Transmembrane Polar Relay Drives the Allosteric Regulation for ABCG5/G8 Sterol Transporter


 and
and
Abstract
1. Introduction
2. Results
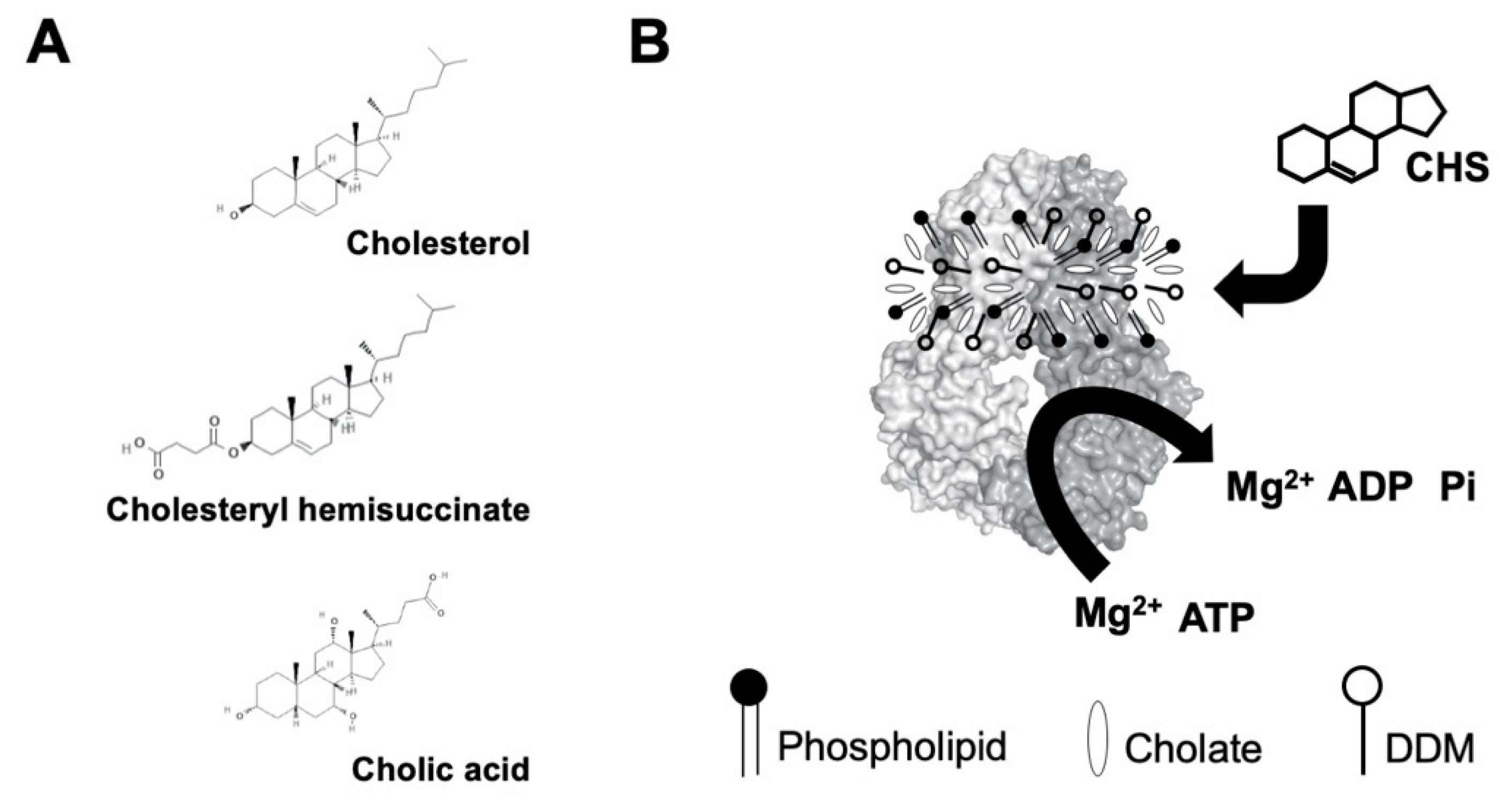
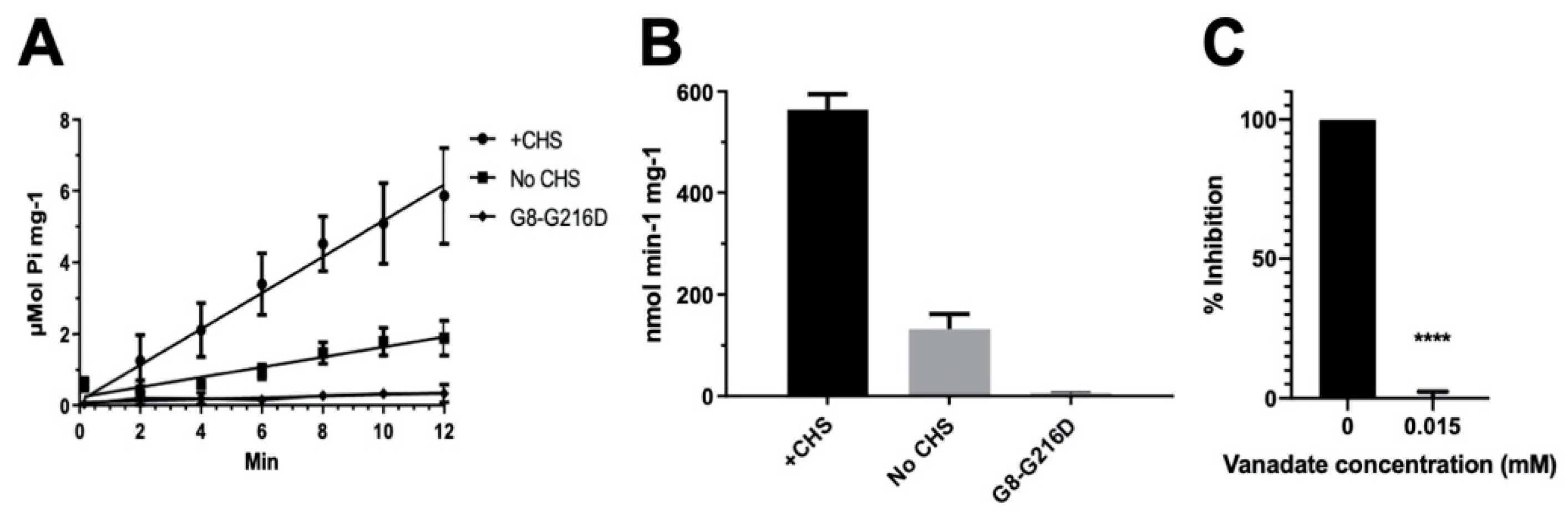
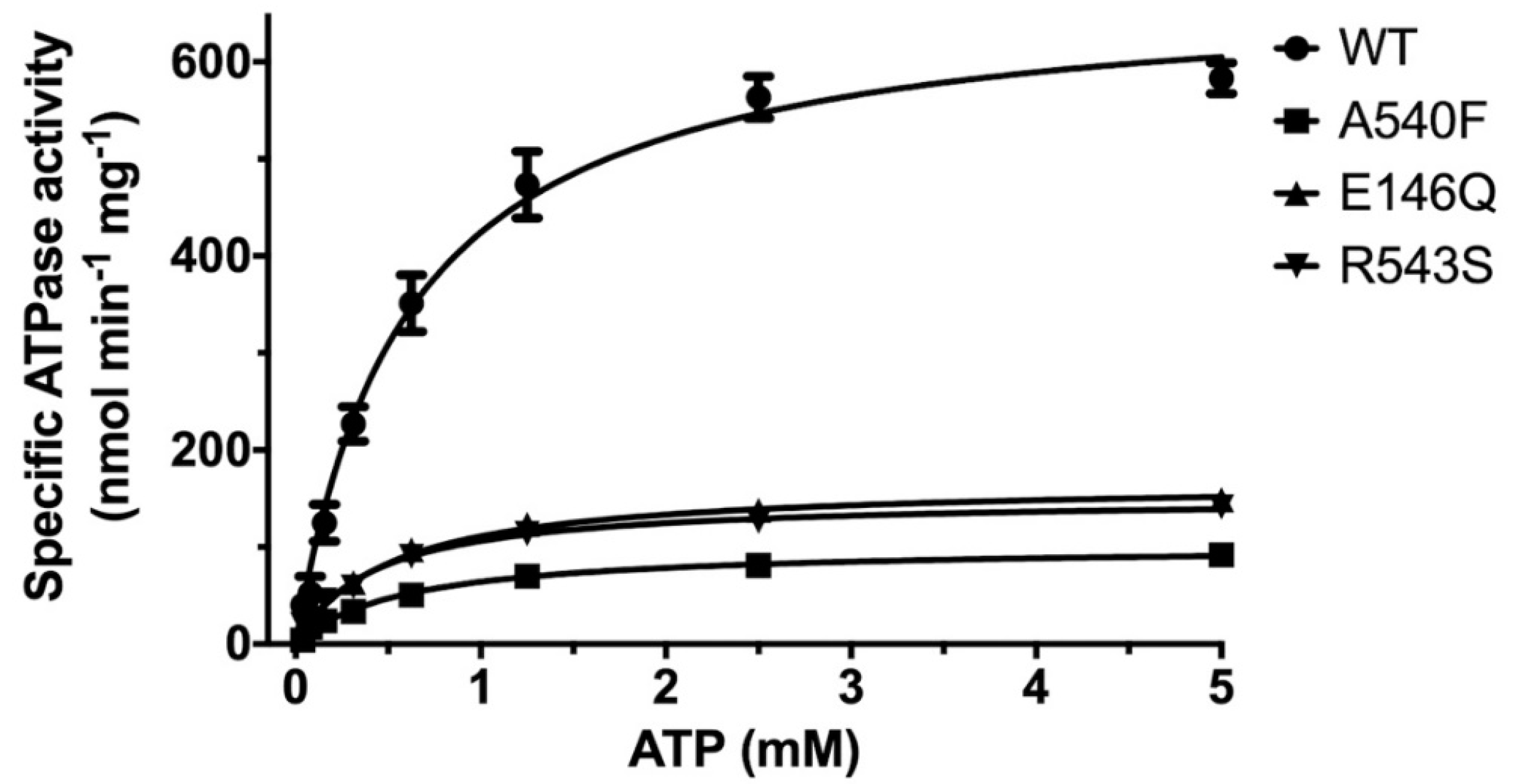
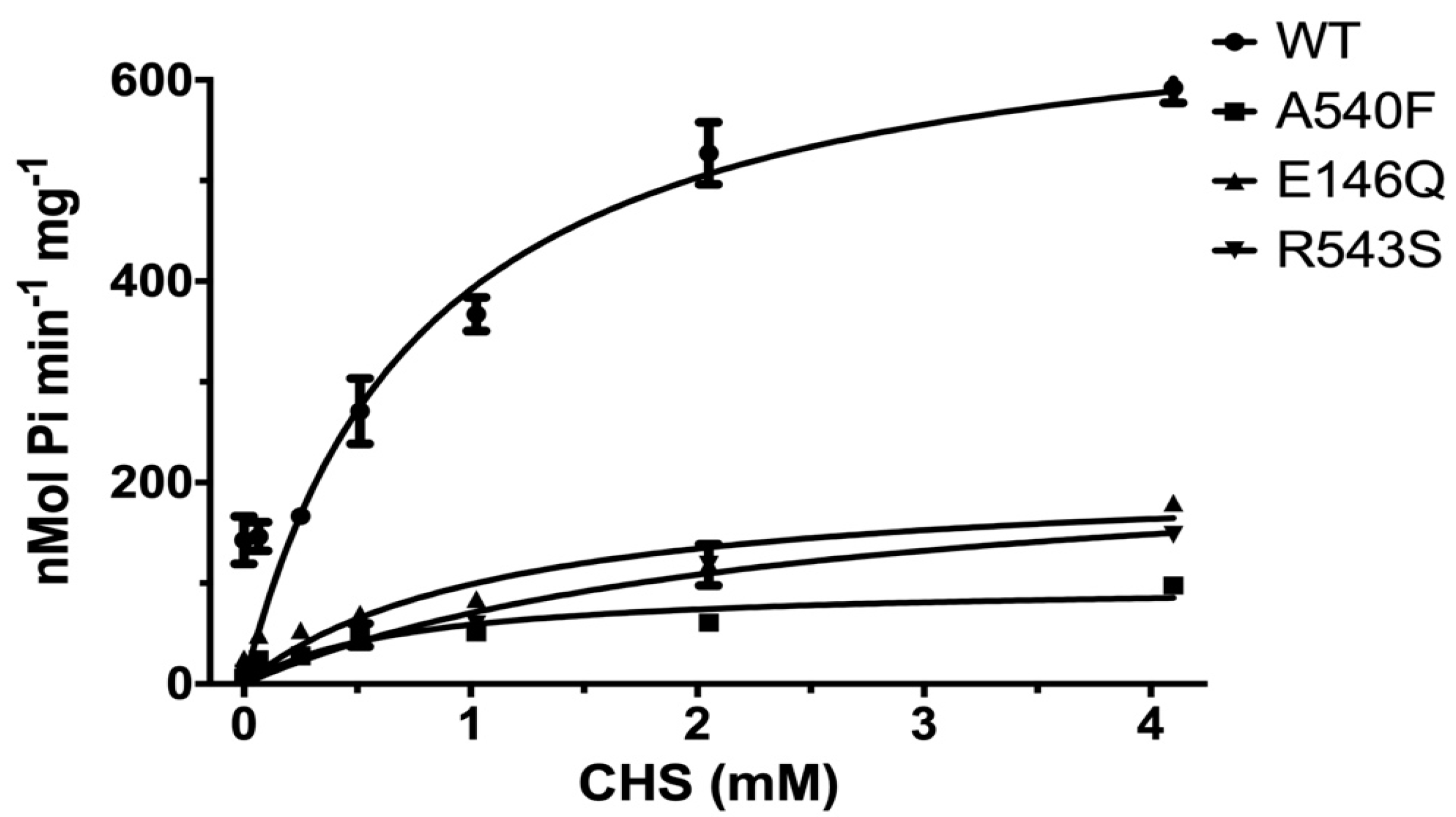
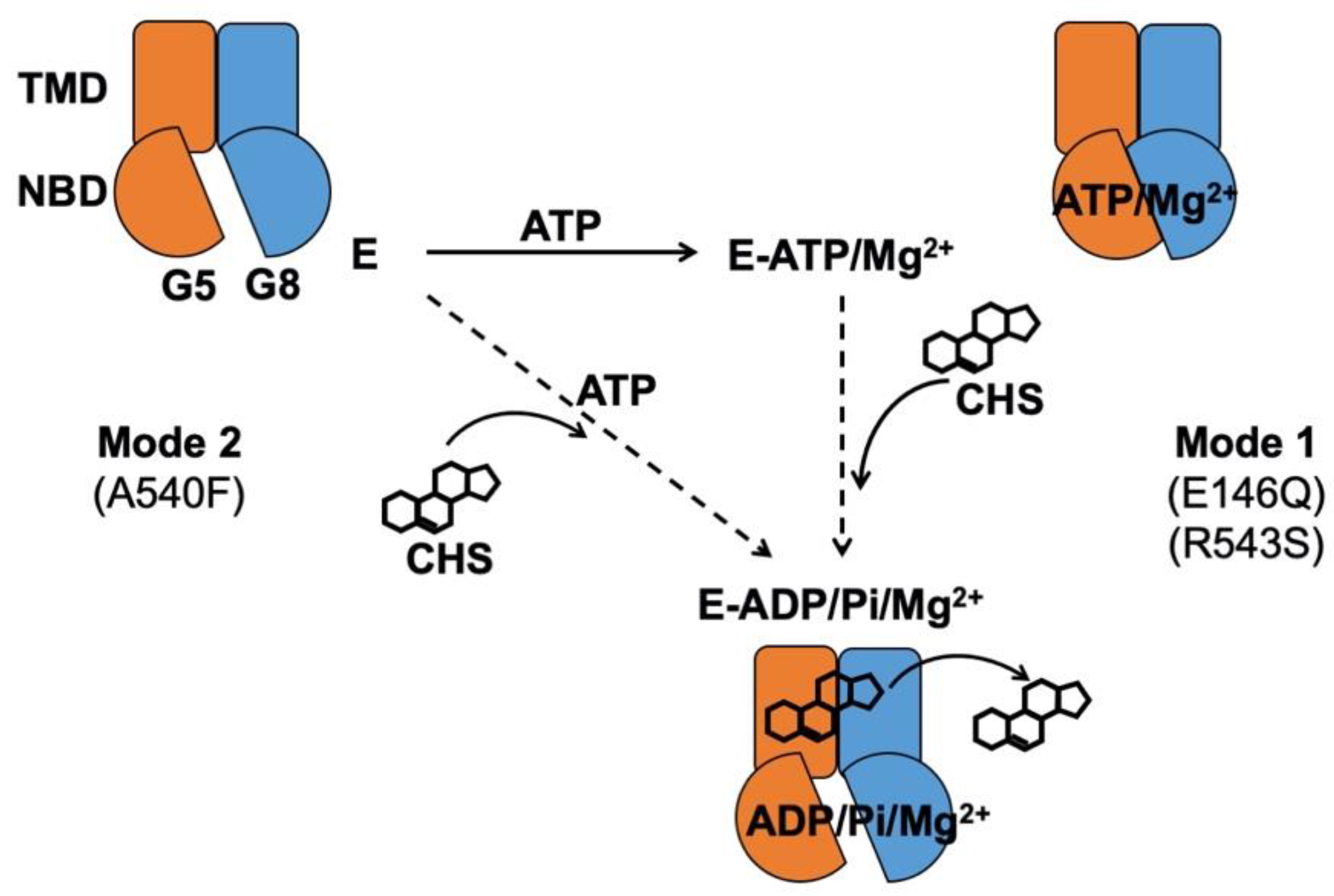
2.1. CHS Stimulates ATP Hydrolysis by Wild-Type (WT) ABCG5/G8
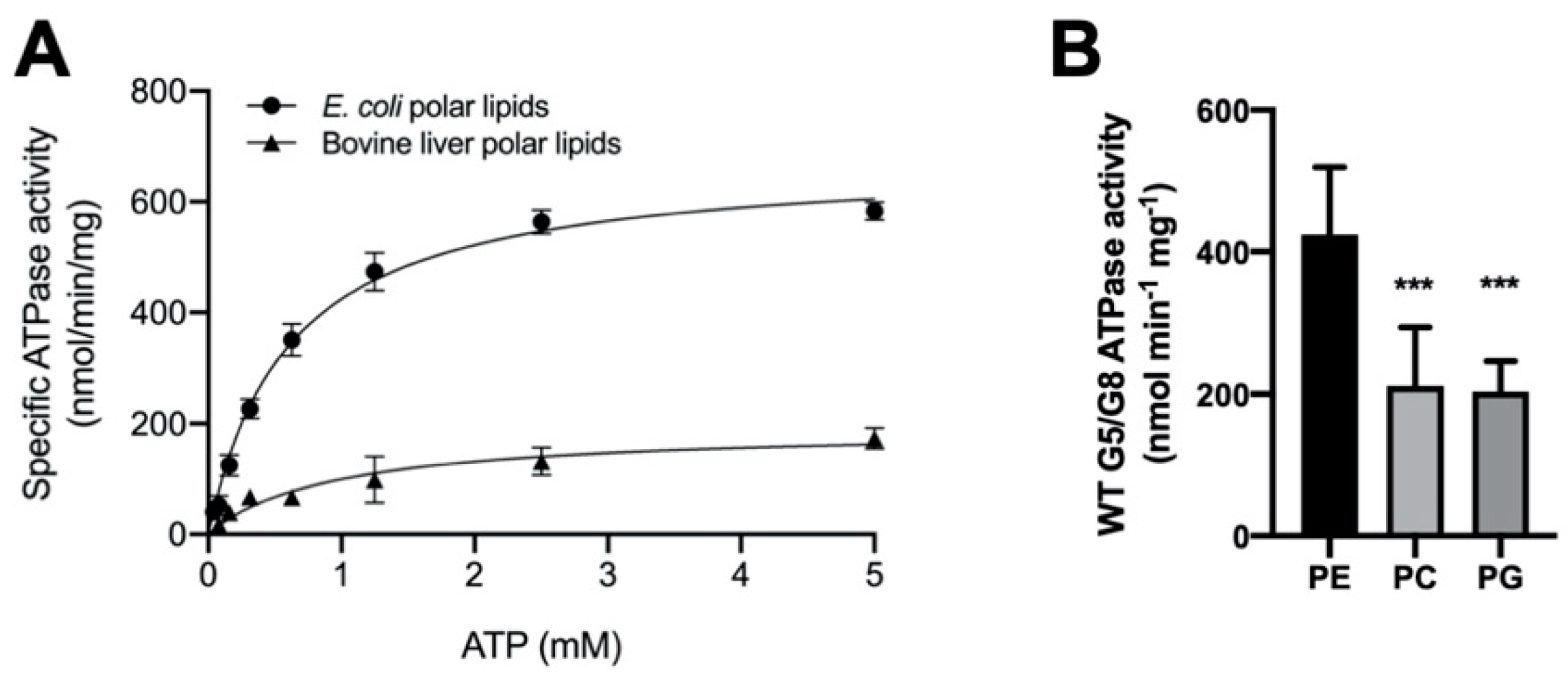
2.2. The Lipid Environments Fine-Tune ABCG5/G8 ATPase Activity
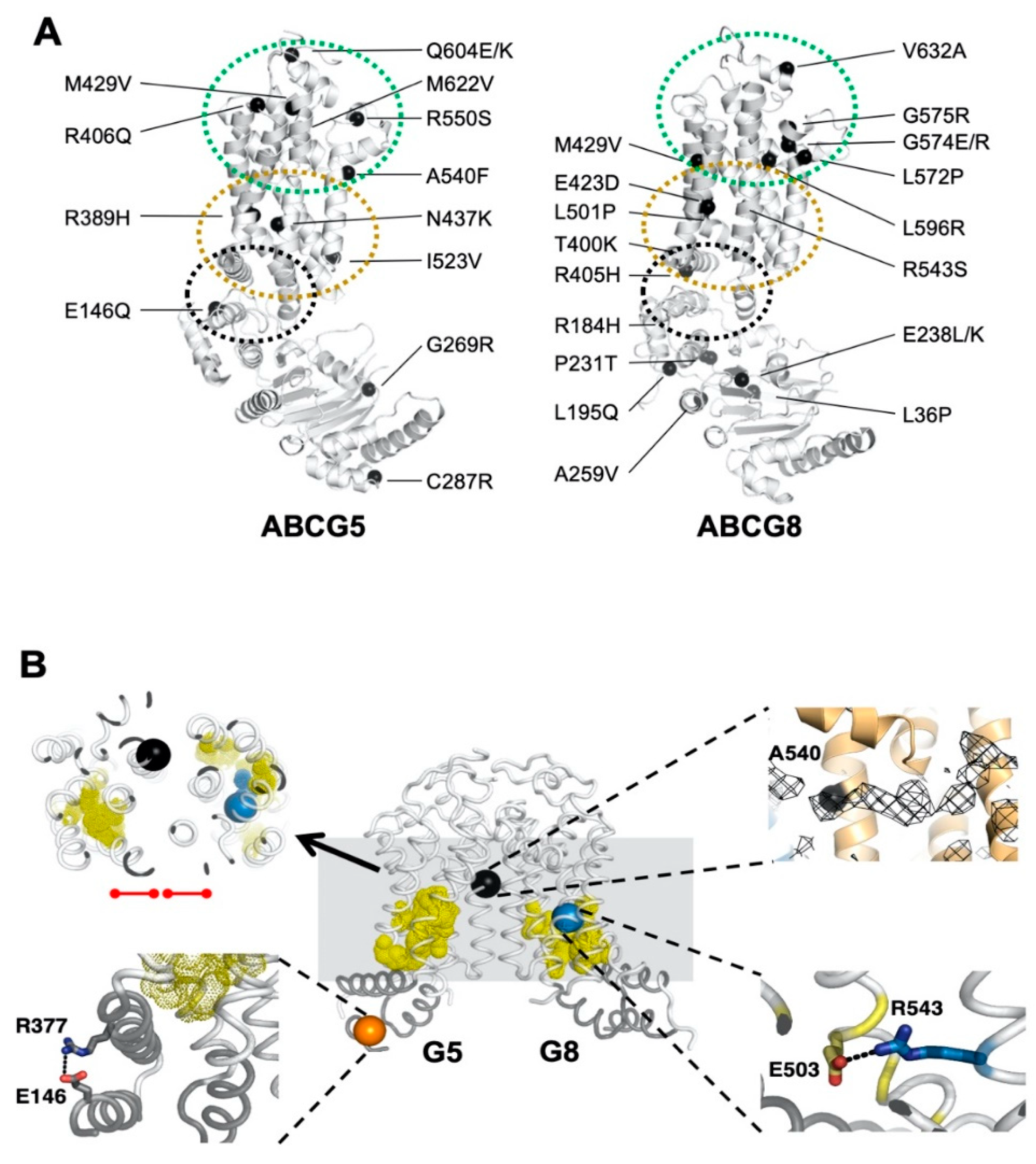
2.3. Missense Mutants Impair CHS-Coupled ATPase Activity of ABCG5/G8
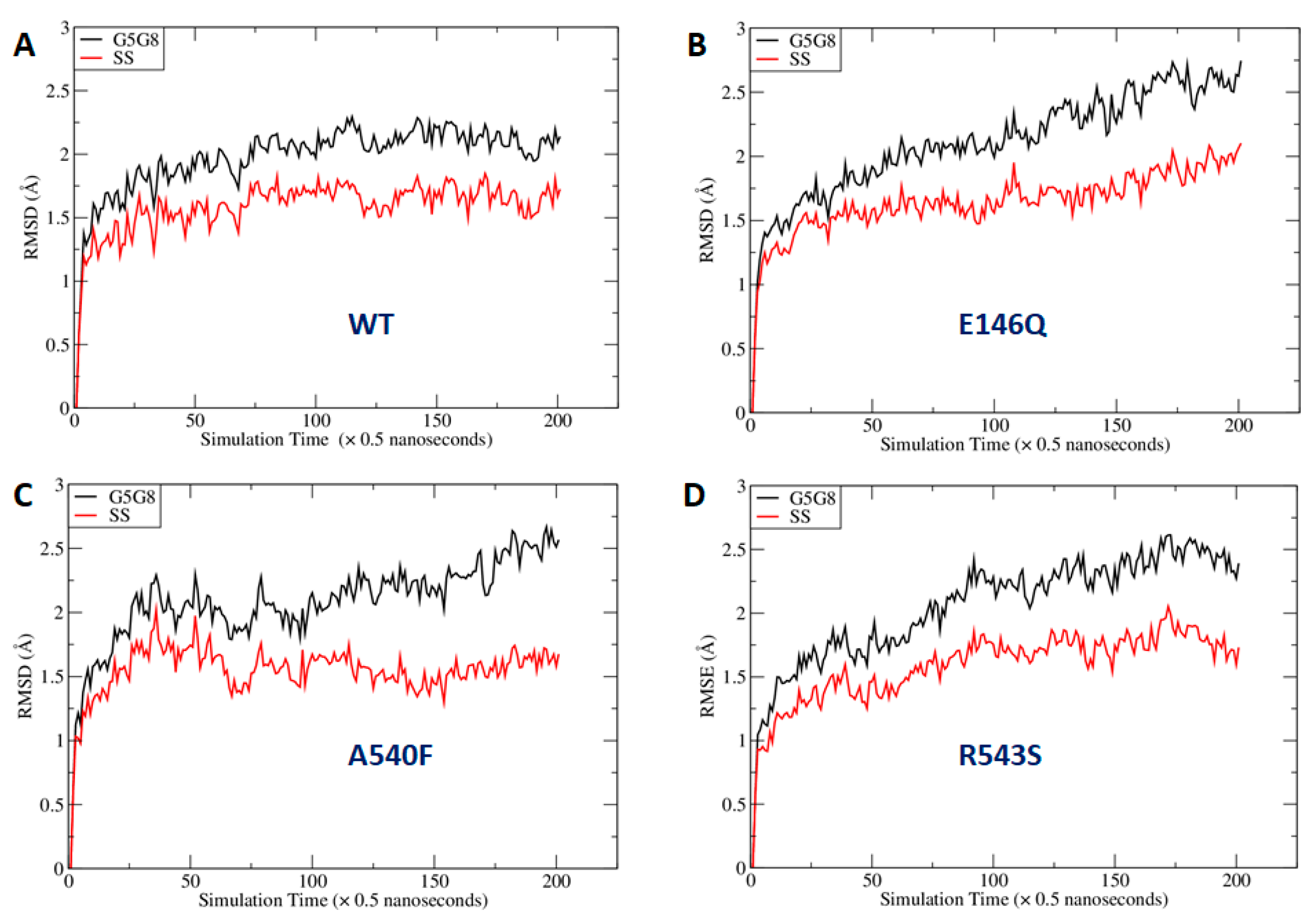
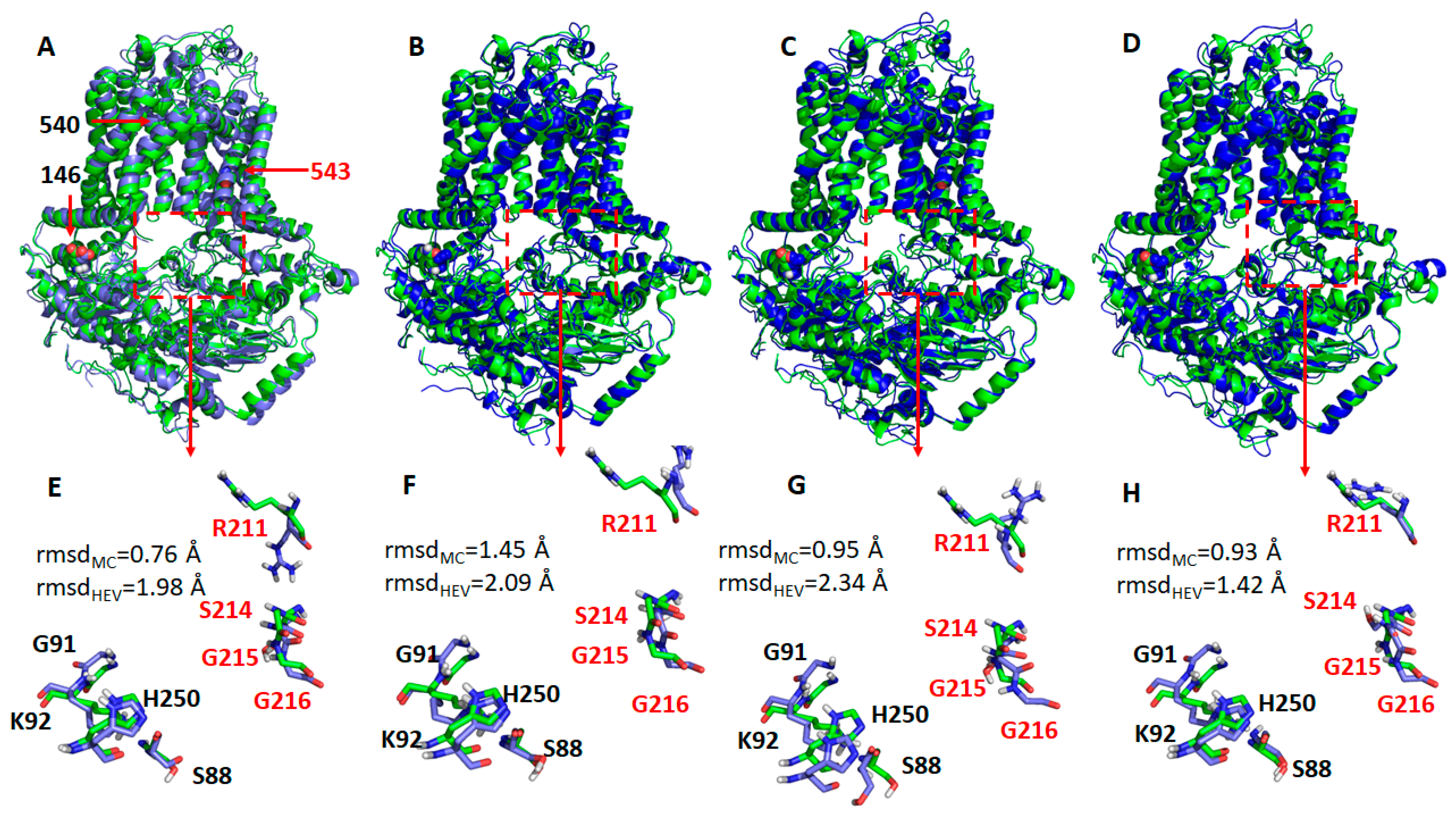
2.4. Missense Mutations Cause Conformational Changes at the ATP-Binding Site
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cloning of ABCG5/G8 Missense Mutants
4.3. Expression of ABCG5/G8 Missense Mutants in Pichia pastoris Yeast
4.4. Cell Culture and Microsomal Membrane Preparation
4.5. Purification of ABCG5/G8 and Its Mutants
4.6. ATPase Assay
4.7. Computational Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| ABCC7 | ATP-binding cassette sub-family C member 7 |
| ABCG5 | ATP-binding cassette sub-family G member 5 |
| ABCG8 | ATP-binding cassette sub-family G member 8 |
| ATP | Adenosine triphosphate |
| CBP | Calmodulin-binding peptide |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| CHS | Cholesteryl hemisuccinate |
| DDM | Dodecyl maltoside or n-dodecyl β-d-maltopyranoside |
| DMPC | 1,2-Dimyristoyl-sn-glycero-3-phosphocholine |
| DNA | Deoxyribonucleic acid |
| DTT | Dithiothreitol |
| EDTA | Ethylenediaminetetraacetic acid |
| EGTA | Ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid |
| ER | Endoplasmic reticulum |
| ICL | Intracellular loop |
| LDL | Low-density lipoprotein |
| LOF | Loss of function |
| LS | Least square |
| MD | Molecular dynamics |
| MGY | Minimal glycerol yeast nitrogen base |
| MM | Minimal methanol |
| mPIB | Minimum protease inhibitor buffer |
| NBD | Nucleotide-binding domain |
| NBS | Nucleotide-binding site |
| Ni-NTA | Nickel–nitrilotriacetic acid |
| PC | Phosphatidylcholine |
| PCR | Polymerase chain reaction |
| PDB | Protein Data Bank |
| PE | Phosphatidylethanolamine |
| PG | Phosphatidylglycerol |
| RCT | Reverse cholesterol transport |
| RMSD | Root-mean-square deviation |
| SDS | Sodium dodecyl sulfate |
| TCEP | Tris-(2-carboxyethyl)-phosphine |
| TICE | Transintestinal cholesterol efflux |
| TMD | Transmembrane domain |
| WT | Wild type |
| YNB | Yeast nitrogen base |
| YPD | Yeast extract peptone dextrose |
| YPDS | Yeast extract peptone dextrose sorbitol |
References
- Dean, M.; Allikmets, R. Evolution of ATP-binding cassette transporter genes. Curr. Opin. Genet. Dev. 1995, 5, 779–785. [Google Scholar] [CrossRef]
- Dean, M.; Hamon, Y.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Linton, K.J.; Higgins, C.F. The Escherichia coli ATP-binding cassette (ABC) proteins. Mol. Microbiol. 1998, 28, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.U.; Song, W.Y.; Hong, D.; Ko, D.; Yamaoka, Y.; Jang, S.; Yim, S.; Lee, E.; Khare, D.; Kim, K.; et al. Plant ABC Transporters Enable Many Unique Aspects of a Terrestrial Plant’s Lifestyle. Mol. Plant 2016, 9, 338–355. [Google Scholar] [CrossRef]
- Li, G.; Gu, H.M.; Zhang, D.W. ATP-binding cassette transporters and cholesterol translocation. IUBMB Life 2013, 65, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.B.; Graf, G.A.; Temel, R.E. ABCG5 and ABCG8: More than a defense against xenosterols. J. Lipid Res. 2018, 59, 1103–1113. [Google Scholar] [CrossRef]
- Borst, P.; Zelcer, N.; Van Helvoort, A. ABC transporters in lipid transport. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2000, 1486, 128–144. [Google Scholar] [CrossRef]
- Xavier, B.M.; Jennings, W.J.; Zein, A.A.; Wang, J.; Lee, J.Y. Structural snapshot of the cholesterol-transport ATP-binding cassette proteins. Biochem. Cell Biol. 2019, 97, 224–233. [Google Scholar] [CrossRef]
- Graf, G.A.; Li, W.-P.; Gerard, R.D.; Gelissen, I.; White, A.; Cohen, J.C.; Hobbs, H.H. Coexpression of ATP-binding cassette proteins ABCG5 and ABCG8 permits their transport to the apical surface. J. Clin. Investig. 2002, 110, 659–669. [Google Scholar] [CrossRef]
- Graf, G.A.; Yu, L.; Li, W.P.; Gerard, R.; Tuma, P.L.; Cohen, J.C.; Hobbs, H.H. ABCG5 and ABCG8 Are Obligate Heterodimers for Protein Trafficking and Biliary Cholesterol Excretion. J. Biol. Chem. 2003, 278, 48275–48282. [Google Scholar] [CrossRef]
- Bhattacharyya, A.K.; Connor, W.E.; Lin, D.S.; McMurry, M.M.; Shulman, R.S. Sluggish sitosterol turnover and hepatic failure to excrete sitosterol into bile cause expansion of body pool of sitosterol in patients with sitosterolemia and xanthomatosis. Arterioscler. Thromb. J. Vasc. Biol. 1991, 11, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Hammer, R.E.; Li-Hawkins, J.; Von Bergmann, K.; Lutjohann, D.; Cohen, J.C.; Hobbs, H.H. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc. Natl. Acad. Sci. USA 2002, 99, 16237–16242. [Google Scholar] [CrossRef] [PubMed]
- Klett, E.L.; Lu, K.; Kosters, A.; Vink, E.; Lee, M.-H.H.; Altenburg, M.; Shefer, S.; Batta, A.K.; Yu, H.; Chen, J.; et al. A mouse model of sitosterolemia: Absence of Abcg8/sterolin-2 results in failure to secrete biliary cholesterol. BMC Med. 2004, 2, 5. [Google Scholar] [CrossRef]
- Plösch, T.; Bloks, V.W.; Terasawa, Y.; Berdy, S.; Siegler, K.; Van Der Sluijs, F.; Kema, I.P.; Groen, A.K.; Shan, B.; Kuipers, F.; et al. Sitosterolemia in ABC-Transporter G5-deficient mice is aggravated on activation of the liver-X receptor. Gastroenterology 2004, 126, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Jakulj, L.; van Dijk, T.H.; de Boer, J.F.; Kootte, R.S.; Schonewille, M.; Paalvast, Y.; Boer, T.; Bloks, V.W.; Boverhof, R.; Nieuwdorp, M.; et al. Transintestinal Cholesterol Transport Is Active in Mice and Humans and Controls Ezetimibe-Induced Fecal Neutral Sterol Excretion. Cell Metab. 2016, 24, 783–794. [Google Scholar] [CrossRef]
- Ford, R.C.; Beis, K. Learning the ABCs one at a time: Structure and mechanism of ABC transporters. Biochem. Soc. Trans. 2019, 47, 23–36. [Google Scholar] [CrossRef]
- Takahashi, K.; Kimura, Y.; Kioka, N.; Matsuo, M.; Ueda, K. Purification and ATPase activity of human ABCA1. J. Biol. Chem. 2006, 281, 10760–10768. [Google Scholar] [CrossRef]
- Zhang, D.W.; Graf, G.A.; Gerard, R.D.; Cohen, J.C.; Hobbs, H.H. Functional asymmetry of nucleotide-binding domains in ABCG5 and ABCG8. J. Biol. Chem. 2006, 281, 4507–4516. [Google Scholar] [CrossRef]
- Hirayama, H.; Kimura, Y.; Kioka, N.; Matsuo, M.; Ueda, K. ATPase activity of human ABCG1 is stimulated by cholesterol and sphingomyelin. J. Lipid Res. 2013, 54, 496–502. [Google Scholar] [CrossRef]
- Wang, J.; Grishin, N.; Kinch, L.; Cohen, J.C.; Hobbs, H.H.; Xie, X.S. Sequences in the nonconsensus nucleotide-binding domain of ABCG5/ABCG8 required for sterol transport. J. Biol. Chem. 2011, 286, 7308–7314. [Google Scholar] [CrossRef]
- Lee, J.-Y.Y.; Kinch, L.N.; Borek, D.M.; Wang, J.J.; Wang, J.J.; Urbatsch, I.L.; Xie, X.-S.S.; Grishin, N.V.; Cohen, J.C.; Otwinowski, Z.; et al. Crystal structure of the human sterol transporter ABCG5/ABCG8. Nature 2016, 533, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Zein, A.A.; Kaur, R.; Hussein, T.O.K.; Graf, G.A.; Lee, J.Y. ABCG5/G8: A structural view to pathophysiology of the hepatobiliary cholesterol secretion. Biochem. Soc. Trans. 2019, 47, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Berge, K.E.; Tian, H.; Graf, G.A.; Yu, L.; Grishin, N.V.; Schultz, J.; Kwiterovich, P.; Shan, B.; Barnes, R.; Hobbs, H.H. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science 2000, 290, 1771–1775. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Lee, M.H.; Hazard, S.; Brooks-Wilson, A.; Hidaka, H.; Kojima, H.; Ose, L.; Stalenhoef, A.F.H.; Mietinnen, T.; Bjorkhem, I.; et al. Two genes that map to the STSL locus cause sitosterolemia: Genomic structure and spectrum of mutations involving sterolin-1 and sterolin-2, encoded by ABCG5 and ABCG8, respectively. Am. J. Hum. Genet. 2001, 69, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Buch, S.; Schafmayer, C.; Völzke, H.; Becker, C.; Franke, A.; Von Eller-Eberstein, H.; Kluck, C.; Bässmann, I.; Brosch, M.; Lammert, F.; et al. A genome-wide association scan identifies the hepatic cholesterol transporter ABCG8 as a susceptibility factor for human gallstone disease. Nat. Genet. 2007, 39, 995–999. [Google Scholar] [CrossRef]
- Kuo, K.-K.; Shin, S.-J.; Chen, Z.-C.; Yang, Y.-H.; Yang, J.F.; Hsiao, P.J. Significant association of ABCG5 604Q and ABCG8 D19H polymorphisms with gallstone disease. Br. J. Surg. 2008, 95, 1005–1011. [Google Scholar] [CrossRef]
- Chen, Z.C.; Shin, S.J.; Kuo, K.K.; Lin, K.D.; Yu, M.L.; Hsiao, P.J. Significant association of ABCG8:D19H gene polymorphism with hypercholesterolemia and insulin resistance. J. Hum. Genet. 2008, 53, 757–763. [Google Scholar] [CrossRef][Green Version]
- Kajinami, K.; Brousseau, M.E.; Ordovas, J.M.; Schaefer, E.J. Interactions between common genetic polymorphisms in ABCG5/G8 and CYP7A1 on LDL cholesterol-lowering response to atorvastatin. Atherosclerosis 2004, 175, 287–293. [Google Scholar] [CrossRef]
- Graf, G.A.; Cohen, J.C.; Hobbs, H.H. Missense mutations in ABCG5 and ABCG8 disrupt heterodimerization and trafficking. J. Biol. Chem. 2004, 279, 24881–24888. [Google Scholar] [CrossRef]
- Lukacs, G.L.; Mohamed, A.; Kartner, N.; Chang, X.B.; Riordan, J.R.; Grinstein, S. Conformational maturation of CFTR but not its mutant counterpart (ΔF508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994, 13, 6076–6086. [Google Scholar] [CrossRef]
- Kizhakkedath, P.; John, A.; Al-Sawafi, B.K.; Al-Gazali, L.; Ali, B.R. Endoplasmic reticulum quality control of LDLR variants associated with familial hypercholesterolemia. FEBS Open Bio 2019, 9, 1994–2005. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Klein, I.; Kopácsi, S.; Remaley, A.T.; Rajnavölgyi, E.; Sarkadi, B.; Váradi, A. Co-expression of human ABCG5 and ABCG8 in insect cells generates an androstan stimulated membrane ATPase activity. FEBS Lett. 2006, 580, 6139–6144. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Johnson, B.J.H.; Lee, J.Y.; Pickert, A.; Urbatsch, I.L. Bile acids stimulate atp hydrolysis in the purified cholesterol transporter ABCG5/G8. Biochemistry 2010, 49, 3403–3411. [Google Scholar] [CrossRef] [PubMed]
- Sarkadi, B.; Price, E.M.; Boucher, R.C.; Germann, U.A.; Scarborough, G.A. Expression of the human multidrug resistance cDNA in insect cells generates a high activity drug-stimulated membrane ATPase. J. Biol. Chem. 1992, 267, 4854–4858. [Google Scholar]
- Cariani, L.; Thomas, L.; Brito, J.; Del Castillo, J.R. Bismuth citrate in the quantification of inorganic phosphate and its utility in the determination of membrane-bound phosphatases. Anal. Biochem. 2004, 324, 79–83. [Google Scholar] [CrossRef]
- Davies, D.R.; Hol, W.G.J. The power of vanadate in crystallographic investigations of phosphoryl transfer enzymes. FEBS Lett. 2004, 577, 315–321. [Google Scholar] [CrossRef]
- Khunweeraphong, N.; Mitchell-White, J.; Szöllősi, D.; Hussein, T.; Kuchler, K.; Kerr, I.D.; Stockner, T.; Lee, J. Picky ABCG5/G8 and promiscuous ABCG2—A tale of fatty diets and drug toxicity. FEBS Lett. 2020. [Google Scholar] [CrossRef]
- Manolaridis, I.; Jackson, S.M.; Taylor, N.M.I.; Kowal, J.; Stahlberg, H.; Locher, K.P. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 2018, 563, 426–430. [Google Scholar] [CrossRef]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.P.; Chien, E.Y.T.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A Specific Cholesterol Binding Site Is Established by the 2.8 Å Structure of the Human β2-Adrenergic Receptor. Structure 2008, 16, 897–905. [Google Scholar] [CrossRef]
- Neumann, J.; Rose-Sperling, D.; Hellmich, U.A. Diverse relations between ABC transporters and lipids: An overview. Biochim. Biophys. Acta Biomembr. 2017, 1859, 605–618. [Google Scholar] [CrossRef]
- Kurumi, Y.; Adachi, Y.; Itoh, T.; Kobayashi, H.; Nanno, T.; Yamamoto, T. Novel high-performance liquid chromatography for determination of membrane phospholipid composition of rat hepatocytes. Gastroenterol. Jpn. 1991, 26, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Hauser, H.; Howell, K.; Dawson, R.M.C.; Bowyer, D.E. Rabbit small intestinal brush border membrane. Preparation and lipid composition. Biochim. Biophys. Acta Biomembr. 1980, 602, 567–577. [Google Scholar] [CrossRef]
- Lee, A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta Biomembr. 2004, 1666, 62–87. [Google Scholar] [CrossRef] [PubMed]
- Feldblum, E.S.; Arkin, I.T. Strength of a bifurcated H bond. Proc. Natl. Acad. Sci. USA 2014, 111, 4085–4090. [Google Scholar] [CrossRef]
- Wang, Z.; Stalcup, L.D.; Harvey, B.J.; Weber, J.; Chloupkova, M.; Dumont, M.E.; Dean, M.; Urbatsch, I.L. Purification and ATP hydrolysis of the putative cholesterol transporters ABCG5 and ABCG8. Biochemistry 2006, 45, 9929–9939. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The amber lipid force field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, I.T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Darden, T.; Perera, L.; Li, L.; Lee, P.; Pedersen, L. New tricks for modelers from the crystallography toolkit: The particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure 1999, 7, R55–R60. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Kong, Y.; Karplus, M. Signaling pathways of PDZ2 domain: A molecular dynamics interaction correlation analysis. Proteins 2009, 74, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.; Li, H.; Wang, J.; Zhao, Z. Landscape of drug-resistance mutations in kinase regulatory hotspots. Brief. Bioinform. 2020. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vmax a (nmol/min/mg) | KM (ATP) (mM) | kcat b (s−1) | kcat/KM (M−1·s−1) | ΔΔGMUT c (kJ/mol) | ne | |
|---|---|---|---|---|---|---|
| WT (liver polar lipids) | 192.8 ± 17.9 | 0.93 ± 0.25 | 0.48 ± 0.04 | 0.52 × 103 | - | 4 |
| WT (E. coli polar lipids) | 677.1 ± 25.6 | 0.60 ± 0.07 | 1.69 ± 0.06 | 2.8 × 103 | - | 6 |
| G5-E146Q d | 167.1 ± 0.05 | 0.51 ± 0.05 | 0.41 ± 0.00 | 0.82 × 103 | 11.7 | 5 |
| G8-R543S d | 150.7 ± 3.7 | 0.42 ± 0.04 | 0.38 ± 0.01 | 0.90 × 103 | 12.3 | 3 |
| G5-A540F d | 101.2 ± 4.2 | 0.58 ± 0.08 | 0.25 ± 0.01 | 0.43 × 103 | 15.8 | 5 |
| Vmax a (nmol/min/mg) | KM (CHS) (mM) | kcat b (s−1) | kcat/KM (M−1·s−1) | ΔΔGMUT c (kJ/mol) | ne | |
|---|---|---|---|---|---|---|
| WT d | 702.9 ± 50.7 | 0.79 ± 0.17 | 1.74 ± 0.13 | 2.2 × 103 | - | 6 |
| G5-E146Q d | 210.0 ± 33.2 | 1.13 ± 0.45 | 0.52 ± 0.08 | 0.46 × 103 | 10.0 | 2 |
| G8-R543S d | 237.1 ± 33.4 | 2.38 ± 0.67 | 0.59 ± 0.08 | 0.25 × 103 | 9.0 | 2 |
| G5-A540F d | 99.8 ± 11.4 | 0.70 ± 0.24 | 0.25 ± 0.03 | 0.36 × 103 | 16.1 | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xavier, B.M.; Zein, A.A.; Venes, A.; Wang, J.; Lee, J.-Y. Transmembrane Polar Relay Drives the Allosteric Regulation for ABCG5/G8 Sterol Transporter. Int. J. Mol. Sci. 2020, 21, 8747. https://doi.org/10.3390/ijms21228747
Xavier BM, Zein AA, Venes A, Wang J, Lee J-Y. Transmembrane Polar Relay Drives the Allosteric Regulation for ABCG5/G8 Sterol Transporter. International Journal of Molecular Sciences. 2020; 21(22):8747. https://doi.org/10.3390/ijms21228747
Chicago/Turabian StyleXavier, Bala M., Aiman A. Zein, Angelica Venes, Junmei Wang, and Jyh-Yeuan Lee. 2020. "Transmembrane Polar Relay Drives the Allosteric Regulation for ABCG5/G8 Sterol Transporter" International Journal of Molecular Sciences 21, no. 22: 8747. https://doi.org/10.3390/ijms21228747
APA StyleXavier, B. M., Zein, A. A., Venes, A., Wang, J., & Lee, J.-Y. (2020). Transmembrane Polar Relay Drives the Allosteric Regulation for ABCG5/G8 Sterol Transporter. International Journal of Molecular Sciences, 21(22), 8747. https://doi.org/10.3390/ijms21228747