL-3. Oncotarget 2018, 9, 33818–33831. [Google Scholar] [CrossRef]
- Dijk, S.N.; Protasoni, M.; Elpidorou, M.; Kroon, A.M.; Taanman, J.W. Mitochondria as target to inhibit proliferation and induce apoptosis of cancer cells: The effects of doxycycline and gemcitabine. Sci. Rep. 2020, 10, 4363. [Google Scholar] [CrossRef]
- Fiorillo, M.; Tóth, F.; Sotgia, F.; Lisanti, M.P. Doxycycline, Azithromycin and vitamin C (DAV): A potent combination therapy for targeting mitochondria and eradicating cancer stem cells (CSCs). Aging 2019, 11, 2202–2216. [Google Scholar] [CrossRef]
- Song, H.; Fares, M.; Maguire, K.R.; Sidén, Å.; Potácová, Z. Cytotoxic effects of tetracycline analogues (doxycycline, minocycline and COL-3) in acute Myeloid leukemia HL-60 cells. PLoS ONE 2014, 9, e114457. [Google Scholar] [CrossRef]
- Rok, J.; Karkoszka, M.; Rzepka, Z.; Respondek, M.; Banach, K.; Beberok, A.; Wrześniok, D. Cytotoxic and proapoptotic effect of doxycycline—An in vitro study on the human skin melanoma cells. Toxicol. In Vitro 2020, 65. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Retamal, S.; Sandoval-Acuña, C.; Peredo-Silva, L.; Guzmán-Rivera, D.; Pavani, M.; Torrealba, N.; Truksa, J.; Castro-Castillo, V.; Catalán, M.; Kemmerling, U.; et al. Complex Mitochondrial Dysfunction Induced by TPP+-Gentisic Acid and Mitochondrial Translation Inhibition by Doxycycline Evokes Synergistic Lethality in Breast Cancer Cells. Cells 2020, 9, 407. [Google Scholar] [CrossRef] [PubMed]
- Scatena, C.; Roncella, M.; Di Paolo, A.; Aretini, P.; Menicagli, M.; Fanelli, G.; Marini, C.; Mazzanti, C.M.; Ghilli, M.; Sotgia, F.; et al. Doxycycline, an inhibitor of mitochondrial biogenesis, effectively reduces cancer stem cells (CSCs) in early breast cancer patients: A clinical pilot study. Front. Oncol. 2018, 8, 452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, L.; Zhang, F.; Vlashi, E. Doxycycline inhibits the cancer stem cell phenotype and epithelial-to-mesenchymal transition in breast cancer. Cell Cycle 2017, 16, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, F.; Chen, D.; Wang, L. Inhibition of mitochondrial respiration by tigecycline selectively targets thyroid carcinoma and increases chemosensitivity. Clin. Exp. Pharmacol. Physiol. 2019, 46, 890–897. [Google Scholar] [CrossRef]
- Jia, X.; Gu, Z.; Chen, W.; Jiao, J. Tigecycline targets nonsmall cell lung cancer through inhibition of mitochondrial function. Fundam. Clin. Pharmacol. 2016, 30, 297–306. [Google Scholar] [CrossRef]
- Tan, J.; Song, M.; Zhou, M.; Hu, Y. Antibiotic tigecycline enhances cisplatin activity against human hepatocellular carcinoma through inducing mitochondrial dysfunction and oxidative damage. Biochem. Biophys. Res. Commun. 2017, 483, 17–23. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, N.; He, B.; Pan, C.; Lan, Y.; Zhou, H.; Liu, X. Inhibition of autophagy enhances the selective anti-cancer activity of tigecycline to overcome drug resistance in the treatment of chronic myeloid leukemia. J. Exp. Clin. Cancer Res. 2017, 36, 43. [Google Scholar] [CrossRef]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemic stem cells. Nat. Med. 2018, 23, 1234–1240. [Google Scholar] [CrossRef]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem. Pharmacol. 2020, 180, 114147. [Google Scholar] [CrossRef]
- Sun, Y.; Huang, L.; Mackenzie, G.G.; Rigas, B. Oxidative stress mediates through apoptosis the anticancer effect of phospho-nonsteroidal anti-inflammatory drugs: Implications for the role of oxidative stress in the action of anticancer agents. J. Pharmacol. Exp. Ther. 2011, 338, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Raza, H.; John, A.; Benedict, S. Acetylsalicylic acid-induced oxidative stress, cell cycle arrest, apoptosis and mitochondrial dysfunction in human hepatoma HepG2 cells. Eur. J. Pharmacol. 2011, 668, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Inoue, T.; Ra, C. NSAIDs, mitochondria and calcium signaling: Special focus on aspirin/salicylates. Pharmaceuticals 2010, 3, 1594–1613. [Google Scholar] [CrossRef] [PubMed]
- Tewari, D.; Majumdar, D.; Vallabhaneni, S.; Bera, A.K. Aspirin induces cell death by directly modulating mitochondrial voltage-dependent anion channel (VDAC). Sci. Rep. 2017, 7, 45184. [Google Scholar] [CrossRef] [PubMed]
- Bank, A.; Yu, J.; Zhang, L. NSAIDs downregulate Bcl-XL and dissociate BAX and Bcl-X L to induce apoptosis in colon cancer cells. Nutr. Cancer 2008, 60, 98–103. [Google Scholar] [CrossRef]
- Chen, J.; Xu, R.; Xia, J.; Huang, J.; Su, B.; Wang, S. Aspirin inhibits hypoxia-mediated lung cancer cell stemness and exosome function. Pathol. Res. Pract. 2019, 215, 152379. [Google Scholar] [CrossRef]
- Chen, Z.; Li, W.; Qiu, F.; Huang, Q.; Jiang, Z.; Ye, J.; Cheng, P.; Low, C.; Guo, Y.; Yi, X.; et al. Aspirin cooperates with p300 to activate the acetylation of H3K9 and promote FasL-mediated apoptosis of cancer stem-like cells in colorectal cancer. Theranostics 2018, 8, 4447–4461. [Google Scholar] [CrossRef]
- Pritchard, R.; Rodríguez-Enríquez, S.; Pacheco-Velázquez, S.C.; Bortnik, V.; Moreno-Sánchez, R.; Ralph, S. Celecoxib inhibits mitochondrial O2 consumption, promoting ROS dependent death of murine and human metastatic cancer cells via the apoptotic signalling pathway. Biochem. Pharmacol. 2018, 154, 318–334. [Google Scholar] [CrossRef]
- Amanullah, A.; Mishra, R.; Upadhyay, A.; Reddy, P.P.; Das, R.; Mishra, A. Indomethacin elicits proteasomal dysfunctions develops apoptosis through mitochondrial abnormalities. J. Cell. Physiol. 2018, 233, 1685–1699. [Google Scholar] [CrossRef]
- Mazumder, S.; De, R.; Debsharma, S.; Bindu, S.; Maity, P.; Sarkar, S.; Saha, S.J.; Siddiqui, A.A.; Banerjee, C.; Nag, S.; et al. Indomethacin impairs mitochondrial dynamics by activating the PKCζ-p38-DRP1 pathway and inducing apoptosis in gastric cancer and normal mucosal cells. J. Biol. Chem. 2019, 294, 8238–8258. [Google Scholar] [CrossRef]
- Scatena, R.; Bottoni, P.; Giardina, B. Mitochondria, PPARs, and cancer: Is receptor-independent action of PPAR agonists a key? PPAR Res. 2008, 2008. [Google Scholar] [CrossRef]
- Wilk, A.; Wyczechowska, D.; Zapata, A.; Dean, M.; Mullinax, J.; Marrero, L.; Parsons, C.; Peruzzi, F.; Culicchia, F.; Ochoa, A.; et al. Molecular mechanisms of fenofibrate-induced metabolic catastrophe and glioblastoma cell death. Mol. Cell. Biol. 2015, 35, 182–198. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Peng, J.; Wang, Y.; Jiang, H.; Wang, W.; Dai, J.; Tang, M.; Wei, Y.; Kuang, H.; Xu, G.; et al. Fenofibrate-induced mitochondrial dysfunction and metabolic reprogramming reversal: The anti-tumor effects in gastric carcinoma cells mediated by the PPAR pathway. Am. J. Transl. Res. 2020, 12, 428–446. [Google Scholar] [PubMed]
- Du Souich, P.; Roederer, G.; Dufour, R. Myotoxicity of statins: Mechanism of action. Pharmacol. Ther. 2017, 175, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Christie, C.F.; Fang, D.; Hunt, E.G.; Morris, M.E.; Rovini, A.; Heslop, K.A.; Beeson, G.C.; Beeson, C.C.; Maldonado, E.N. Statin-dependent modulation of mitochondrial metabolism in cancer cells is independent of cholesterol content. FASEB J. 2019, 33, 8186–8201. [Google Scholar] [CrossRef]
- Lee, M.-H.; Cho, Y.S.; Han, Y.-M. Simvastatin Suppresses Self-Renewal of Mouse Embryonic Stem Cells by Inhibiting RhoA Geranylgeranylation. Stem Cells 2007, 25, 1654–1663. [Google Scholar] [CrossRef]
- Wang, C.; Li, P.; Xuan, J.; Zhu, C.; Liu, J.; Shan, L.; Du, Q.; Ren, Y.; Ye, J. Cholesterol Enhances Colorectal Cancer Progression via ROS Elevation and MAPK Signaling Pathway Activation. Cell. Physiol. Biochem. 2017, 42, 729–742. [Google Scholar] [CrossRef]
- Gauthaman, K.; Fong, C.Y.; Bongso, A. Statins, stem cells, and cancer. J. Cell. Biochem. 2009, 106, 975–983. [Google Scholar] [CrossRef]
- Likus, W.; Siemianowicz, K.; Bieńk, K.; Pakuła, M.; Pathak, H.; Dutta, C.; Wang, Q.; Shojaei, S.; Assaraf, Y.G.; Ghavami, S.; et al. Could drugs inhibiting the mevalonate pathway also target cancer stem cells? Drug Resist. Updates 2016, 25, 13–25. [Google Scholar] [CrossRef]
- Mok, E.H.K.; Lee, T.K.W. The pivotal role of the dysregulation of cholesterol homeostasis in cancer: Implications for therapeutic targets. Cancers 2020, 12, 1410. [Google Scholar] [CrossRef]
- Hwang, H.; Ryu, D.H.; Hwang, G.; Cheong, J. Integrated omics-analysis reveals Wnt-mediated metabolic reprogramming in cancer stem-like cells. Oncotarget 2016, 7, 48562–48576. [Google Scholar]
- Shin, M.K.; Cheong, J.H. Mitochondria-centric bioenergetic characteristics in cancer stem-like cells. Arch. Pharm. Res. 2019, 42, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Bridges, H.R.; Jones, A.J.Y.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Scott Budigner, G.R.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front. Endocrinol. 2018, 9, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Droguett, D.; Castillo, C.; Leiva, E.; Theoduloz, C.; Schmeda, G.; Kemmerling, U. Efficacy of quercetin against chemically induced murine oral squamous cell carcinoma. Oncol. Lett. 2015, 10, 2432–2438. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef]
- Sonnenblick, A.; Agbor-Tarh, D.; Bradbury, I.; Di Cosimo, S.; Azim, H.A.; Fumagalli, D.; Sarp, S.; Wolff, A.C.; Andersson, M.; Kroep, J.; et al. Impact of diabetes, insulin, and metformin use on the outcome of patients with human epidermal growth factor receptor 2-positive primary breast cancer: Analysis from the ALTTO phase III randomized trial. J. Clin. Oncol. 2017, 35, 1421–1429. [Google Scholar] [CrossRef]
- Chae, Y.K.; Arya, A.; Malecek, M.K.; Shin, D.S.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Altman, J.K.; Platanias, L.; et al. Repurposing metformin for cancer treatment: Current clinical studies. Oncotarget 2016, 7, 40767–40780. [Google Scholar] [CrossRef]



| Drug | Family Drug | Chemical Structure | Mechanism of Action | Effects in CSC | Mitochondrial Mechanism |
|---|---|---|---|---|---|
| Doxycycline | Tetracycline/Antibiotics |  | 30S ribosomal subunit inhibition in bacteria | ↓OXPHOS protein translation leading to OXPHOS inhibition | 28S mito-ribosomal subunit inhibition |
| Tigecycline | Glycylglycine |  | 30S ribosomal subunit inhibition in bacteria | ↓ OXPHOS proteins translation leading to OXPHOS inhibition | 28S mito-ribosomal subunit inhibition |
| Acetylsalicylic acid | Salicylates/NSAIDs |  | Inhibition of COX1 and COX2 | Inhibition COX-2. Histone H3K9 acetylation, leading to FasL-mediated apoptosis | VDAC inhibition |
| Indomethacin | Indole derivative/NSAIDs |  | Inhibition of COX1-COX2 | Apoptosis through mitochondrial hyper fission. Proteosome inhibition | Activation of DRP1 |
| Celecoxib | Pyrazole derivative/NSAIDs |  | Selectively inhibition of COX2 | OXPHOS inhibition | Inhibition of Complex I and III from ETC |
| Fenofibrate | Fibrate/Antilipemic |  | Activation of PPAR-α Receptor | OXPHOS inhibition | Inhibition of Complex I from ETC |
| Simvastatin | Statin/Antilipemic |  | HMG CoA reductase inhibition | OXPHOS inhibition | Unknown |
| Lovastatin | Statin/Antilipemic |  | HMG CoA reductase inhibition | OXPHOS inhibition | Unknown |
| Metformin | Biguanide/Anti-hyperglycemic |  | Activation of AMPK | OXPHOS inhibition leading to AMPK activation | Inhibition of Complex I of ETC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catalán, M.; Olmedo, I.; Faúndez, J.; Jara, J.A. Medicinal Chemistry Targeting Mitochondria: From New Vehicles and Pharmacophore Groups to Old Drugs with Mitochondrial Activity. Int. J. Mol. Sci. 2020, 21, 8684. https://doi.org/10.3390/ijms21228684
Catalán M, Olmedo I, Faúndez J, Jara JA. Medicinal Chemistry Targeting Mitochondria: From New Vehicles and Pharmacophore Groups to Old Drugs with Mitochondrial Activity. International Journal of Molecular Sciences. 2020; 21(22):8684. https://doi.org/10.3390/ijms21228684
Chicago/Turabian StyleCatalán, Mabel, Ivonne Olmedo, Jennifer Faúndez, and José A. Jara. 2020. "Medicinal Chemistry Targeting Mitochondria: From New Vehicles and Pharmacophore Groups to Old Drugs with Mitochondrial Activity" International Journal of Molecular Sciences 21, no. 22: 8684. https://doi.org/10.3390/ijms21228684
APA StyleCatalán, M., Olmedo, I., Faúndez, J., & Jara, J. A. (2020). Medicinal Chemistry Targeting Mitochondria: From New Vehicles and Pharmacophore Groups to Old Drugs with Mitochondrial Activity. International Journal of Molecular Sciences, 21(22), 8684. https://doi.org/10.3390/ijms21228684