Can We Treat Neuroinflammation in Alzheimer’s Disease?



 ,
, 
Abstract
:1. Introduction
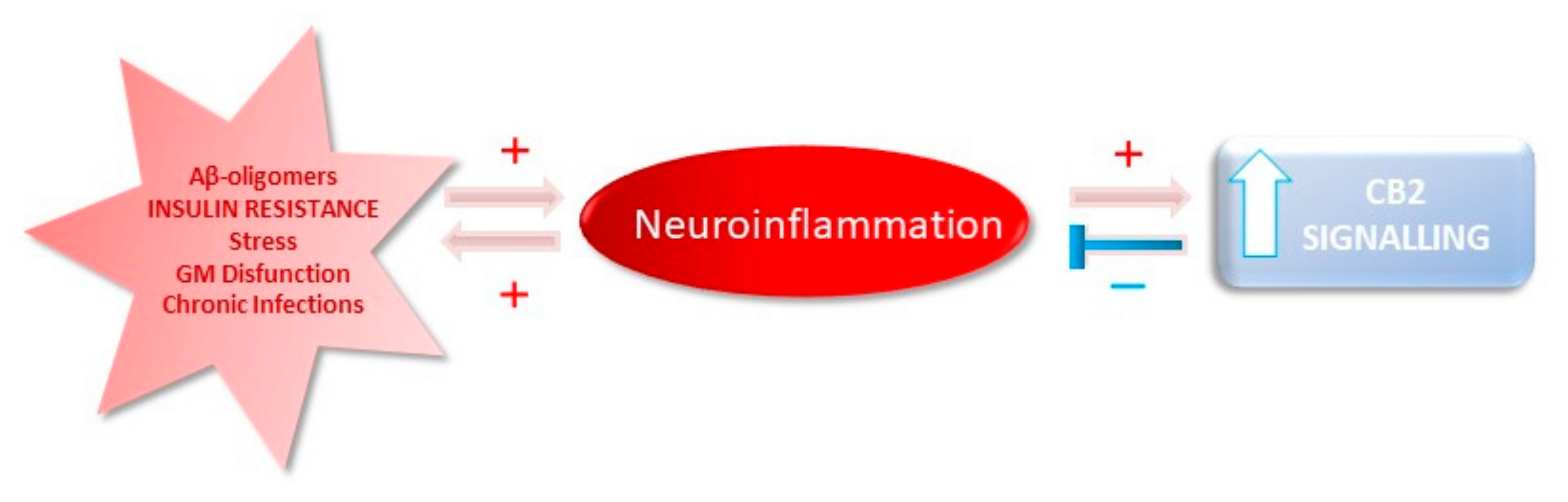
2. Neuroinflammation in AD
3. Targeting Insulin Resistance to Treat AD
4. Nutraceuticals as a Treatment of AD
5. Targeting the Endocannabinoid System in Preclinical Models of AD
6. Gut Microbiota, Neuroinflammation and AD
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2-AG | 2-arachidonilglycerol |
| 5-HT | 5-hydroxytryptamine (serotonin) |
| Aβ | Amyloid beta |
| ABA | Abscisic acid |
| AD | Alzheimer’s disease |
| ADAS-Cog | Alzheimer disease assessment scale–cognitive |
| ADAPT | Alzheimer’s disease anti-inflammatory prevention trial |
| ADCS-ADL | Alzheimer’s disease cooperative study-activities of daily living |
| AEA | N-arachidonil-ethanolamine |
| AMPK | Adenosine monophosphate-activated protein kinase |
| APP | Amyloid protein precursor |
| APS | Alzheimer progression score |
| AVL | Auditory Verbal Learning Test |
| BACE1 | β-site APP-cleaving enzyme 1 secretase |
| BADLS | Bristol Activities of Daily Living Scale; |
| BBB | Blood brain barrier |
| BDNF | Brain-derived neurotrophic factor |
| CB1 | G-protein-coupled cannabinoid 1 receptor |
| CB2 | G-protein-coupled cannabinoid 2 receptor |
| CBD | Cannabidiol |
| CCR | Cambridge Contextual Reading Test |
| CD68 | Cluster of Differentiation 68 |
| CDT | Clock Drawing Test |
| CGI-I | Clinical Global Impression-improvement |
| CN | Category naming test |
| CNS | Central nervous system |
| COX | Cyclooxygenase |
| COWA | Controlled Oral Word Association Test |
| CSF | Cerebrospinal fluid |
| DAGLα | Diacylglycerol Lipase alpha |
| DAMPs | Damage-associated molecular patterns |
| DMS | Delayed Matching to Sample |
| DSS | Digit Symbol Substitution |
| ECS | Endocannabinoid system |
| EGCG | Epigallocatechin gallate |
| FAAH | Fatty acid amide hydrolase |
| FMT | Fecal microbiota transplant |
| GABA | Gamma-aminobutyric acid |
| GM | Gut microbiota |
| IFN-γ | Interferon gamma |
| IL-1β | Interleukin 1 beta |
| IN | Intranasal |
| IRS1 | Insulin receptor substate 1 |
| LanCL2 | Lanthionine synthetase C-like protein 2 |
| LDL | Low density lipoprotein |
| LPS | Lipopolysaccharides |
| MAGL | Monoacylglycerol lipase |
| MAPT | MultiDomain Alzheimer preventive trial |
| MCI | Mild cognitive impairment |
| MD | Mediterranean diet |
| MMSE | Mini-mental state examination |
| MoCA | Montreal Cognitive Assessment |
| mTOR | Mammalian target of rapamycin |
| NAPE | N-acyl-phosphatidylethanolamine |
| NTB | Neuropsychological Test Battery |
| NTB | Neuropsychological Test Battery NFTs Neurofibrillary tangles |
| NGF | Nerve growth factor |
| NLR | NOD-like receptors |
| NLRP3 | NLR family pyrin domain containing 3 |
| NPI | Neuropsychiatric Inventory |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| p70S6K | p70 ribosomal S6 kinase |
| PAL | Paired associate learning scale |
| PAMPs | Pathogen-associated molecular patterns |
| PEA | Palmitoylethanolamide |
| PG | Prostaglandin |
| PPAR-α | Peroxisome proliferator-activated receptor alpha |
| PRRs | Pattern recognition receptors |
| PUFAs | Polyunsaturated fatty acids |
| RANTES | Regulated on Activation, Normal T cell Expressed and Secreted |
| RBANS | Repeatable Battery for the Assessment of Neuropsychological Status |
| RV | Resveratrol |
| SMMSE | Standardized mini-mental state examination |
| T2DM | Type 2 diabetes mellitus |
| TGF-β | Transforming growth factor beta |
| THC | Tetrahidrocannabinol |
| TLRs | Toll-like receptors |
| TNF-α | Tumor necrosis factor alpha |
| WAIS-R | Wechsler Adults Intelligence Scale |
| WMS-R | Wechsler Memory Scale |
| WT | Wild-type |
References
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A Review: Inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012, 2012, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Szabo, L.; Eckert, A.; Grimm, A. Insights into disease-associated tau impact on mitochondria. Int. J. Mol. Sci. 2020, 21, 6344. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.W.; Initiative, A.D.N.; Iturria-Medina, Y.; Strandberg, O.T.; Smith, R.; Levitis, E.; Evans, A.C.; Hansson, O.; The Swedish BioFINDER Study. Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Kayed, R.; Lasagna-Reeves, C.A. Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimer’s Dis. 2012, 33, S67–S78. [Google Scholar] [CrossRef] [Green Version]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S.S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Lee, C.Y.D.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef] [Green Version]
- Birla, H.; Minocha, T.; Kumar, G.; Misra, A.; Singh, S.K. Role of oxidative stress and metal toxicity in the progression of Alzheimer’s disease. Curr. Neuropharmacol. 2020, 18, 552–562. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. PubMed. Oxid. Med. Cell. Longev. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Skaper, S.D. Alzheimer’s disease and amyloid: Culprit or coincidence? Int. Rev. Neurobiol. 2012, 102, 277–316. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Chung, J.H.; Choi, T.K.; Suh, S.Y.; Oh, B.H.; Hong, C.H. Peripheral cytokines and chemokines in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2009, 28, 281–287. [Google Scholar] [CrossRef] [PubMed]
- DaRocha-Souto, B.; Scotton, T.C.; Coma, M.; Serrano-Pozo, A.; Hashimoto, T.; Serenó, L.; Rodríguez, M.; Sánchez, B.; Hyman, B.T.; Gómez-Isla, T. Brain oligomeric β-amyloid but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in amyloid precursor protein/tau transgenic mice. J. Neuropathol. Exp. Neurol. 2011, 70, 360–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, C.F.; Santos, A.E.; Moreira, P.I.; Pereira, A.C.; Sousa, F.J.; Cardoso, S.M.; Cruz, M.T. Is Alzheimer’s disease an inflammasomopathy? Ageing Res. Rev. 2019, 56, 100966. [Google Scholar] [CrossRef]
- Tarkowski, E.; Andreasen, N.; Blennow, K. Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1200–1205. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Neuroscience: Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Kanneganti, T.-D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 2019, 39, 1053–1090. [Google Scholar] [CrossRef] [Green Version]
- Albornoz, E.A.; Woodruff, T.M.; Gordon, R. Inflammasomes in CNS Diseases. Experientia Supplementum 2018, 108, 41–60. [Google Scholar] [CrossRef]
- Bagyinszky, E.; Van Giau, V.; Shim, K.; Suk, K.; An, S.S.A.; Kim, S. Role of inflammatory molecules in the Alzheimer’s disease progression and diagnosis. J. Neurol. Sci. 2017, 376, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Obulesu, M.; Lakshmi, M.J. Neuroinflammation in Alzheimer’s disease: An understanding of physiology and pathology. Int. J. Neurosci. 2013, 124, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Tournier, B.B.; Tsartsalis, S.; Ceyzériat, K.; Valentina, G.; Millet, P. In vivo TSPO signal and neuroinflammation in Alzheimer’s disease. Cells 2020, 9, 1941. [Google Scholar] [CrossRef] [PubMed]
- Meda, L.; Cassatella, M.A.; Szendrei, G.I.; Otvos, L.; Baron, P.; Villalba, M.; Ferrari, D.; Rossi, F. Activation of microglial cells by β-amyloid protein and interferon-γ. Nat. Cell Biol. 1995, 374, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Perea, J.R.; Bolós, M.; Avila, J. Microglia in Alzheimer’s disease in the context of Tau pathology. Biomolecules 2020, 10, 1439. [Google Scholar] [CrossRef]
- François, A.; Bilan, A.R.; Quellard, N.; Fernandez, B.; Janet, T.; Chassaing, D.; Paccalin, M.; Terro, F.; Page, G. Longitudinal follow-up of autophagy and inflammation in brain of APPswePS1dE9 transgenic mice. J. Neuroinflamm. 2014, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nat. Cell Biol. 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Hanslik, K.L.; Ulland, T.K. The Role of microglia and the Nlrp3 inflammasome in Alzheimer’s disease. Front. Neurol. 2020, 11, 11. [Google Scholar] [CrossRef]
- Feng, Y.-S.; Tan, Z.-X.; Wu, L.-Y.; Dong, F.; Zhang, F. The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Res. Rev. 2020, 101192. [Google Scholar] [CrossRef]
- Clement, A.; Wiborg, O.; Asuni, A.A. Steps towards developing effective treatments for neuropsychiatric disturbances in Alzheimer’s disease: Insights from preclinical models, clinical data, and future directions. Front. Aging Neurosci. 2020, 12, 56. [Google Scholar] [CrossRef]
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Inflammation, antiinflammatory agents, and Alzheimer’s disease: The last 22 years. J. Alzheimer’s Dis. 2016, 54, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Pasinetti, G.M. From epidemiology to therapeutic trials with anti-inflammatory drugs in Alzheimer’s disease: The role of NSAIDs and cyclooxygenase in β-amyloidosis and clinical dementia1. J. Alzheimer’s Dis. 2002, 4, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; Fitzgerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Breitner, J.; Meyer, P.-F. Author response: INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology 2020, 94, 594. [Google Scholar] [CrossRef] [PubMed]
- Breitner, J.; Baker, L.; Drye, L.; Evans, D.; Lyketsos, C.G.; Ryan, L.; Zandi, P.; Saucedo, H.H.; Anau, J.; Cholerton, B. Follow-up evaluation of cognitive function in the randomized Alzheimer’s disease anti-inflammatory prevention trial and its follow-up study. Alzheimer’s Dement. 2015, 11, 216–225. [Google Scholar]
- Butchart, J.; Brook, L.; Hopkins, V.; Teeling, J.L.; Püntener, U.; Culliford, D.; Sharples, R.; Sharif, S.; McFarlane, B.; Raybould, R.; et al. Etanercept in Alzheimer disease: A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 2015, 84, 2161–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najem, D.; Bamji-Mirza, M.; Chang, N.; Liu, Q.Y.; Zhang, W. Insulin resistance, neuroinflammation, and Alzheimer’s disease. Rev. Neurosci. 2014, 25, 509–525. [Google Scholar] [CrossRef]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a type 3 diabetes? A critical appraisal. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms linking inflammation to insulin resistance. Int. J. Endocrinol. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- De La Monte, S.M. Brain insulin resistance and deficiency as therapeutic targets in Alzheimers disease. Curr. Alzheimer Res. 2012, 9, 35–66. [Google Scholar] [CrossRef]
- Cukierman, T.; Gerstein, H.C.; Williamson, J.D. Cognitive decline and dementia in diabetes-systematic overview of prospective observational studies. Diabetologia 2005, 48, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Avgerinos, K.I.; Kalaitzidis, G.; Malli, A.; Kalaitzoglou, D.; Myserlis, P.G.; Lioutas, V.-A. Intranasal insulin in Alzheimer’s dementia or mild cognitive impairment: A systematic review. J. Neurol. 2018, 265, 1497–1510. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Haapasalo, A.; Soininen, H.; Hiltunen, M. AMP-activated protein kinase: A potential player in Alzheimer’s disease. J. Neurochem. 2011, 118, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Hartley, D.; Cooper, G.M. Role of mTOR in the degradation of IRS-1: Regulation of PP2A activity. J. Cell. Biochem. 2002, 85, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, Y.; Inamitsu, T.; Chida, K.; Iemura, S.-I.; Natsume, T.; Maeda, T.; Hakuno, F.; Takahashi, S.-I. Serine phosphorylation by mTORC1 promotes IRS-1 degradation through SCFβ-TRCP E3 ubiquitin ligase. iScience 2018, 5, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.M.; Mechanic-Hamilton, D.; Xie, S.X.; Combs, M.F.; Cappola, A.R.; Xie, L.; Detre, J.A.; Wolk, D.A.; Arnold, S.E. Effects of the insulin sensitizer metformin in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2017, 31, 107–113. [Google Scholar] [CrossRef]
- Fernández-Real, J.M.; García-Fuentes, E.; Moreno-Navarrete, J.M.; Murri-Pierri, M.; Garrido-Sánchez, L.; Ricart, W.; Tinahones, F. Fat overload induces changes in circulating lactoferrin that are associated with postprandial lipemia and oxidative stress in severely obese subjects. Obesity 2010, 18, 482–488. [Google Scholar] [CrossRef]
- Artym, J. A remedy against obesity? The role of lactoferrin in the metabolism of glucose and lipids. Postęp. Higien. Med. Dośw. 2012, 66, 937–953. [Google Scholar] [CrossRef]
- Brizzio, E.; Castro, M.; Narbaitz, M.; Borda, N.; Carbia, C.D.; Correa, L.; Mengarelli, R.; Merelli, A.; Brizzio, V.; Sosa, M.; et al. Ulcerated hemosiderinic dyschromia and iron deposits within lower limbs treated with a topical application of biological chelator. Veins Lymphat. 2012, 1, e6. [Google Scholar] [CrossRef]
- Mohamed, W.A.; Salama, R.M.; Schaalan, M.F. A pilot study on the effect of lactoferrin on Alzheimer’s disease pathological sequelae: Impact of the p-Akt/PTEN pathway. Biomed. Pharmacother. 2019, 111, 714–723. [Google Scholar] [CrossRef]
- Manzanares, W.; Hardy, G. Vitamin B12: The forgotten micronutrient for critical care. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Kouroglou, E.; Anagnostis, P.; Daponte, A.; Bargiota, A. Vitamin B12 insufficiency is associated with increased risk of gestational diabetes mellitus: A systematic review and meta-analysis. Endocrine 2019, 66, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gueant-Rodriguez, R.-M.; Quilliot, D.; Sirveaux, M.-A.; Meyre, D.; Gueant, J.-L.; Brunaud, L. Folate and vitamin B12 status is associated with insulin resistance and metabolic syndrome in morbid obesity. Clin. Nutr. 2018, 37, 1700–1706. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhou, X.; Li, Q.; Zhao, J.; Song, A.; An, P.; Du, Y.; Xu, W.; Huang, G. Effects of folic acid and vitamin B12, alone and in combination on cognitive function and inflammatory factors in the elderly with mild cognitive impairment: A single-blind experimental design. Curr. Alzheimer Res. 2019, 16, 622–632. [Google Scholar] [CrossRef]
- Vakilian, A.; Razavi-Nasab, S.M.; Ravari, A.; Mirzaei, T.; Moghadam-Ahmadi, A.; Jalali, N.; Bahramabadi, R.; Rezayati, M.; Yazdanpanah-Ravari, A.; Bahmaniar, F.; et al. Vitamin B12 in association with antipsychotic drugs can modulate the expression of pro-/anti-inflammatory cytokines in Alzheimer disease patients. Neuroimmunomodulation 2018, 24, 310–319. [Google Scholar] [CrossRef]
- Bahramabadi, R.; Samadi, M.; Vakilian, A.; Jafari, E.; Fathollahi, M.S.; Arababadi, M.K. Evaluation of the effects of anti-psychotic drugs on the expression of CD68 on the peripheral blood monocytes of Alzheimer patients with psychotic symptoms. Life Sci. 2017, 179, 73–79. [Google Scholar] [CrossRef]
- Ramprasad, M.P.; Terpstra, V.; Kondratenko, N.; Quehenberger, O.; Steinberg, D. Cell surface expression of mouse macrosialin and human CD68 and their role as macrophage receptors for oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1996, 93, 14833–14838. [Google Scholar] [CrossRef] [Green Version]
- Farah, R.R.; Khamisy-Farah, R.S.-S. Calcium channel blocker effect on insulin resistance and inflammatory markers in essential hypertension patients. PubMed. Int. Angiol. 2013, 32, 85–93. [Google Scholar]
- Cabezas-Cerrato, J.; García-Estévez, D.A.; Araújo, D.; Iglesias, M. Insulin sensitivity, glucose effectiveness, and β-cell function in obese males with essential hypertension: Investigation of the effects of treatment with a calcium channel blocker (diltiazem) or an angiotensin-converting enzyme inhibitor (quinapril). Metabolism 1997, 46, 173–178. [Google Scholar] [CrossRef]
- Weitz-Schmidt, G. Statins as anti-inflammatory agents. Trends Pharmacol. Sci. 2002, 23, 482–487. [Google Scholar] [CrossRef]
- Wang, Q.; Yan, J.; Chen, X.; Li, J.; Yang, Y.; Weng, J.; Deng, C.; Yenari, M.A. Statins: Multiple neuroprotective mechanisms in neurodegenerative diseases. Exp. Neurol. 2011, 230, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, B.A.; Segurado, R.; Kennelly, S.; Rikkert, M.G.M.O.; Howard, R.; Pasquier, F.; Börjesson-Hanson, A.; Tsolaki, M.; Lucca, U.; Molloy, D.W.; et al. Nilvadipine in mild to moderate Alzheimer disease: A randomised controlled trial. PLoS Med. 2018, 15, e1002660. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Li, Z.; Zhao, L.; Zhao, W. Simvastatin ameliorate memory deficits and inflammation in clinical and mouse model of Alzheimer’s disease via modulating the expression of miR-106b. Biomed. Pharmacother. 2017, 92, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Bell, K.L.; Galasko, D.; Galvin, J.E.; Thomas, R.G.; Van Dyck, C.H.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology 2011, 77, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Zhao, Q.; Zhou, Y.; Zhao, Y.; Wan, Q. Atorvastatin may correct dyslipidemia in adult patients at risk for Alzheimer’s disease through an anti-inflammatory pathway. CNS Neurol. Disord. Drug Targets 2016, 15, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Feldman, H.; Doody, R.S.; Kivipelto, M.; Sparks, D.L.; Waters, D.D.; Jones, R.W.; Schwam, E.; Schindler, R.; Hey-Hadavi, J.; Demicco, D.A.; et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology 2010, 74, 956–964. [Google Scholar] [CrossRef]
- Elsen, G.V.D.; Ahmed, A.I.; Verkes, R.-J.; Kramers, C.; Feuth, T.; Rosenberg, P.B.; Van Der Marck, M.A.; Rikkert, M.G.O. Tetrahydrocannabinol for neuropsychiatric symptoms in dementia: A randomized controlled trial. Neurology 2015, 84, 2338–2346. [Google Scholar] [CrossRef] [Green Version]
- Gera, M.; Sharma, N.; Ghosh, M.; Huynh, D.L.; Lee, S.J.; Min, T.; Kwon, T.; Jeong, D.K. Nanoformulations of curcumin: An emerging paradigm for improved remedial application. Oncotarget 2017, 8, 66680–66698. [Google Scholar] [CrossRef] [Green Version]
- Fidelis, E.M.; Savall, A.S.P.; Abreu, E.D.L.; Carvalho, F.; Teixeira, F.E.G.; Haas, S.E.; Sampaio, T.B.; Pinton, S. Curcumin-loaded nanocapsules reverses the depressant-like behavior and oxidative stress induced by β-amyloid in mice. Neuroscience 2019, 423, 122–130. [Google Scholar] [CrossRef]
- Teter, B.; Morihara, T.; Lim, G.; Chu, T.; Jones, M.; Zuo, X.; Paul, R.; Frautschy, S.A.; Cole, G.M. Curcumin restores innate immune Alzheimer’s disease risk gene expression to ameliorate Alzheimer pathogenesis. Neurobiol. Dis. 2019, 127, 432–448. [Google Scholar] [CrossRef]
- Panahi, Y.; Hosseini, M.S.; Khalili, N.; Naimi, E.; Simental-Mendía, L.E.; Majeed, M.; Sahebkar, A. Effects of curcumin on serum cytokine concentrations in subjects with metabolic syndrome: A post-hoc analysis of a randomized controlled trial. Biomed. Pharmacother. 2016, 82, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Rainey-Smith, S.R.; Brown, B.M.; Sohrabi, H.R.; Shah, T.; Goozee, K.G.; Gupta, V.B.; Martins, R.N. Curcumin and cognition: A randomised, placebo-controlled, double-blind study of community-dwelling older adults. Br. J. Nutr. 2016, 115, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Baum, L.; Lam, C.W.K.; Cheung, S.K.-K.; Kwok, T.; Lui, V.; Tsoh, J.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; et al. Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastianetto, S.; Ménard, C.; Quirion, R. Neuroprotective action of resveratrol. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 1195–1201. [Google Scholar] [CrossRef] [Green Version]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Turner, R.S.; Thomas, R.G.; Craft, S.; Van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S.; et al. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, Y.K.; Ban, J.O.; Ha, T.Y.; Yun, Y.P.; Han, S.-B.; Oh, K.W.; Hong, J.T. Green tea (-)-epigallocatechin-3-gallate inhibits β-amyloid-induced cognitive dysfunction through modification of secretase activity via inhibition of ERK and NF-κB pathways in mice. J. Nutr. 2009, 139, 1987–1993. [Google Scholar] [CrossRef]
- Cascella, M.; Bimonte, S.; Muzio, M.R.; Schiavone, V.; Cuomo, A. The efficacy of Epigallocatechin-3-gallate (green tea) in the treatment of Alzheimer’s disease: An overview of pre-clinical studies and translational perspectives in clinical practice. Infect. Agents Cancer 2017, 12, 1–7. [Google Scholar] [CrossRef]
- Mori, T.; Koyama, N.; Guillot-Sestier, M.-V.; Tan, J.; Town, T. Ferulic Acid Is a Nutraceutical β-secretase modulator that improves behavioral impairment and Alzheimer-like pathology in transgenic mice. PLoS ONE 2013, 8, e55774. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; Koyama, N.; Tan, J.; Segawa, T.; Maeda, M.; Town, T. Combined treatment with the phenolics (−)-epigallocatechin-3-gallate and ferulic acid improves cognition and reduces Alzheimer-like pathology in mice. J. Biol. Chem. 2018, 294, 2714–2731. [Google Scholar] [CrossRef] [Green Version]
- Jin, G.; Bai, D.; Yin, S.; Yang, Z.; Zou, D.; Zhang, Z.; Li, X.; Sun, Y.; Zhu, Q. Silibinin rescues learning and memory deficits by attenuating microglia activation and preventing neuroinflammatory reactions in SAMP8 mice. Neurosci. Lett. 2016, 629, 256–261. [Google Scholar] [CrossRef]
- Hostetler, G.L.; Ralston, R.A.; Schwartz, S.J. Flavones: Food sources, bioavailability, metabolism, and bioactivity. Adv. Nutr. 2017, 8, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Wang, J.-L.; Liu, R.; Li, X.-X.; Li, J.-F.; Zhang, L. Neuroprotective, anti-amyloidogenic and neurotrophic effects of Apigenin in an Alzheimer’s disease mouse model. Molecules 2013, 18, 9949–9965. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Tong, T.; Wan, S.; Yan, T.; Ren, F.; Bi, K.; Jia, Y. Protective effects of Puerarin against A β 1-42-induced learning and memory impairments in mice. Planta Med. 2017, 83, 224–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.-J.; Huang, C.-Y.; Lee, M.; Lin, J.-Y.; Hsieh-Li, H.M. Puerariae radix prevents anxiety and cognitive deficits in mice under oligomeric Aβ-induced stress. Am. J. Chin. Med. 2019, 47, 1459–1481. [Google Scholar] [CrossRef] [PubMed]
- Pandareesh, M.D.; Chauhan, V.; Chauhan, A. Walnut supplementation in the diet reduces oxidative damage and improves antioxidant status in transgenic mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 64, 1295–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, N.; Wang, K.C.; Wegiel, J.; Malik, M.N. Walnut extract inhibits the fibrillization of amyloid beta-protein, and also defibrillizes its preformed fibrils. Curr. Alzheimer Res. 2004, 1, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Verme, J.L.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-α mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Beggiato, S.; Tomasini, M.C.; Ferraro, L. Palmitoylethanolamide (PEA) as a potential therapeutic agent in Alzheimer’s disease. Front. Pharmacol. 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrieu, S.; Guyonnet, S.; Coley, N.; Cantet, C.; Bonnefoy, M.; Bordes, S.; Bories, L.; Noëlle-Cuffi, M.; Dantoine, T.; Dartigues, J.-F.; et al. Effect of long-term omega 3 polyunsaturated fatty acid supplementation with or without multidomain intervention on cognitive function in elderly adults with memory complaints (MAPT): A randomised, placebo-controlled trial. Lancet Neurol. 2017, 16, 377–389. [Google Scholar] [CrossRef]
- Im, D.-S. Pro-Resolving Effect of ginsenosides as an anti-inflammatory mechanism of Panax ginseng. Biomololecules 2020, 10, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, K.; Wong, A.S.T. Pharmacology of ginsenosides: A literature review. Chin. Med. 2010, 5, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.-H.; Lee, S.-T.; Chu, K.; Oh, M.J.; Park, H.J.; Shim, J.-Y.; Kim, M. Heat-processed ginseng enhances the cognitive function in patients with moderately severe Alzheimer’s disease. Nutr. Neurosci. 2012, 15, 278–282. [Google Scholar] [CrossRef]
- Sachdeva, A.K.; Chopra, K. Lycopene abrogates Aβ (1–42)-mediated neuroinflammatory cascade in an experimental model of Alzheimer’s disease. J. Nutr. Biochem. 2015, 26, 736–744. [Google Scholar] [CrossRef]
- Von Arnim, C.A.F.; Herbolsheimer, F.; Nikolaus, T.; Peter, R.; Biesalski, H.K.; Ludolph, A.C.; Riepe, M.; Nagel, G.; The ActiFE Ulm Study Group. Dietary antioxidants and dementia in a population-based case-control study among older people in South Germany. J. Alzheimer’s Dis. 2012, 31, 717–724. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, F.S.; Villar, A.; Mulà, A.; Zangara, A.; Risco, E.; Smidt, C.R.; Hontecillas, R.; Leber, A.; Bassaganya-Riera, J. Abscisic acid standardized fig (Ficus carica) extracts ameliorate postprandial glycemic and insulinemic responses in healthy adults. Nutrients 2019, 11, 1757. [Google Scholar] [CrossRef] [Green Version]
- Bassaganya-Riera, J.; Skoneczka, J.; Kingston, D.G.I.; Krishnan, A.; Misyak, S.; Guri, A.; Pereira, A.; Carter, A.; Minorsky, P.; Tumarkin, R.; et al. Mechanisms of action and medicinal applications of abscisic acid. Curr. Med. Chem. 2010, 17, 467–478. [Google Scholar] [CrossRef]
- Bi, B.; Tang, J.; Han, S.; Guo, J.; Miao, Y. Sinapic acid or its derivatives interfere with abscisic acid homeostasis during Arabidopsis thaliana seed germination. BMC Plant Biol. 2017, 17, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Li, H.-H.; Hao, R.-L.; Wu, S.-S.; Guo, P.-C.; Chen, C.-J.; Pan, L.-P.; Ni, H. Occurrence, function and potential medicinal applications of the phytohormone abscisic acid in animals and humans. Biochem. Pharmacol. 2011, 82, 701–712. [Google Scholar] [CrossRef]
- Sánchez-Sarasúa, S.; Moustafa, S.; García-Avilés, Á.; López-Climent, M.F.; Gómez-Cadenas, A.; Olucha-Bordonau, F.E.; Sánchez-Pérez, A.M. The effect of abscisic acid chronic treatment on neuroinflammatory markers and memory in a rat model of high-fat diet induced neuroinflammation. Nutr. Metab. 2016, 13, 73. [Google Scholar] [CrossRef] [Green Version]
- Ribes-Navarro, A.; Atef, M.; Sánchez-Sarasúa, S.; Beltrán-Bretones, M.T.; Olucha-Bordonau, F.; Sánchez-Pérez, A.M. Abscisic acid supplementation rescues high fat diet-induced alterations in hippocampal inflammation and IRSs expression. Mol. Neurobiol. 2019, 56, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Fernández, V.; Mañas-Ojeda, A.; Pacheco-Herrero, M.; Castro-Salazar, E.; Ros-Bernal, F.; Sánchez-Pérez, A.M. Early intervention with ABA prevents neuroinflammation and memory impairment in a triple transgenic mice model of Alzheimer’s disease. Behav. Brain Res. 2019, 374, 112106. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Kumar, U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef] [Green Version]
- Watt, G.; Karl, T. In vivo evidence for therapeutic properties of cannabidiol (CBD) for Alzheimer’s disease. Front. Pharmacol. 2017, 8, 20. [Google Scholar] [CrossRef] [Green Version]
- Murataeva, N.; Straiker, A.; Mackie, K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br. J. Pharmacol. 2014, 171, 1379–1391. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G. Receptors and channels targeted by synthetic cannabinoid receptor agonists and antagonists. Curr. Med. Chem. 2010, 17, 1360–1381. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Anderson, H.D. Cannabinoid signaling in health and disease. Can. J. Physiol. Pharmacol. 2017, 95, 311–327. [Google Scholar] [CrossRef]
- Ahmed, A.I.A.; Elsen, G.A.H.V.D.; Colbers, A.; Kramers, C.; Burger, D.M.; Van Der Marck, M.A.; Rikkert, M.G.M.O. Safety, pharmacodynamics, and pharmacokinetics of multiple oral doses of delta-9-tetrahydrocannabinol in older persons with dementia. Psychopharmacology 2015, 232, 2587–2595. [Google Scholar] [CrossRef] [Green Version]
- Cassano, T.; Calcagnini, S.; Pace, L.; De Marco, F.; Romano, A.; Gaetani, S. Cannabinoid receptor 2 signaling in neurodegenerative disorders: From pathogenesis to a promising therapeutic target. Front. Neurosci. 2017, 11, 30. [Google Scholar] [CrossRef] [Green Version]
- Kendall, D.A.; Yudowski, G. Cannabinoid receptors in the central nervous system: Their signaling and roles in disease. Front. Cell. Neurosci. 2017, 10, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulder, J.; Zilberter, M.; Pasquaré, S.J.; Alpár, A.; Schulte, G.; Ferreira, S.G.; Köfalvi, A.; Martín-Moreno, A.M.; Keimpema, E.; Tanila, H.; et al. Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain 2011, 134, 1041–1060. [Google Scholar] [CrossRef] [PubMed]
- Solas, M.; Francis, P.T.; Franco, R.; Ramirez, M.J. CB2 receptor and amyloid pathology in frontal cortex of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Tolón, R.M.; Núñez, E.; Pazos, M.R.; Benito, C.; Castillo, A.I.; Martínez-Orgado, J.A.; Romero, J. The activation of cannabinoid CB2 receptors stimulates in situ and in vitro beta-amyloid removal by human macrophages. Brain Res. 2009, 1283, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, A.E.; Marchalant, Y. Potential therapeutical contributions of the endocannabinoid system towards aging and Alzheimer’s disease. Aging Dis. 2015, 6, 400–405. [Google Scholar] [CrossRef] [Green Version]
- Aso, E.; Juvés, S.; Maldonado, R.; Ferrer, I. CB2 Cannabinoid receptor agonist ameliorates Alzheimer-like phenotype in AβPP/PS1 mice. J. Alzheimer’s Dis. 2013, 35, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Pihlaja, R.; Takkinen, J.; Eskola, O.; Vasara, J.; López-Picón, F.R.; Haaparanta-Solin, M.; Rinne, J.O. Monoacylglycerol lipase inhibitor JZL184 reduces neuroinflammatory response in APdE9 mice and in adult mouse glial cells. J. Neuroinflamm. 2015, 12, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Janefjord, E.; Mååg, J.L.V.; Harvey, B.S.; Smid, S.D. Cannabinoid effects on β amyloid fibril and aggregate formation, neuronal and microglial-activated neurotoxicity in vitro. Cell. Mol. Neurobiol. 2013, 34, 31–42. [Google Scholar] [CrossRef]
- Scuderi, C.; Steardo, L.; Esposito, G. Cannabidiol promotes amyloid precursor protein ubiquitination and reduction of beta amyloid expression in SHSY5Y APP + cells through PPARγ involvement. Phytother. Res. 2014, 28, 1007–1013. [Google Scholar] [CrossRef]
- Cassano, T.; Villani, R.; Pace, L.; Carbone, A.; Bukke, V.N.; Orkisz, S.; Avolio, C.; Serviddio, G. From cannabis sativa to cannabidiol: Promising therapeutic candidate for the treatment of neurodegenerative diseases. Front. Pharmacol. 2020, 11, 124. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.; Spiro, A.S.; Jenner, A.M.; Garner, B.; Karl, T. Long-term cannabidiol treatment prevents the development of social recognition memory deficits in Alzheimer’s disease transgenic mice. J. Alzheimer’s Dis. 2014, 42, 1383–1396. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.J.; Tobin, A.B. Crosstalk between the M1 muscarinic acetylcholine receptor and the endocannabinoid system: A relevance for Alzheimer’s disease? Cell. Signal. 2020, 70, 109545. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, S.M. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.; Gasbarrini, A.; Mele, M.C. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis in health and disease. Gastroenterol. Clin. N. Am. 2017, 46, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-X.; Wang, Y.-P. Gut Microbiota-brain Axis. Chin. Med. J. 2016, 129, 2373–2380. [Google Scholar] [CrossRef]
- Mayer, E.A.; Tillisch, K.; Gupta, A. Gut/brain axis and the microbiota. J. Clin. Investig. 2015, 125, 926–938. [Google Scholar] [CrossRef]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar]
- Strandwitz, P. Neurotransmitter modulation by the gut microbiota. Brain Res. 2018, 1693, 128–133. [Google Scholar] [CrossRef]
- Liu, S.; Gao, J.; Zhu, M.; Liu, K.; Zhang, H.-L. Gut microbiota and dysbiosis in Alzheimer’s disease: Implications for pathogenesis and treatment. Mol. Neurobiol. 2020, 57, 5026–5043. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.M.; Surette, M.G.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Genet. 2012, 10, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Mazmanian, S.K. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe 2015, 17, 565–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalski, K.; Mulak, A. Brain-gut-microbiota axis in Alzheimer’s disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Goyal, D.; Ali, S.A.; Singh, R.K. Emerging role of gut microbiota in modulation of neuroinflammation and neurodegeneration with emphasis on Alzheimer’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 110112. [Google Scholar] [CrossRef]
- Angelucci, F.; Cechova, K.; Amlerova, J.; Hort, J. Antibiotics, gut microbiota, and Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 1–10. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Schwartz, K.; Boles, B.R. Microbial amyloids-functions and interactions within the host. Curr. Opin. Microbiol. 2013, 16, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Sochocka, M.; Donskow-Łysoniewska, K.; Diniz, B.S.; Kurpas, D.; Brzozowska, E.; Leszek, J. The gut microbiome alterations and inflammation-driven pathogenesis of Alzheimer’s disease-A critical review. Mol. Neurobiol. 2019, 56, 1841–1851. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Zhao, S.; Zhu, Y.; Fan, Z.; Wang, J.; Zhang, B.; Chen, Y. Microbiota-gut-brain axis and toll-like receptors in Alzheimer’s disease. Comput. Struct. Biotechnol. J. 2019, 17, 1309–1317. [Google Scholar] [CrossRef]
- Gąsiorowski, K.; Brokos, B.; Echeverria, V.; Barreto, G.E.; Leszek, J. RAGE-TLR crosstalk sustains chronic inflammation in neurodegeneration. Mol. Neurobiol. 2018, 55, 1463–1476. [Google Scholar] [CrossRef] [PubMed]
- Cerovic, M.; Forloni, G.; Balducci, C. Neuroinflammation and the gut microbiota: Possible alternative therapeutic targets to counteract Alzheimer’s disease? Front. Aging Neurosci. 2019, 11, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, X.; Stamova, B.; Jin, L.-W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Wang, Q.; Zheng, M.; Hao, S.; Lum, J.S.; Chen, X.; Huang, X.-F.; Yu, Y.; Zheng, K. Supplement of microbiota-accessible carbohydrates prevents neuroinflammation and cognitive decline by improving the gut microbiota-brain axis in diet-induced obese mice. J. Neuroinflamm. 2020, 17, 1–21. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, F.; Pellegrini, N.; Vannini, L.; Jeffery, I.B.; La Storia, A.; Laghi, L.; Serrazanetti, D.I.; Di Cagno, R.; Ferrocino, I.; Lazzi, C.; et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 2015, 65, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Neth, B.J.; Wang, S.; Craft, S.; Yadav, H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019, 47, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Lapiscina, E.H.; Clavero, P.; Toledo, E.; Estruch, R.; Salas-Salvadó, J.; Julián, B.S.; Sanchez-Tainta, A.; Ros, E.; Valls-Pedret, C.; Martinez-Gonzalez, M. Á Mediterranean diet improves cognition: The PREDIMED-NAVARRA randomised trial. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1318–1325. [Google Scholar] [CrossRef] [Green Version]
- Probiotics: What You Need To Know. National Center for Complementary and Integrative Health (NIH). 2019 August. Available online: https://www.nccih.nih.gov/health/probiotics-what-you-need-to-know#:~%20:%20text=Probiotics%20are%20live%20microorganisms%20that,dietary%20supplements%2C%20and%20beauty%20products (accessed on 17 November 2020).
- Bonfili, L.; Cecarini, V.; Berardi, S.; Scarpona, S.; Suchodolski, J.S.; Nasuti, C.; Fiorini, D.; Boarelli, M.C.; Rossi, G.; Eleuteri, A.M. Microbiota modulation counteracts Alzheimer’s disease progression influencing neuronal proteolysis and gut hormones plasma levels. Sci. Rep. 2017, 7, 1–21. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Sugahara, H.; Shimada, K.; Mitsuyama, E.; Kuhara, T.; Yasuoka, A.; Kondo, T.; Abe, K.; Xiao, J.-Z. Therapeutic potential of Bifidobacterium breve strain A1 for preventing cognitive impairment in Alzheimer’s disease. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kuhara, T.; Oki, M.; Xiao, J.-Z. Effects of Bifidobacterium breve A1 on the cognitive function of older adults with memory complaints: A randomised, double-blind, placebo-controlled trial. Benef. Microbes 2019, 10, 511–520. [Google Scholar] [CrossRef]
- Chen, D.-L.; Yang, X.; Yang, J.; Lai, G.; Yong, T.; Tang, X.; Shuai, O.; Zhou, G.; Xie, Y.; Wu, Q. Prebiotic effect of Fructooligosaccharides from Morinda officinalis on Alzheimer’s disease in rodent models by targeting the microbiota-gut-brain axis. Front. Aging Neurosci. 2017, 9, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Liu, S.; Ling, Z.; Wang, F.; Ling, Y.; Gong, T.; Fang, N.; Ye, S.; Si, J.; Liu, J. Fructooligosaccharides ameliorating cognitive deficits and neurodegeneration in APP/PS1 transgenic mice through modulating gut microbiota. J. Agric. Food Chem. 2019, 67, 3006–3017. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Yanckello, L.M.; Chlipala, G.; Hammond, T.C.; McCulloch, S.D.; Parikh, I.; Sun, S.; Morganti, J.M.; Green, S.; Lin, A.-L. Dietary inulin alters the gut microbiome, enhances systemic metabolism and reduces neuroinflammation in an APOE4 mouse model. PLoS ONE 2019, 14, e0221828. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xu, J.; Ling, Y.; Wang, F.; Gong, T.; Yang, C.; Ye, S.; Ye, K.; Wei, D.; Song, Z.; et al. Fecal microbiota transplantation alleviated Alzheimer’s disease-like pathogenesis in APP/PS1 transgenic mice. Transl. Psychiatry 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.-S.; Kim, Y.; Choi, H.; Kim, W.; Park, S.; Lee, D.; Kim, D.K.; Kim, H.J.; Choi, H.; Hyun, D.-W.; et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut 2020, 69, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Moreira, S.C.; Jansen, A.K.; Silva, F.M. Dietary interventions and cognition of Alzheimer’s disease patients: A systematic review of randomized controlled trial. Dement. Neuropsychol. 2020, 14, 258–282. [Google Scholar] [CrossRef]
- Ngandu, T.; Lehtisalo, J.; Solomon, A.; Levälahti, E.; Ahtiluoto, S.; Antikainen, R.; Bäckman, L.; Hänninen, T.; Jula, A.; Laatikainen, T.; et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): A randomised controlled trial. Lancet 2015, 385, 2255–2263. [Google Scholar] [CrossRef]
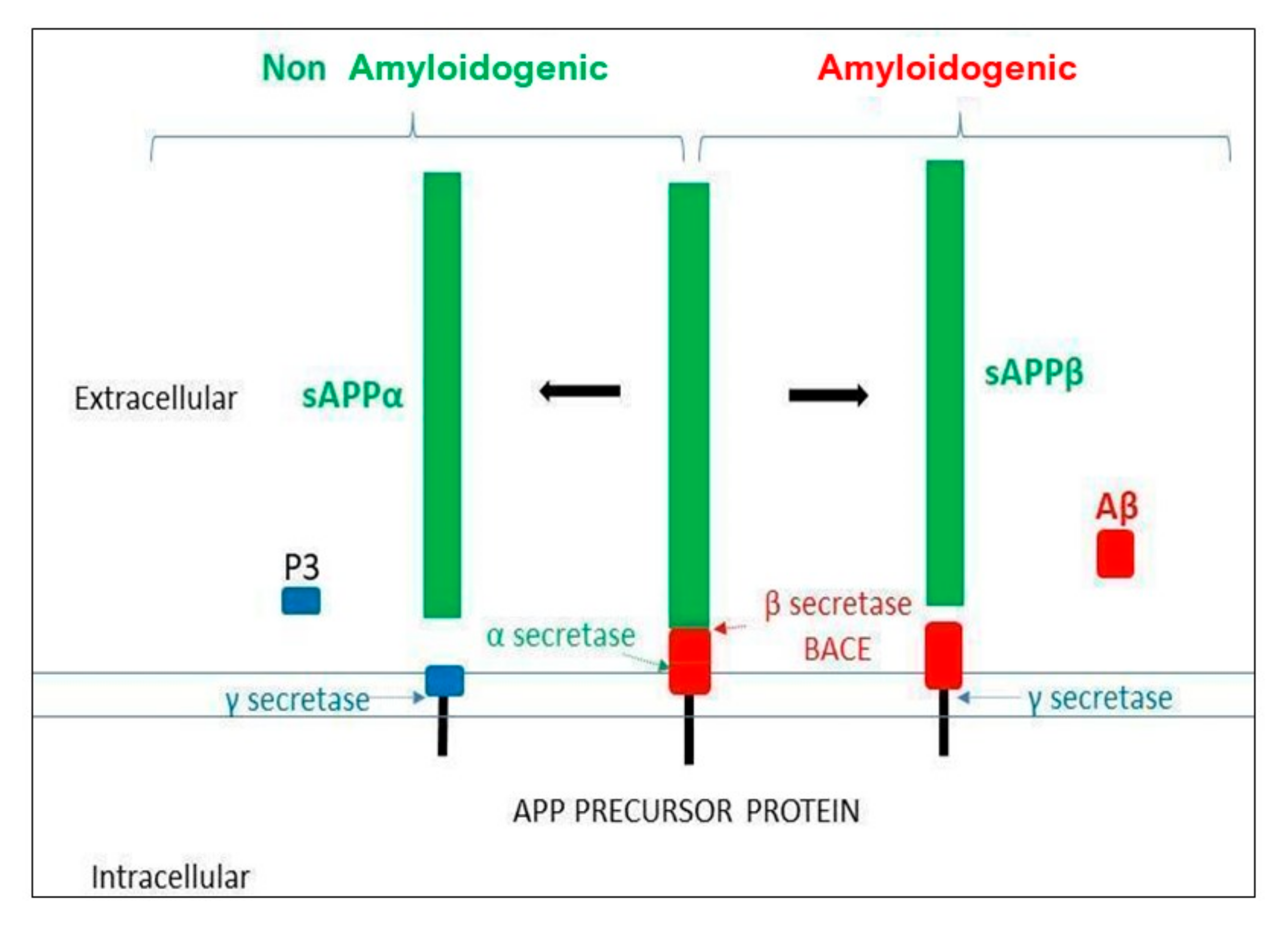
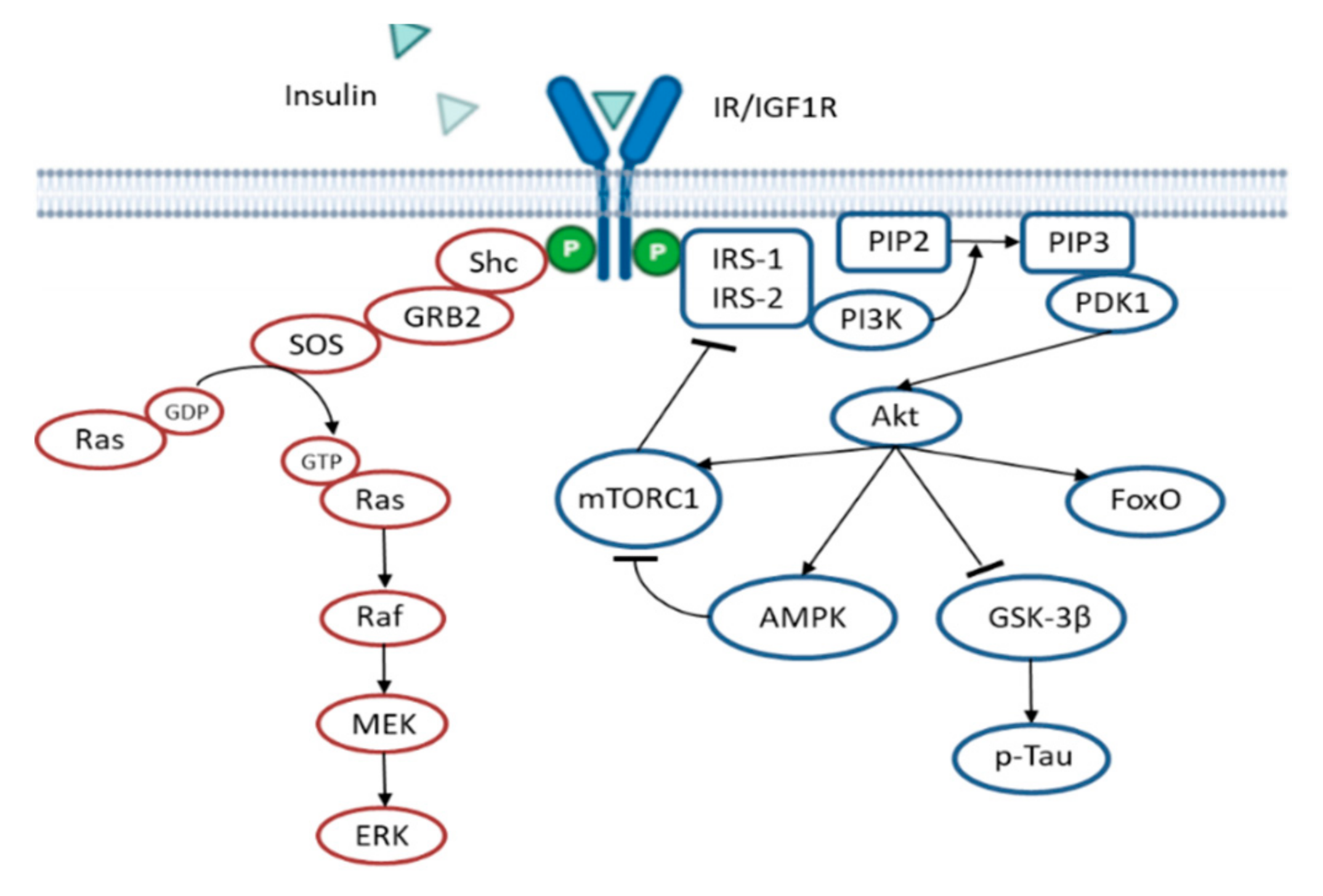
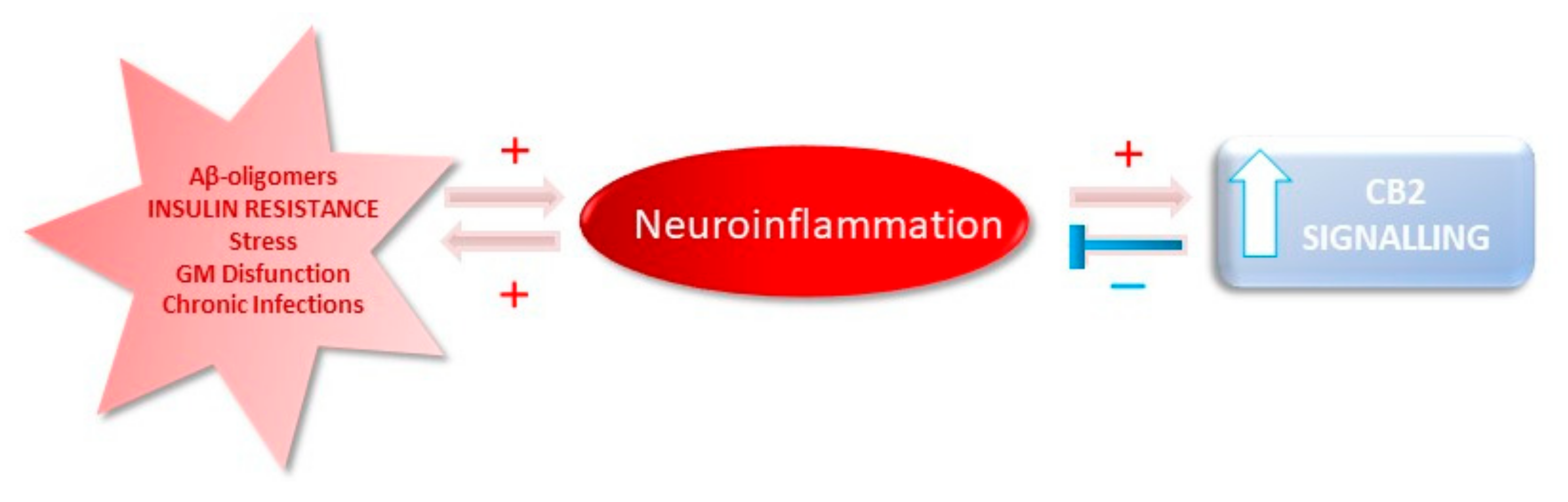

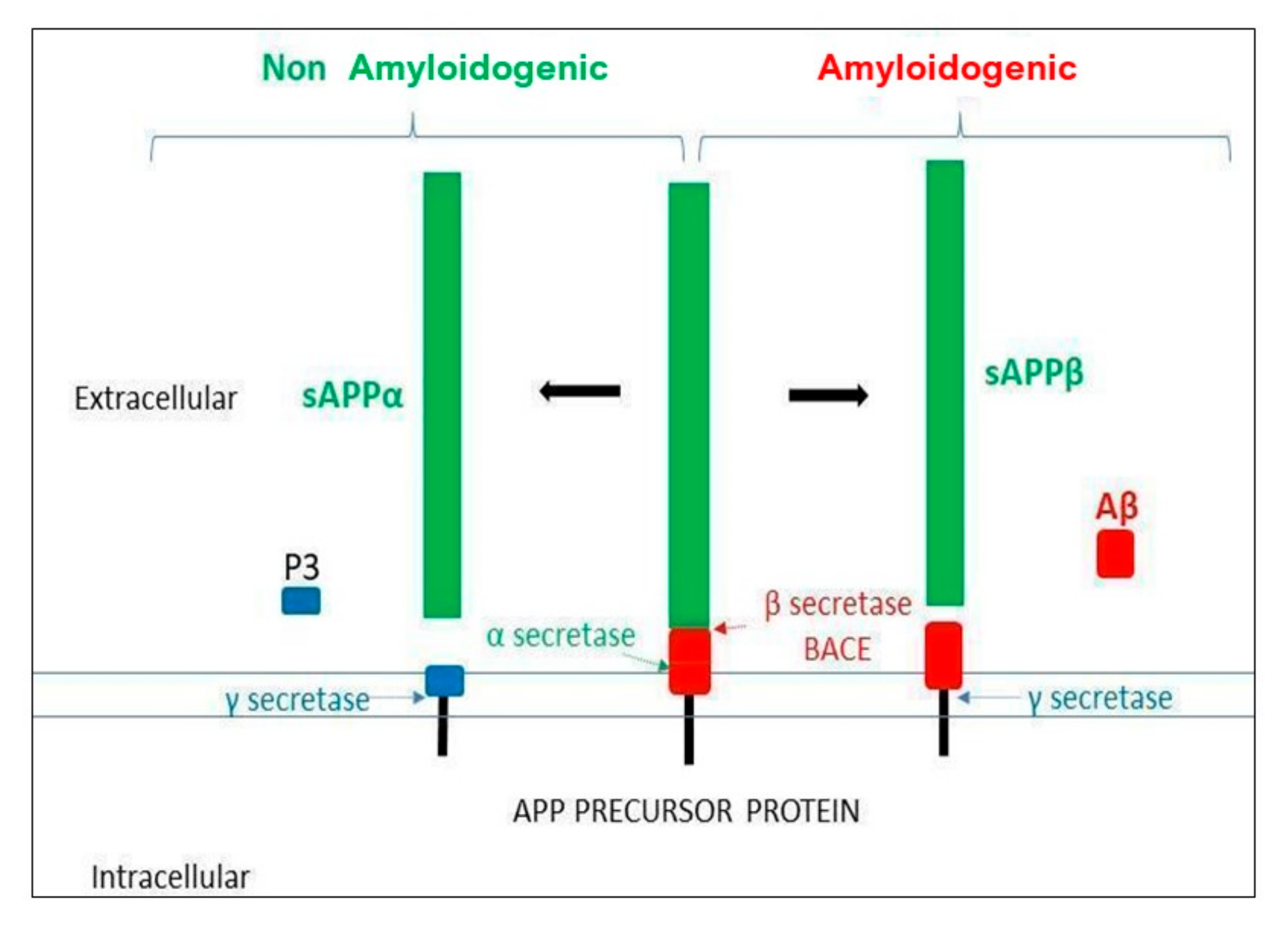{kind=link}
{kind=link}
{kind=link}
| Compound (Dose). | Source | Patients (Years Old) | Study Design | Inflammatory/AD Biomarkers | Cognitive Effect | Other | Citation Year |
|---|---|---|---|---|---|---|---|
| Naproxen (220 mg/twice daily) | Derived from propionic acid | 195 (>55) | 2 years | NT | = APS progression | - | [34] 2020 |
| Celecoxib (200 mg/twice daily) | Derived from propionic acid | 2356 (70–85) | 3 years | NT | = ADAPT score | - | [35] 2015 |
| Etanercept (50 mg/once weekly subcutaneous) | U | 41 (70–74) | 24 weeks | = TNF-α levels = IL-6 levels = IL-10 levels = IL-12p70 levels = CRP levels | = ADAS-cog score = BADLS score = CGI-I = Cornell Scale score = MMSE score = NPI score | - | [36] 2015 |
| Compound (Dose) | Source | Patients (Years Old) | Study Design | Inflammatory/AD Biomarkers | Cognitive Effect | Other | Citation Year |
|---|---|---|---|---|---|---|---|
| Metformin (200 mg/day) | Galega officinalis plant | 20 (55–80) | 8 weeks | = Aβ42 levels | ↑ Learning and memory (CANTAB PAL scale) | Crosses the BBB | [46] 2017 |
| = phospho-TAU levels | ↑ Attention (DMS Percent Correct Simultaneous) | ||||||
| = total TAU levels | |||||||
| Lactoferrin (250 mg/day) | Milk | 50 (>65) | 3 months | ↑ IL-10 levels | [50] 2019 | ||
| ↑ GSH levels | ↑ Ach levels | ||||||
| ↓ IL-6 levels | ↑ 5-HT levels | ||||||
| ↓ Aβ42 levels | ↑ AKT levels | ||||||
| ↓ Caspase-3 levels | ↑ MMSE score | ↑ phospho- AKT(S473) levels | |||||
| ↓ Cholesterol levels | ↑ ADAS-Cog11 score | ↑ PI3K levels | |||||
| ↓ HSP90 levels | |||||||
| ↓ TAU pTAU(181) | ↓ PTEN levels | ||||||
| ↓ NO levels | ↓ MAPK1 levels | ||||||
| ↓ MDA levels | |||||||
| Vitamin B12 (25 μg/day) + Folic acid (800 μg/day) | Vitamin B12: animal products Folic acid: broccoli, peas, chickpeas, leafy green vegetables | 240 (>65) | 6 months | ↓ IL-6 levels | - | [54] 2019 | |
| ↓ TNF-α levels | ↑ Full Scale IQ (FSIQ) score | ||||||
| ↓ MCP-1 levels | ↑ Verbal intelligence quotient (VIQ) score | ||||||
| ↓ Homocysteine levels | ↑ Information and Digit Span | ||||||
| Vitamin B12 + Risperidone and Quetiapine (atypical antipsychotic drugs) | Risperidone: and Quetiapine are synthetic | 102 (>65) | U | ↑ TGF-β score ↓ IL-8 levels ↓ TNF-α levels ↓ CD68 levels | NT | ↓ Pain (VAS scale) | [55,56] 2018 |
| Nilvadipine (8 mg/day) | Pyridine (crude coal tar) | 511 (>50) | 18 months | NT | = ADAS-Cog 12 = CDR-sb = DAD | - | [62] 2018 |
| Simvastatin 80 mg/day | Statins (Fungus Aspergillus terreus) | 80 (>50) | 18 months | ↓ IL-6 levels | - | [63] 2017 | |
| ↓ IL-1β levels | ↑ ADAS-Cog score | ||||||
| ↓ ACT levels | ↑ MMSE score | ||||||
| ↓ TNF-α levels | ↑ Dependence Scale score | ||||||
| ↓ APP levels | ↑ ADCS-ADL score | ||||||
| ↓ BACE1 levels | ↑ NPI score | ||||||
| ↓ Aβ levels | |||||||
| Simvastatin 40 mg/day | 406 (>50) | 18 months | ↓ CRP levels | = ADAS-Cog score | ↑ HDL levels | [64] 2011 | |
| = MMSE score | |||||||
| = Dependence Scale score | ↓ Total cholesterol levels | ||||||
| = ADCS-ADL score | |||||||
| = NPI score | ↓ LDL levels | ||||||
| Atorvastatin 40 mg/day | Statins | 178 (45–60) | 18 months | ↓ IL-1β levels ↓ IL-6 levels ↓ TNF-α levels ↓ CRP levels ↓ MCP-1 levels | NT | ↓ Lipid levels | [65] 2016 |
| Atorvastatin 80 mg/day | 640 (50–90) | 18 months | NT | = ADAS-Cog score | [66] 2010 | ||
| = ADCS-CGIC score | ↓ Total cholesterol levels | ||||||
| = MMSE score | |||||||
| = CDR-SB score | ↓ LDL-C levels | ||||||
| = ADFACS score | ↓ Triglycerides levels | ||||||
| = NPI score | |||||||
| Tetrahydrocannabinol (4.5 mg/day) | Cannabis plant | 50 (78–79) | 3 weeks | NT | = NPI score = Cohen-Mansfield Agitation Inventory = Quality of Life-Alzheimer’s Disease = Barthel Index score = PAL WMS-R score | [67] 2015 |
| Compound (Dose) | Source/Study | Patients (Years Old) | Study Design | Inflammatory/AD Biomarkers | Cognitive Effect | Other | Citation | |
|---|---|---|---|---|---|---|---|---|
| Curcumin | (1.5–4 g/day) | Turmeric | 34 (>50) | 6 months | ↓ Aβ aggregation | = MMSE score | - | [72] 2008 |
| (1500 mg/day) | 160 (40–90) | 12 months | NT | ↑ MoCA score = CCR Test score = DASS score = AVL Test score = COWA Test score = WAIS-R score = CogState score | - | [73] 2016 | ||
| Resveratrol (500 mg/day) | Red grapes, peanuts and other plant species | 119 (>49) | 52 weeks | ↑ MDC levels ↑ IL-4 levels ↑ FGF-2 levels ↑ MMP10 levels ↓ MMP9 levels ↓ IL-12 levels ↓ RANTES levels | ↑ ADCS-ADL score | - | [76,77] 2015 | |
| PUFA 800 mg docosahexaenoic acid 225 mg eicosapentaenoic acid/day. | Multimodal The MAPT study | 1680 Non demented (>70) | 3 years | NT | = MMSE score = DSS Test score = CN Test | Safe | [91] 2017 | |
| Heat processed Ginseng (4.5 g/day) | 40 (U) | 6 months | NT | ↑ ADAS-Cog score ↑ MMSE score | - | [94] 2012 | ||
| Abscisic acid (40/80 µg) | Fig fruit extract | 10 Non-demented (18–45) | 4 non-consecutive sessions | NT | NT | Safe ↓Postprandial glycemic responses. | [97] 2019 | |
| Compound (Dose) | Patients (Years Old) | Study Design | Inflammatory/AD Biomarkers | Cognitive Effect | Other | Citation Year |
|---|---|---|---|---|---|---|
| Mediterranean-Ketogenic diet (MMKD) (<10% carbohydrate, 60–65% fat, and 30–35% protein) American Heart Association diet (AHAD) (55–65% carbohydrate, 15–20% fat, and 20–30% protein) | 17 (64.6 ± 6.4) | 6 weeks | ↓ Aß42 ↓ Tau-p181 | NT | ↑ Enterobacteriaceae, Akkermansia, Slackia, Christensenellaceae and Erysipelotriaceae ↑ Propionate and butyrate ↓ Bifidobacterium and Lachnobacterium ↓ Fecal acetate and lactate | [146] 2019 |
| = Aß-42 = Tau-p181 | NT | ↑ Mollicutes ↑ Acetate and propionate ↓ Butyrate | ||||
| Mediterranean diet (extra-virgin olive oil 1 l/week) or 30 g/day nuts | 522 (55–80) | 6.5 years | NT | ↑ MMSE score ↑ CDT score | - | [147] 2013 |
| Bifidobacterium breve A1 (daily) | 117 (50–80) | 12 weeks | NT | ↑ RBANS score ↑ MMSE score | Safe | [151] 2019 |
| Compound (Dose) and Treatment | Source | Patients (Years Old) | Study Design | Inflammatory/AD Biomarkers | Cognitive Effect | Other | Citation |
|---|---|---|---|---|---|---|---|
| Nutritional intervention (10–20% of daily energy (E%) from proteins, 25–35E% from fat, 45–55 E% from carbohydrates, 25–35 g/day dietary fiber) + Physical exercise training + Cognitive training (executive processes, working memory, episodic memory and mental speed) + Social activities | Multimodal The FINGER study | 1260 (60–77) | 2 years | NT | ↑ NTB score ↑ NTB Executive functioning domain score ↑ NTB Processing speed domain score = NTB Memory domain score | - | [158] 2015 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Sarasúa, S.; Fernández-Pérez, I.; Espinosa-Fernández, V.; Sánchez-Pérez, A.M.; Ledesma, J.C. Can We Treat Neuroinflammation in Alzheimer’s Disease? Int. J. Mol. Sci. 2020, 21, 8751. https://doi.org/10.3390/ijms21228751
Sánchez-Sarasúa S, Fernández-Pérez I, Espinosa-Fernández V, Sánchez-Pérez AM, Ledesma JC. Can We Treat Neuroinflammation in Alzheimer’s Disease? International Journal of Molecular Sciences. 2020; 21(22):8751. https://doi.org/10.3390/ijms21228751
Chicago/Turabian StyleSánchez-Sarasúa, Sandra, Iván Fernández-Pérez, Verónica Espinosa-Fernández, Ana María Sánchez-Pérez, and Juan Carlos Ledesma. 2020. "Can We Treat Neuroinflammation in Alzheimer’s Disease?" International Journal of Molecular Sciences 21, no. 22: 8751. https://doi.org/10.3390/ijms21228751
APA StyleSánchez-Sarasúa, S., Fernández-Pérez, I., Espinosa-Fernández, V., Sánchez-Pérez, A. M., & Ledesma, J. C. (2020). Can We Treat Neuroinflammation in Alzheimer’s Disease? International Journal of Molecular Sciences, 21(22), 8751. https://doi.org/10.3390/ijms21228751