Developmental Programming and Reprogramming of Hypertension and Kidney Disease: Impact of Tryptophan Metabolism



Abstract
:1. Introduction
2. Tryptophan Metabolism
2.1. Tryptophan Metabolic Pathways
2.2. Tryptophan Metabolism in Pregnancy
3. Tryptophan Metabolism in Hypertension and Kidney Disease
4. Tryptophan Metabolic Pathways: Programming versus Reprogramming Effects
4.1. Tryptophan-Related Metabolites-Induced Hypertension and CKD of Developmental Origin
4.2. Targeting on Tryptophan Metabolic Pathway as Reprogramming Strategies in Animal Models
5. Common Mechanisms Link Tryptophan Metabolism to Developmental Programming of Hypertension and Kidney Disease
5.1. Oxidative Stress
5.2. Gut Microbiota
5.3. Renin–Angiotensin System
5.4. Immunity and Inflammation
5.5. Others
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AANAT | Arylalkylamine N-acetyltransferase |
| ACE | Angiotensin converting enzyme |
| AhR | Aryl hydrocarbon receptor |
| AMPK | Adenosine monophosphate activated protein kinase |
| ArAT | Acromatic amino acid aminotransferase |
| ASMT | N-acetylserotonin O-methyltransferase |
| AT1R | Angiotensin type 1 receptor |
| AT2R | Angiotensin type 2 receptor |
| CKD | Chronic kidney disease |
| CVD | Cardiovascular disease |
| NAD+ | Nicotinamide adenine dinucleotide |
| DOHaD | Developmental origins of health and disease |
| GC | Glucocorticoid |
| IAA | Indoleacetic acid |
| IAM | Indole-3-acetamide |
| IAlD | Indole-3-aldehyde |
| IDG | Indoxyl-β-D glucuronide |
| IDO | Indoleamine 2,3-dioxygenase |
| ILA | Indolelactic acid |
| IPA | Indole-3-propionic acid |
| KAT | Kynurenine aminotransferase |
| KMO | Kynurenine-3-monooxygenase |
| KYNU | Kynureninase |
| L-NAME | NG-nitro-l-arginine-methyester |
| MAO | monoamine oxidase |
| Mas | Angiotensin-(1-7) receptor |
| NO | Nitric oxide |
| PGC-1α | PPARγ coactivator-1α |
| RAS | Renin-angiotensin system |
| ROS | Reactive oxygen species |
| SHR | Spontaneously hypertensive rat |
| SIRT | Silent information regulator transcript |
| SSRI | Selective serotonin reuptake inhibitor |
| TCDD | 2,3,7,8-Tetrachlorodibenzo-p-dioxin |
| TDO | Tryptophan 2,3-dioxygenase |
| TPH | Tryptophan hydroxylase |
| 3-HK | 3-hydroxykynurenine |
| 5-HIAA | 5-hydroxyindoleacetic acid |
| 5-HTP | 5-hydroxytryptophan |
References
- Mills, K.T.; Bundy, J.D.; Kelly, T.N.; Reed, J.E.; Kearney, P.M.; Reynolds, K.; Chen, J.; He, J. Global Disparities of Hypertension Prevalence and Control: A Systematic Analysis of Population-Based Studies From 90 Countries. Circulation 2016, 134, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.T.; Stefanescu, A.; He, J. The epidemiology of hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Couser, W.G.; Remuzzi, G.; Mendis, S.; Tonelli, M. The contribution of chronic kidney disease to the global burden of major noncommunicable diseases. Kidney Int. 2011, 80, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Luyckx, V.A.; Bertram, J.F.; Brenner, B.M.; Fall, C.; Hoy, W.E.; Ozanne, S.E.; Vikse, B.E. Effect of fetal and child health on kidney development and long-term risk of hypertension and kidney disease. Lancet 2013, 382, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Chong, E.; Yosypiv, I.V. Developmental programming of hypertension and kidney disease. Int. J. Nephrol. 2012, 2012, 760580. [Google Scholar] [CrossRef] [Green Version]
- Hanson, M.; Gluckman, P. Developmental origins of noncommunicable disease: Population and public health implications. Am. J. Clin. Nutr. 2011, 94, 1754S–1758S. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Tain, Y.L. The Good, the Bad, and the Ugly of Pregnancy Nutrients and Developmental Programming of Adult Disease. Nutrients 2019, 11, 894. [Google Scholar] [CrossRef] [Green Version]
- Kett, M.M.; Denton, K.M. Renal programming: Cause for concern? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R791–R803. [Google Scholar] [CrossRef]
- Nüsken, E.; Dötsch, J.; Weber, L.T.; Nüsken, K. Developmental Programming of Renal Function and Re-Programming Approaches. Front. Pediatr. 2018, 6, 36. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Joles, J.A. Reprogramming: A preventive strategy in hypertension focusing on the kidney. Int. J. Mol. Sci. 2015, 17, 23. [Google Scholar] [CrossRef] [Green Version]
- Richard, D.M.; Dawes, M.A.; Mathias, C.W.; Acheson, A.; Hill-Kapturczak, N.; Dougherty, D.M. L-tryptophan: Basic metabolic functions, behavioral research and therapeutic indications. Int. J. Tryptophan Res. 2009, 2, 45–60. [Google Scholar] [CrossRef] [Green Version]
- Fernstrom, J.D. A perspective on the safety of supplemental tryptophan based on its metabolic fates. J. Nutr. 2016, 146, 2601S–2608S. [Google Scholar] [CrossRef] [Green Version]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [Green Version]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Badawy, A. Tryptophan metabolism, disposition and utilization in pregnancy. Biosci. Rep. 2015, 35, e00261. [Google Scholar] [CrossRef]
- Institute of Medicine, Food and Nutrition Board. Dietary Reference Intakes: Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein and Amino Acids; National Academies Press: Washington, DC, USA, 2005. [Google Scholar]
- Addi, T.; Dou, L.; Burtey, S. Tryptophan-Derived Uremic Toxins and Thrombosis in Chronic Kidney Disease. Toxins 2018, 10, 412. [Google Scholar] [CrossRef] [Green Version]
- Friedman, M. Analysis, Nutrition, and Health Benefits of Tryptophan. Int. J. Tryptophan Res. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Yao, K.; Fang, J.; Yin, Y.L.; Feng, Z.M.; Tang, Z.R.; Wu, G. Tryptophan metabolism in animals: Important roles in nutrition and health. Front. Biosci. (Schol. Ed.) 2011, 3, 286–297. [Google Scholar]
- Stone, T.W.; Darlington, L.G. Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discov. 2002, 1, 609–620. [Google Scholar] [CrossRef]
- Meyer, T.W.; Hostetter, T.H. Uremic solutes from colon microbes. Kidney Int. 2012, 81, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef] [Green Version]
- Sallée, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef]
- Moehn, S.; Pencharz, P.B.; Ball, R.O. Lessons learned regarding symptoms of tryptophan deficiency and excess from animal requirement studies. J. Nutr. 2012, 142, 2231S–2235S. [Google Scholar] [CrossRef] [Green Version]
- WHO. Protein and Amino Acid Requirements in Human Nutrition. Report of a Joint WHO/FAO/UNU Expert Consultation; WHO Technical Report Series 935; WHO Press: Geneva, Switzerland, 2007. [Google Scholar]
- Schoengold, D.M.; DeFiore, R.H.; Parlett, R.C. Free amino acids in plasma throughout pregnancy. Am. J. Obstet. Gynecol. 1978, 131, 490–499. [Google Scholar] [CrossRef]
- Duggleby, S.L.; Jackson, A.A. Protein, amino acid and nitrogen metabolism during pregnancy: How might the mother meet the needs of her fetus? Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 503–509. [Google Scholar] [CrossRef]
- Elango, R.; Ball, R.O. Protein and amino acid requirements during pregnancy. Adv. Nutr. 2016, 7, 839S–844S. [Google Scholar] [CrossRef] [Green Version]
- Gao, H. Amino Acids in Reproductive Nutrition and Health. Adv. Exp. Med. Biol. 2020, 1265, 111–131. [Google Scholar]
- Hsu, C.N.; Tain, Y.L. Amino Acids and Developmental Origins of Hypertension. Nutrients 2020, 12, 1763. [Google Scholar] [CrossRef]
- Tricklebank, M.D.; Pickard, F.J.; de Souza, S.W. Free and bound tryptophan in human plasma during the perinatal period. Acta Paediatr. Scand. 1979, 68, 199–204. [Google Scholar] [CrossRef]
- Blaschitz, A.; Gauster, M.; Fuchs, D.; Lang, I.; Maschke, P.; Ulrich, D.; Karpf, E.; Takikawa, O.; Schimek, M.G.; Dohr, G.; et al. Vascular endothelial expression of indoleamine 2,3-dioxygenase 1 forms a positive gradient towards the feto-maternal interface. PLoS ONE 2011, 6, e21774. [Google Scholar] [CrossRef] [Green Version]
- Fukuwatari, T.; Murakami, M.; Ohta, M.; Kimura, N.; Jin-No, Y.; Sasaki, R.; Shibata, K. Changes in the urinary excretion of the metabolites of the tryptopan-niacin pathway during pregnancy in japanese women and rats. J. Nutr. Sci. Vitaminol. 2004, 50, 392–398. [Google Scholar] [CrossRef] [Green Version]
- Bonnin, A.; Levitt, P. Fetal, maternal, and placental sources of serotonin and new implications for developmental programming of the brain. Neuroscience 2011, 197, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Tain, Y.L. Light and Circadian Signaling Pathway in Pregnancy: Programming of Adult Health and Disease. Int. J. Mol. Sci. 2020, 21, 2232. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Huang, L.T.; Tain, Y.L. Perinatal Use of Melatonin for Offspring Health: Focus on Cardiovascular and Neurological Diseases. Int. J. Mol. Sci. 2019, 20, 5681. [Google Scholar] [CrossRef] [Green Version]
- Glover, M.E.; Clinton, S.M. Of rodents and humans: A comparative review of the neurobehavioral effects of early life SSRI exposure in preclinical and clinical research. Int. J. Dev. Neurosci. 2016, 51, 50–72. [Google Scholar] [CrossRef]
- Mendez, N.; Abarzua-Catalan, L.; Vilches, N.; Galdames, H.A.; Spichiger, C.; Richter, H.G.; Valenzuela, G.J.; Seron-Ferre, M.; Torres-Farfan, C. Timed maternal melatonin treatment reverses circadian disruption of the fetal adrenal clock imposed by exposure to constant light. PLoS ONE 2012, 7, e42713. [Google Scholar] [CrossRef] [Green Version]
- Louca, P.; Mompeo, O.; Leeming, E.R.; Berry, S.E.; Mangino, M.; Spector, T.D.; Padmanabhan, S.; Menni, C. Dietary Influence on Systolic and Diastolic Blood Pressure in the TwinsUK Cohort. Nutrients 2020, 12, 2130. [Google Scholar] [CrossRef]
- Altorf-van der Kuil, W.; Engberink, M.F.; De Neve, M.; van Rooij, F.J.; Hofman, A.; van’tVeer, P.; Witteman, J.C.; Franco, O.H.; Geleijnse, J.M. Dietary amino acids and the risk of hypertension in a Dutch older population: The Rotterdam Study. Am. J. Clin. Nutr. 2013, 97, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Fregly, M.J.; Sumners, C.; Cade, J.R. Effect of chronic dietary treatment with L-tryptophan on the maintenance of hypertension in spontaneously hypertensive rats. Can. J. Physiol. Pharmacol. 1989, 67, 656–662. [Google Scholar] [CrossRef]
- Lark, L.A.; Witt, P.A.; Becker, K.B.; Studzinski, W.M.; Weyhenmeyer, J.A. Effect of dietary tryptophan on the development of hypertension in the Dahl salt-sensitive rat. Clin. Exp. Hypertens. A 1990, 12, 1–13. [Google Scholar] [CrossRef]
- Fregly, M.J.; Lockley, O.E.; Cade, J.R. Effect of chronic dietary treatment with L-tryptophan on the development of renal hypertension in rats. Pharmacology 1988, 36, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, H.; McKenzie, G.; Witting, P.K.; Stasch, J.P.; Hahn, M.; Changsirivathanathamrong, D.; Wu, B.J.; Ball, H.J.; Thomas, S.R.; et al. Kynurenine is an endothelium-derived relaxing factor produced during inflammation. Nat. Med. 2010, 16, 279–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartosiewicz, J.; Kaminski, T.; Pawlak, K.; Karbowska, M.; Tankiewicz-Kwedlo, A.; Pawlak, D. The activation of the kynurenine pathway in a rat model with renovascular hypertension. Exp. Biol. Med. 2017, 242, 750–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Christou, H.; Liu, L.; Visner, G.; Mitsialis, S.A.; Kourembanas, S.; Liu, H. Endothelial indoleamine 2,3-dioxygenase protects against development of pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2013, 188, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Watts, S.W.; Morrison, S.F.; Davis, R.P.; Barman, S.M. Serotonin and blood pressure regulation. Pharmacol. Rev. 2012, 64, 59–88. [Google Scholar] [CrossRef] [Green Version]
- Itskovitz, H.D.; Werber, J.L.; Sheridan, A.M.; Brewer, T.F.; Stier, C.T., Jr. 5-Hydroxytryptophan and carbidopa in spontaneously hypertensive rats. J. Hypertens. 1989, 7, 311–315. [Google Scholar] [CrossRef]
- Brenner, B.; Harney, J.T.; Ahmed, B.A.; Jeffus, B.C.; Unal, R.; Mehta, J.L.; Kilic, F. Plasma serotonin levels and the platelet serotonin transporter. J. Neurochem. 2007, 102, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Topsakal, R.; Kalay, N.; Gunturk, E.E.; Dogan, A.; Inanc, M.T.; Kaya, M.G.; Ergin, A.; Yarlioglues, M. The relation between serotonin levels and insufficient blood pressure decrease during night-time in hypertensive patients. Blood Press 2009, 18, 367–371. [Google Scholar] [CrossRef]
- Baron, A.; Riesselmann, A.; Fregly, M.J. Reduction in the elevated blood pressure of Dahl salt-sensitive rats treated chronically with L-5-hydroxytryptophan. Pharmacology 1991, 42, 15–22. [Google Scholar] [CrossRef]
- Jelen, I.; Fananapazir, L.; Crawford, T.B. The possible relation between late pregnancy hypertension and 5-hydroxytryptamine levels in maternal blood. Br. J. Obstet. Gynaecol. 1979, 86, 468–471. [Google Scholar] [CrossRef]
- Tain, Y.L.; Huang, L.T.; Chan, J.Y. Transcriptional regulation of programmed hypertension by melatonin: An epigenetic perspective. Int. J. Mol. Sci. 2014, 15, 18484–18495. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Fujigaki, S.; Heyes, M.P.; Shibata, K.; Takemura, M.; Fujii, H.; Wada, H.; Noma, A.; Seishima, M. Mechanism of increases in L-kynurenine and quinolinic acid in renal insufficiency. Am. J. Physiol. Renal Physiol. 2000, 279, F565–F572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlak, D.; Pawlak, K.; Malyszko, J.; Mysliwiec, M.; Buczko, W. Accumulation of toxic products degradation of kynurenine in hemodialyzed patients. Int. Urol. Nephrol. 2001, 33, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Domaniewski, T.; Mysliwiec, M.; Pawlak, D. The kynurenines are associated with oxidative stress, inflammation and the prevalence of cardiovascular disease in patients with end-stage renal disease. Atherosclerosis 2009, 204, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.C.; Sirich, T.L. Indoxyl sulfate-review of toxicity and therapeutic strategies. Toxins 2016, 8, 358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N. The role of endogenous aryl hydrocarbon receptor signaling in cardiovascular physiology. J. Cardiovasc. Dis. Res. 2011, 2, 91–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.N.; Lin, Y.J.; Lu, P.C.; Tain, Y.L. Maternal Resveratrol Therapy Protects Male Rat Offspring against Programmed Hypertension Induced by TCDD and Dexamethasone Exposures: Is It Relevant to Aryl Hydrocarbon Receptor? Int. J. Mol. Sci. 2018, 19, 2459. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Lin, Y.J.; Tain, Y.L. Maternal Exposure to Bisphenol A Combined with High-Fat Diet-Induced Programmed Hypertension in Adult Male Rat Offspring: Effects of Resveratrol. Int. J. Mol. Sci. 2019, 20, 4382. [Google Scholar] [CrossRef] [Green Version]
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mähler, A.; Balogh, A.; Markó, L.; et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 2017, 551, 585–589. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. The Double-Edged Sword Effects of Maternal Nutrition in the Developmental Programming of Hypertension. Nutrients 2018, 10, 1917. [Google Scholar] [CrossRef] [Green Version]
- McMillen, I.C.; Robinson, J.S. Developmental origins of the metabolic syndrome: Prediction, plasticity, and programming. Physiol. Rev. 2005, 85, 571–633. [Google Scholar] [CrossRef]
- Herring, C.M.; Bazer, F.W.; Johnson, G.A.; Wu, G. Impacts of maternal dietary protein intake on fetal survival, growth, and development. Exp. Biol. Med. 2018, 243, 525–533. [Google Scholar] [CrossRef]
- Penatti, E.M.; Barina, A.E.; Raju, S.; Li, A.; Kinney, H.C.; Commons, K.G.; Nattie, E.E. Maternal dietary tryptophan deficiency alters cardiorespiratory control in rat pups. J. Appl. Physiol. 2011, 110, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Omstedt, P.T.; von der Decken, A. Dietary amino acids: Effect of depletion and recovery on protein synthesis in vitro in rat skeletal muscle and liver. Br. J. Nutr. 1974, 31, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Lenis, N.P.; van Diepen, J.T.M.; Goedhart, P.W. Amino acid requirements of pigs. 1. Requirements for methionine + cystine, threonine and tryptophan for fast growing boars and gilts, fed ad libitum. Neth. J. Agric. Sci. 1990, 38, 577–595. [Google Scholar] [CrossRef]
- Tain, Y.L.; Lin, Y.J.; Chan, J.Y.H.; Lee, C.T.; Hsu, C.N. Maternal melatonin or agomelatine therapy prevents programmed hypertension in male offspring of mother exposed to continuous light. Biol. Reprod. 2017, 97, 636–643. [Google Scholar] [CrossRef]
- Hsu, C.N.; Lin, I.C.; Yu, H.R.; Huang, L.T.; Tiao, M.M.; Tain, Y.L. Maternal Tryptophan Supplementation Protects Adult Rat Offspring against Hypertension Programmed by Maternal Chronic Kidney Disease: Implication of Tryptophan-Metabolizing Microbiome and Aryl Hydrocarbon Receptor. Int. J. Mol. Sci. 2020, 21, 4552. [Google Scholar] [CrossRef] [PubMed]
- Young, S.N. Behavioral effects of dietary neurotransmitter precursors: Basic and clinical aspects. Neurosci. Biobehav. Rev. 1996, 20, 313–323. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Developmental Origins of Chronic Kidney Disease: Should We Focus on Early Life? Int. J. Mol. Sci. 2017, 18, 381. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.K.; Sirajudeen, K.N.; Sundaram, A.; Zakaria, R.; Singh, H.J. Effects of antenatal, postpartum and post-weaning melatonin supplementation on blood pressure and renal antioxidant enzyme activities in spontaneously hypertensive rats. J. Physiol. Biochem. 2011, 67, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T.; Hsu, C.N.; Lee, C.T. Melatonin therapy prevents programmed hypertension and nitric oxide deficiency in offspring exposed to maternal caloric restriction. Oxidative Med. Cell Longev. 2014, 2014, 283180. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, C.T.; Chan, J.Y.; Hsu, C.N. Maternal melatonin or N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and renal transcriptome to prevent prenatal N(G)-Nitro-L-arginine-methyl ester (L-NAME)-induced fetal programming of hypertension in adult male offspring. Am. J. Obstet. Gynecol. 2016, 215, 636. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Leu, S.; Wu, K.L.; Lee, W.C.; Chan, J.Y. Melatonin prevents maternal fructose intake-induced programmed hypertension in the offspring: Roles of nitric oxide and arachidonic acid metabolites. J. Pineal Res. 2014, 57, 80–89. [Google Scholar] [CrossRef]
- Tain, Y.L.; Chan, J.Y.H.; Lee, C.T.; Hsu, C.N. Maternal Melatonin Therapy Attenuates Methyl-Donor Diet-Induced Programmed Hypertension in Male Adult Rat Offspring. Nutrients 2018, 10, 1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tain, Y.L.; Leu, S.; Lee, W.C.; Wu, K.L.H.; Chan, J.Y.H. Maternal Melatonin Therapy Attenuated Maternal High-Fructose Combined with Post-Weaning High-Salt Diets-Induced Hypertension in Adult Male Rat Offspring. Molecules 2018, 23, 886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tain, Y.L.; Chen, C.C.; Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Kuo, H.C.; Huang, L.T. Melatonin attenuates prenatal dexamethasone-induced blood pressure increase in a rat model. J. Am. Soc. Hypertens. 2014, 8, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Sheen, J.M.; Yu, H.R.; Chen, C.C.; Tiao, M.M.; Hsu, C.N.; Lin, Y.J.; Kuo, K.C.; Huang, L.T. Maternal Melatonin Therapy Rescues Prenatal Dexamethasone and Postnatal High-Fat Diet Induced Programmed Hypertension in Male Rat Offspring. Front. Physiol. 2015, 6, 377. [Google Scholar] [CrossRef] [Green Version]
- Care, A.S.; Sung, M.M.; Panahi, S.; Gragasin, F.S.; Dyck, J.R.; Davidge, S.T.; Bourque, S.L. Perinatal Resveratrol Supplementation to Spontaneously Hypertensive Rat Dams Mitigates the Development of Hypertension in Adult Offspring. Hypertension 2016, 67, 1038–1044. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Lee, W.C.; Wu, K.L.H.; Leu, S.; Chan, J.Y.H. Resveratrol Prevents the Development of Hypertension Programmed by Maternal Plus Post-Weaning High-Fructose Consumption through Modulation of Oxidative Stress, Nutrient-Sensing Signals, and Gut Microbiota. Mol. Nutr. Food Res. 2018, 30, e1800066. [Google Scholar] [CrossRef]
- Chen, H.E.; Lin, Y.J.; Lin, I.C.; Yu, H.R.; Sheen, J.M.; Tsai, C.C.; Huang, L.T.; Tain, Y.L. Resveratrol prevents combined prenatal NG-nitro-L-arginine-methyl ester (L-NAME) treatment plus postnatal high-fat diet induced programmed hypertension in adult rat offspring: Interplay between nutrient-sensing signals, oxidative stress and gut microbiota. J. Nutr. Biochem. 2019, 70, 28–37. [Google Scholar] [CrossRef]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Sheen, J.M.; Tiao, M.M.; Tain, Y.L.; Huang, L.T. Roles of melatonin in fetal programming in compromised pregnancies. Int. J. Mol. Sci. 2013, 14, 5380–5401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, H.; Nakamura, Y.; Terron, M.P.; Flores, L.J.; Manchester, L.C.; Tan, D.X.; Sugino, N.; Reiter, R.J. Melatonin and pregnancy in the human. Reprod. Toxicol. 2008, 25, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Luyckx, V.A.; Shukha, K.; Brenner, B.M. Low nephron number and its clinical consequences. Rambam. Maimonides. Med. J. 2011, 2, e0061. [Google Scholar] [CrossRef]
- Ortiz, L.A.; Quan, A.; Weinberg, A.; Baum, M. Effect of prenatal dexamethasone on rat renal development. Kidney Int. 2001, 59, 1663–1669. [Google Scholar] [CrossRef] [Green Version]
- Esteban, S.; Nicolaus, C.; Garmundi, A.; Rial, R.V.; Rodríguez, A.B.; Ortega, E.; Ibars, C.B. Effect of orally administered L-tryptophan on serotonin, melatonin, and the innate immune response in the rat. Mol. Cell Biochem. 2004, 267, 39–46. [Google Scholar] [CrossRef]
- Oberlander, T.F. Fetal serotonin signaling: Setting pathways for early childhood development and behavior. J. Adolesc. Health 2012, 51, S9–S16. [Google Scholar] [CrossRef]
- Siemann, J.K.; Green, N.H.; Reddy, N.; McMahon, D.G. Sequential photoperiodic programing of serotonin neurons, signaling and behaviors during prenatal and postnatal development. Front. Neurosci. 2019, 13, 459. [Google Scholar] [CrossRef]
- Savouret, J.F.; Berdeaux, A.; Casper, R.F. The aryl hydrocarbon receptor and its xenobiotic ligands: A fundamental trigger for cardiovascular diseases. Nutr. Metab. Cardiovasc. Dis. 2003, 13, 104–113. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Developmental programming of the metabolic syndrome: Can we reprogram with resveratrol? Int. J. Mol. Sci. 2018, 19, 2584. [Google Scholar] [CrossRef] [Green Version]
- Peter Stein, T.; Scholl, T.O.; Schluter, M.D.; Leskiw, M.J.; Chen, X.; Spur, B.W.; Rodriguez, A. Oxidative stress early in pregnancy and pregnancy outcome. Free Radic. Res. 2008, 42, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N. Interplay between oxidative stress and nutrient sensing signaling in the developmental origins of cardiovascular disease. Int. J. Mol. Sci. 2017, 18, 841. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, W.C.; Hsu, C.N.; Lee, W.C.; Huang, L.T.; Lee, C.T.; Lin, C.Y. Asymmetric dimethylarginine is associated with developmental programming of adult kidney disease and hypertension in offspring of streptozotocin-treated mothers. PLoS ONE 2013, 8, e55420. [Google Scholar] [CrossRef] [PubMed]
- Forrest, C.M.; Mackay, G.M.; Stoy, N.; Egerton, M.; Christofides, J.; Stone, T.W.; Darlington, L.G. Tryptophan loading induces oxidative stress. Free Radic. Res. 2004, 38, 1167–1171. [Google Scholar] [CrossRef] [PubMed]
- Reyes Ocampo, J.; Huitr, L.R.; Gonzalez-Esquivel, D.; Ugalde-Muniz, P.; Jimenez-Anguiano, A.; Pineda, B.; Pedraza-Chaverri, J.; Rios, C.; Perez de la Cruz, V. Kynurenines with neuroactive and redox properties: Relevance to aging and brain diseases. Oxid. Med. Cell. Longev. 2014, 2014, 22. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine, an endogenous oxidative stress generator, causes neuronal cell death with apoptotic features and region selectivity. J. Neurochem. 1998, 70, 299–307. [Google Scholar] [CrossRef]
- Stone, T.W. Kynurenines in the CNS: From endogenous obscurity to therapeutic importance. Prog. Neurobiol. 2001, 64, 185–218. [Google Scholar] [CrossRef]
- Xu, K.; Liu, H.; Bai, M.; Gao, J.; Wu, X.; Yin, Y. Redox properties of tryptophan metabolism and the concept of tryptophan use in pregnancy. Int. J. Mol. Sci. 2017, 18, 1595. [Google Scholar] [CrossRef] [Green Version]
- Bjørklund, G.; Chirumbolo, S. Role of oxidative stress and antioxidants in daily nutrition and human health. Nutrition 2017, 33, 311–321. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Regulation of Nitric Oxide Production in the Developmental Programming of Hypertension and Kidney Disease. Int. J. Mol. Sci. 2019, 60, 681. [Google Scholar] [CrossRef] [Green Version]
- Alberati-Giani, D.; Malherbe, P.; Ricciardi-Castagnoli, P.; Köhler, C.; Denis-Donini, S.; Cesura, A.M. Differential regulation of indoleamine 2,3-dioxygenase expression by nitric oxide and inflammatory mediators in IFN-γ-activated murine macrophages and microglial cells. J. Immunol. 1997, 159, 419–426. [Google Scholar] [PubMed]
- Kuhn, D.M.; Arthur, R., Jr. Molecular mechanism of the inactivation of tryptophan hydroxylase by nitric oxide: Attack on critical sulfhydryls that spare the enzyme iron center. J. Neurosci. 1997, 17, 7245–7251. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S. Novel perspectives on the molecular crosstalk mechanisms of serotonin and melatonin in plants. Plant Physiol. Biochem. 2018, 132, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Kharait, S.; Haddad, D.J.; Springer, M.L. Nitric oxide counters the inhibitory effects of uremic toxin indoxyl sulfate on endothelial cells by governing ERK MAP kinase and myosin light chain activation. Biochem. Biophys. Res. Commun. 2011, 409, 758–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, D.M.; Meyer, K.M.; Prince, A.L.; Aagaard, K.M. Impact of maternal nutrition in pregnancy and lactation on offspring gut microbial composition and function. Gut Microbes 2016, 7, 459–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijers, B.; Jouret, F.; Evenepoel, P. Linking gut microbiota to cardiovascular disease and hypertension: Lessons from chronic kidney disease. Pharmacol. Res. 2018, 133, 101–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Khodor, S.; Reichert, B.; Shatat, I.F. The microbiome and blood pressure: Can microbes regulate our blood pressure? Front. Pediatr. 2017, 5, 138. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef]
- Liang, H.; Dai, Z.; Kou, J.; Sun, K.; Chen, J.; Yang, Y.; Wu, G.; Wu, Z. Dietary l-Tryptophan supplementation enhances the intestinal mucosal barrier function in weaned piglets: Implication of Tryptophan-metabolizing microbiota. Int. J. Mol. Sci. 2019, 20, 20. [Google Scholar] [CrossRef] [Green Version]
- Velasquez, M.T.; Centron, P.; Barrows, I.; Dwivedi, R.; Raj, D.S. Gut Microbiota and Cardiovascular Uremic Toxicities. Toxins 2018, 10, 287. [Google Scholar] [CrossRef] [Green Version]
- Lankelma, J.M.; Nieuwdorp, M.; de Vos, W.M.; Wiersinga, W.J. The gut microbiota in internal medicine: Implications for health and disease. Neth. J. Med. 2015, 73, 61–68. [Google Scholar] [PubMed]
- McFarlane, C.; Ramos, C.I.; Johnson, D.W.; Campbell, K.L. Prebiotic, probiotic, and synbiotic supplementation in chronic kidney disease: A systematic review and meta-analysis. J. Ren. Nutr. 2019, 29, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Lin, Y.J.; Hou, C.Y.; Tain, Y.L. Maternal administration of probiotic or prebiotic prevents male adult rat offspring against developmental programming of hypertension induced by high fructose consumption in pregnancy and lactation. Nutrients 2018, 10, 1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.N.; Chang-Chien, G.P.; Lin, S.; Hou, C.Y.; Tain, Y.L. Targeting on gut microbial metabolite trimethylamine-n-oxide and short-chain fatty acid to prevent maternal high-fructose-diet-induced developmental programming of hypertension in adult male offspring. Mol. Nutr. Food Res. 2019, 63, e1900073. [Google Scholar] [CrossRef]
- Hsu, C.N.; Hou, C.Y.; Chan, J.Y.H.; Lee, C.T.; Tain, Y.L. Hypertension programmed by perinatal high-fat diet: Effect of maternal gut microbiota-targeted therapy. Nutrients 2019, 11, 2908. [Google Scholar] [CrossRef] [Green Version]
- Te Riet, L.; van Esch, J.H.; Roks, A.J.; van den Meiracker, A.H.; Danser, A.H. Hypertension: Renin-angiotensin aldosterone system alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef]
- Santos, P.C.; Krieger, J.E.; Pereira, A.C. Renin-angiotensin system, hypertension, and chronic kidney disease: Pharmacogenetic implications. J. Pharmacol. Sci. 2012, 120, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Sherman, R.C.; Langley-Evans, S.C. Antihypertensive treatment in early postnatal life modulates prenatal dietary influences upon blood pressure in the rat. Clin. Sci. 2000, 98, 269–275. [Google Scholar] [CrossRef]
- Hsu, C.N.; Lee, C.T.; Huang, L.T.; Tain, Y.L. Aliskiren in early postnatal life prevents hypertension and reduces asymmetric dimethylarginine in offspring exposed to maternal caloric restriction. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 506–513. [Google Scholar] [CrossRef] [Green Version]
- Khedr, S.; Deussen, A.; Kopaliani, I.; Zatschler, B.; Martin, M. Effects of tryptophan-containing peptides on angiotensin-converting enzyme activity and vessel tone ex vivo and in vivo. Eur. J. Nutr. 2018, 57, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Hirose, Y.; Goto, S.; Nishijima, F.; Zrelli, H.; Zghonda, N.; Niwa, T.; Miyazaki, H. Indoxyl sulfate enhances angiotensin II signaling through upregulation of epidermal growth factor receptor expression in vascular smooth muscle cells. Life Sci. 2012, 91, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.Y.; Yisireyili, M.; Saito, S.; Lee, C.T.; Adelibieke, Y.; Nishijima, F.; Niwa, T. Indoxyl sulfate downregulates expression of Mas receptor via OAT3/AhR/Stat3 pathway in proximal tubular cells. PLoS ONE 2014, 9, e91517. [Google Scholar] [CrossRef] [PubMed]
- Challis, J.R.; Lockwood, C.J.; Myatt, L.; Norman, J.E.; Strauss, J.F.; Petraglia, F. Inflammation and pregnancy. Reprod. Sci. 2009, 16, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Le Floc’h, N.; Melchior, D.; Seve, B. Dietary tryptophan helps to preserve tryptophan homeostasis in pigs suffering from lung inflammation. J. Anim. Sci. 2008, 86, 3473–3479. [Google Scholar] [CrossRef] [Green Version]
- Asp, L.; Johansson, A.S.; Mann, A.; Owe-Larsson, B.; Urbanska, E.M.; Kocki, T.; Kegel, M.; Engberg, G.; Lundkvist, G.B.; Karlsson, H. Effects of pro-inflammatory cytokines on expression of kynurenine pathway enzymes in human dermal fibroblasts. J. Inflamm. 2011, 8, 1476–9255. [Google Scholar] [CrossRef] [Green Version]
- McMaster, W.G.; Kirabo, A.; Madhur, M.S.; Harrison, D.G. Inflammation, immunity, and hypertensive end-organ damage. Circ. Res. 2015, 116, 1022–1033. [Google Scholar] [CrossRef]
- Ren, J.; Crowley, S.D. Role of T-cell activation in salt-sensitive hypertension. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1345–H1353. [Google Scholar] [CrossRef]
- Zhang, J.; Hua, G.; Zhang, X.; Tong, R.; Du, X.; Li, Z. Regulatory T cells/T-helper cell 17 functional imbalance in uraemic patients on maintenance haemodialysis: A pivotal link between microinflammation and adverse cardiovascular events. Nephrology 2010, 15, 33–41. [Google Scholar] [CrossRef]
- Brito, J.S.; Borges, N.A.; Esgalhado, M.; Magliano, D.C.; Soulage, C.O.; Mafra, D. Aryl hydrocarbon receptor activation in chronic kidney disease: Role of uremic toxins. Nephron 2017, 137, 1–7. [Google Scholar] [CrossRef]
- Stevens, E.A.; Mezrich, J.D.; Bradfield, C.A. The aryl hydrocarbon receptor: A perspective on potential roles in the immune system. Immunology 2009, 127, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Chan, J.Y.; Huang, L.T. Renal transcriptome analysis of programmed hypertension induced by maternal nutritional insults. Int. J. Mol. Sci. 2015, 16, 17826–17837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugden, M.C.; Caton, P.W.; Holness, M.J. PPAR control: It’s SIRTainly as easy as PGC. J. Endocrinol. 2010, 204, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Efeyan, A.; Comb, W.C.; Sabatini, D.M. Nutrient-sensing mechanisms and pathways. Nature 2015, 517, 302–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]

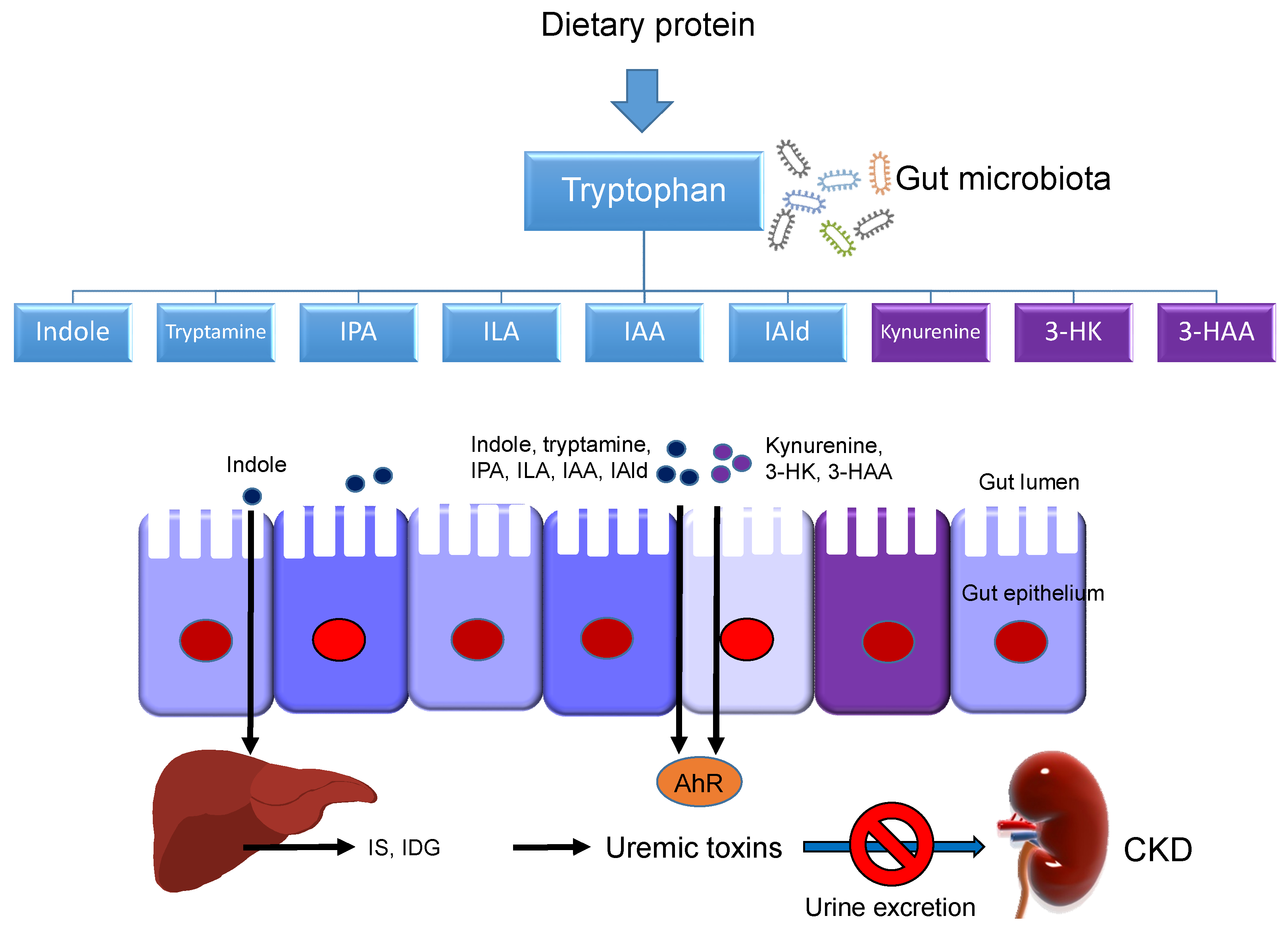
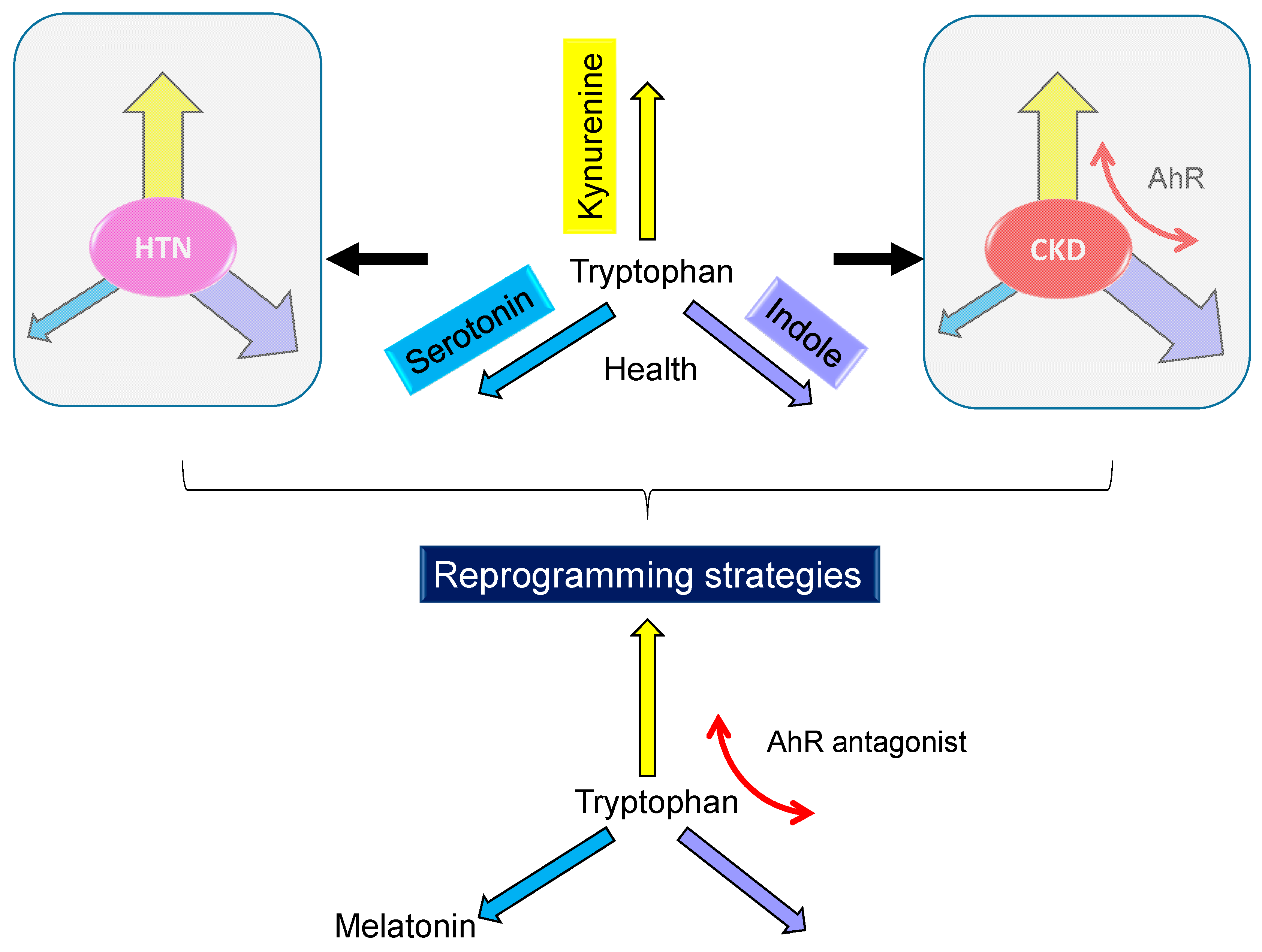

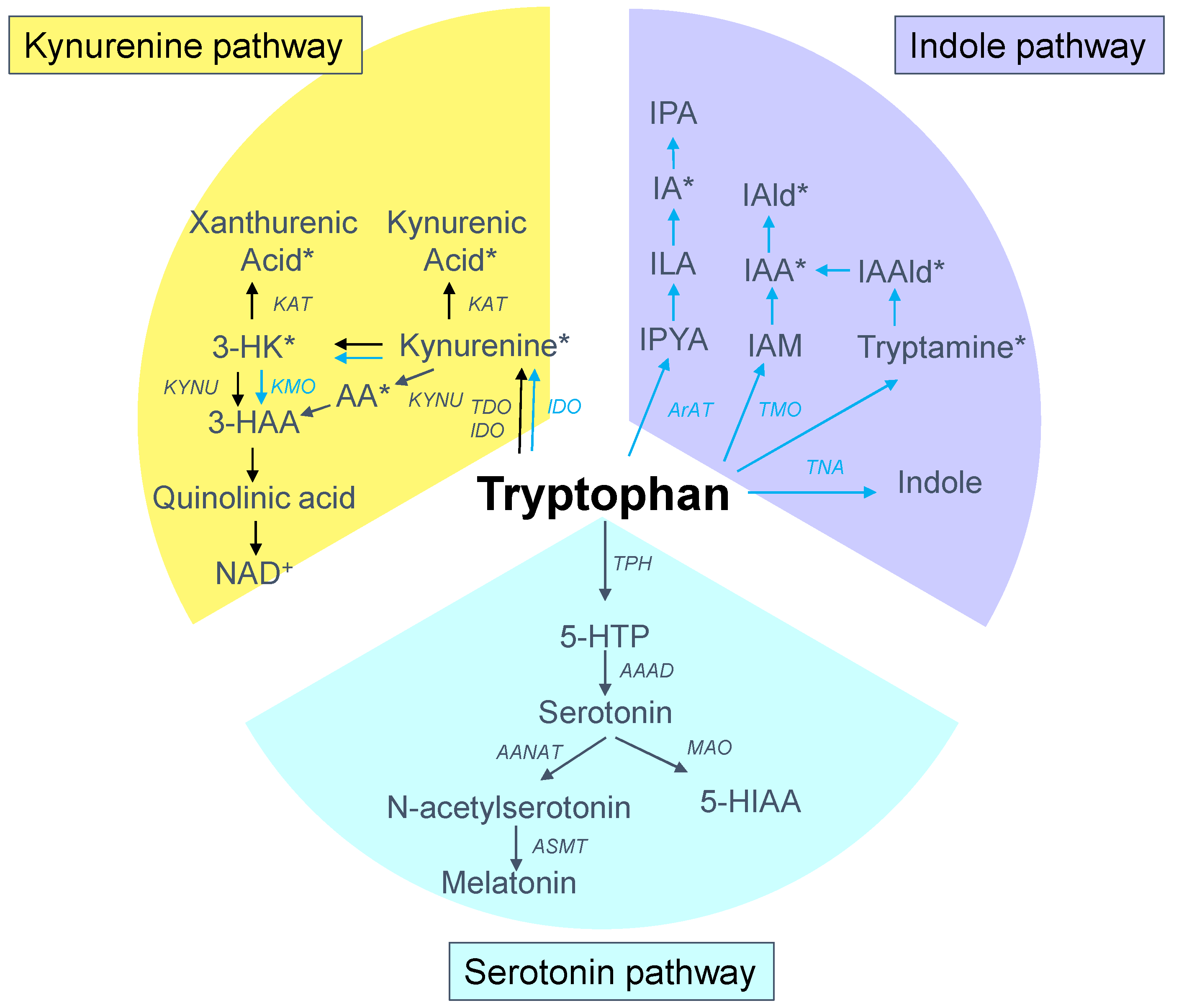{kind=link}
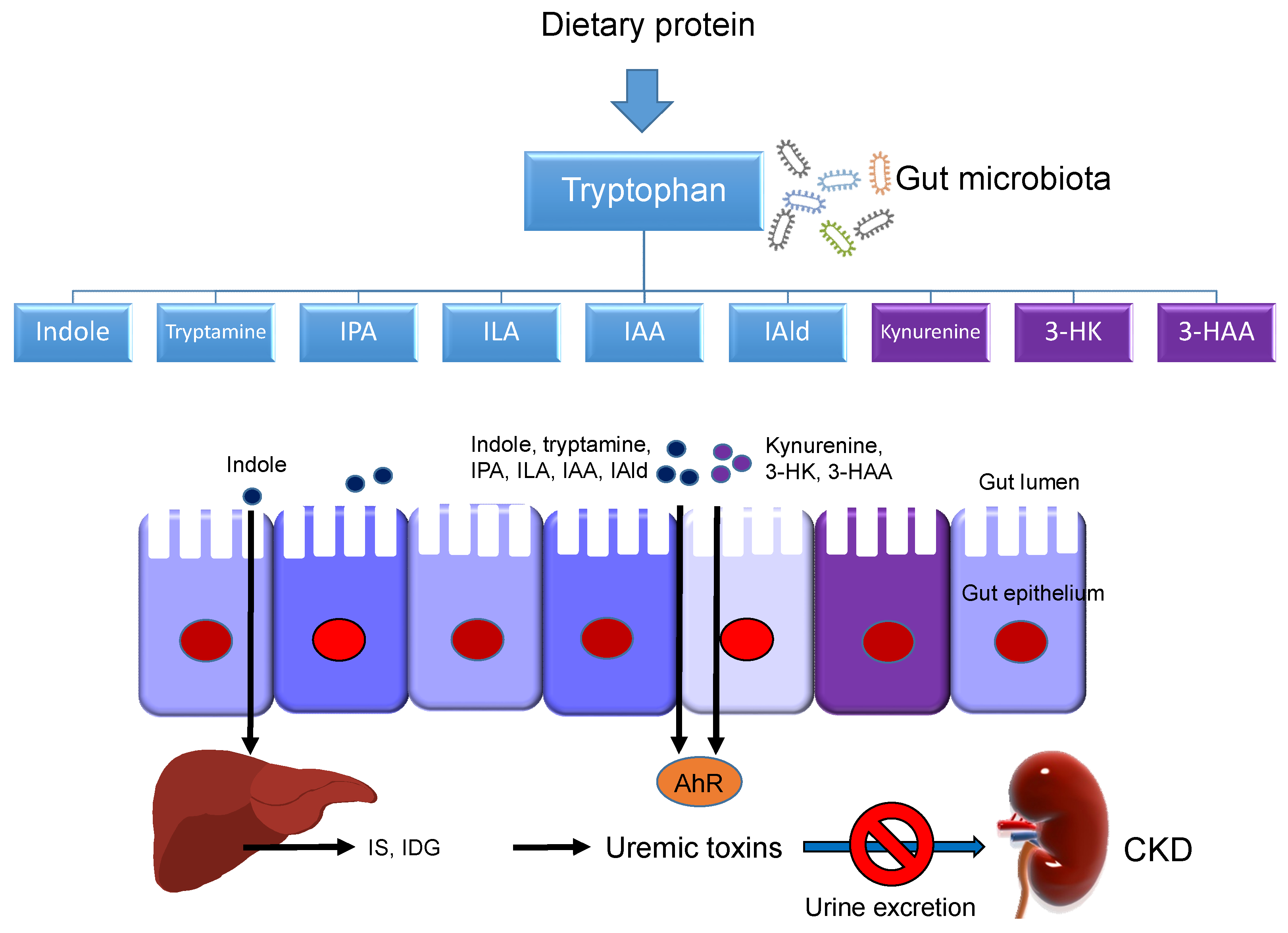{kind=link}
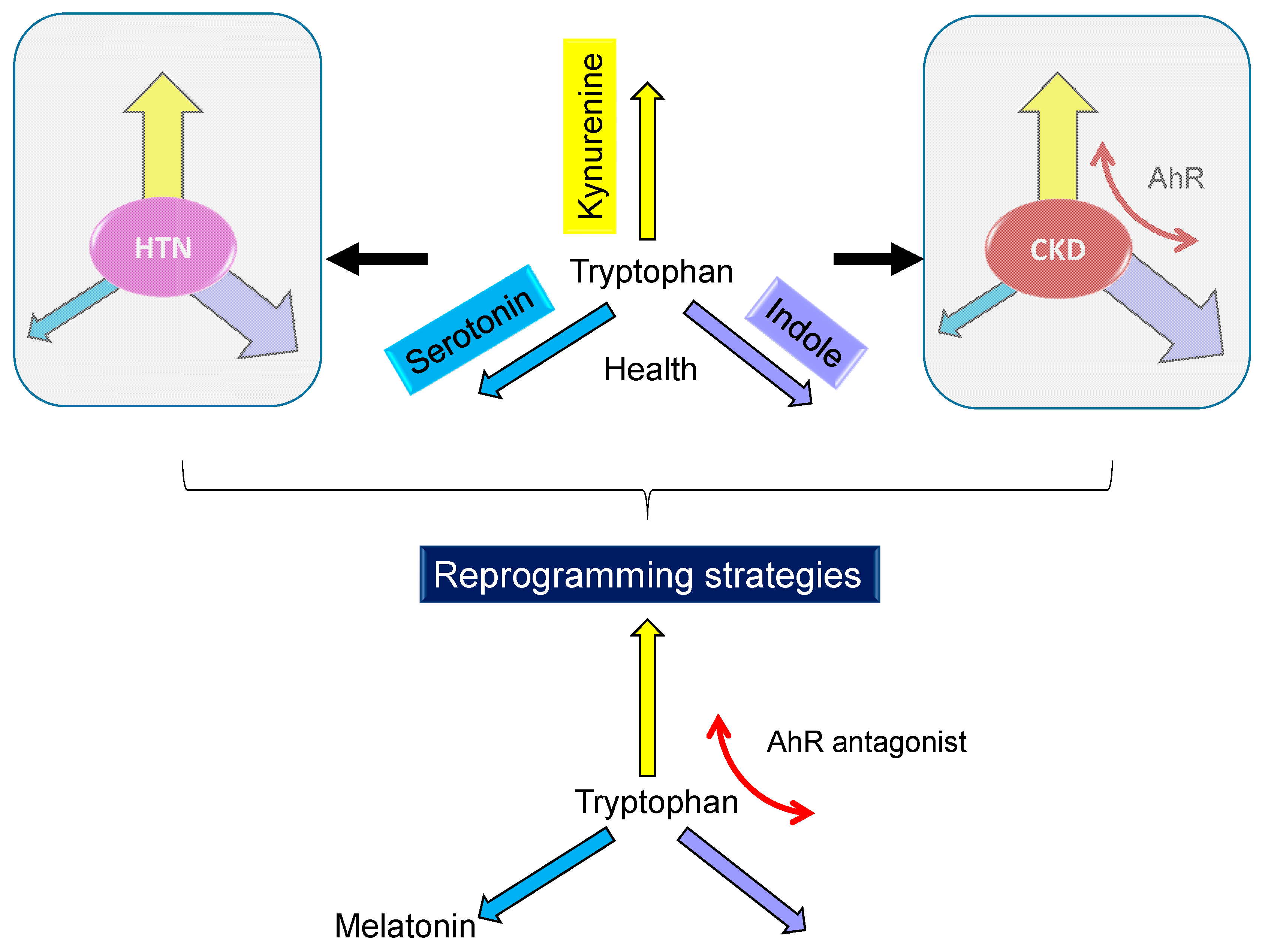{kind=link}
| Interventions | Animal Models | Species/Gender | Age at Measure | Reprogramming Effects |
|---|---|---|---|---|
| Tryptophan | ||||
| Tryptophan 200 mg/kg BW/day via oral gavage during pregnancy | Maternal adenosine-induced CKD | SD rat/M | 12 weeks | Prevented hypertension [69] |
| Melatonin | ||||
| 10 mg/kg BW/day melatonin in drinking water during pregnancy | Genetic hypertension model | SHR/M | 16 weeks | Prevented hypertension [72] |
| 0.01% melatonin in drinking water during pregnancy and lactation | Maternal caloric restriction | SD rat/M | 12 weeks | Prevented hypertension [73] |
| 0.01% melatonin in drinking water during pregnancy and lactation | Maternal L-NAME exposure | SD rat/M | 12 weeks | Prevented hypertension [74] |
| 0.01% melatonin in drinking water during pregnancy and lactation | Maternal high-fructose diet | SD rat/M | 12 weeks | Prevented hypertension [75] |
| 0.01% melatonin in drinking water during pregnancy and lactation | Maternal constant light exposure | SD rat/M | 12 weeks | Prevented hypertension [68] |
| 0.01% melatonin in drinking water during pregnancy and lactation | Maternal methyl-donor diet | SD rat/M | 12 weeks | Attenuated hypertension and altered renal transcriptome [76] |
| 0.01% melatonin in drinking water during pregnancy and lactation | Maternal high-fructose diet plus post-weaning high-salt diet | SD rat/M | 12 weeks | Attenuated hypertension [77] |
| 0.01% melatonin in drinking water during pregnancy and lactation | Prenatal GC exposure | SD rat/M | 16 weeks | Prevented hypertension and increased nephron number [78] |
| 0.01% melatonin in drinking water during pregnancy and lactation | Prenatal GC exposure plus post-weaning high-fat diet | SD rat/M | 16 weeks | Prevented hypertension [79] |
| AhR antagonist | ||||
| 4 g/kg diet resveratrol during pregnancy and lactation | Genetic hypertension model | SHR/M and F | 20 weeks | Prevented hypertension [80] |
| 50 mg/L resveratrol in drinking water during pregnancy and lactation | Maternal plus post-weaning high-fructose diet | SD rat/M | 12 weeks | Prevented hypertension [81] |
| 0.05% resveratrol in drinking water during pregnancy and lactation | Maternal TCDD and GC exposures | SD rat/M | 16 weeks | Prevented hypertension [59] |
| 50 mg/L resveratrol in drinking water during pregnancy and lactation | Maternal bisphenol A exposure and high-fat diet | SD rat/M | 16 weeks | Prevented hypertension [60] |
| 50 mg/L resveratrol in drinking water during pregnancy and lactation | Maternal L-NAME plus postnatal high-fat diet | SD rat/M | 16 weeks | Prevented hypertension [82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, C.-N.; Tain, Y.-L. Developmental Programming and Reprogramming of Hypertension and Kidney Disease: Impact of Tryptophan Metabolism. Int. J. Mol. Sci. 2020, 21, 8705. https://doi.org/10.3390/ijms21228705
Hsu C-N, Tain Y-L. Developmental Programming and Reprogramming of Hypertension and Kidney Disease: Impact of Tryptophan Metabolism. International Journal of Molecular Sciences. 2020; 21(22):8705. https://doi.org/10.3390/ijms21228705
Chicago/Turabian StyleHsu, Chien-Ning, and You-Lin Tain. 2020. "Developmental Programming and Reprogramming of Hypertension and Kidney Disease: Impact of Tryptophan Metabolism" International Journal of Molecular Sciences 21, no. 22: 8705. https://doi.org/10.3390/ijms21228705
APA StyleHsu, C.-N., & Tain, Y.-L. (2020). Developmental Programming and Reprogramming of Hypertension and Kidney Disease: Impact of Tryptophan Metabolism. International Journal of Molecular Sciences, 21(22), 8705. https://doi.org/10.3390/ijms21228705