Candidate Gene Discovery in Hereditary Colorectal Cancer and Polyposis Syndromes–Considerations for Future Studies

 , , and
, , and
Abstract
1. Introduction
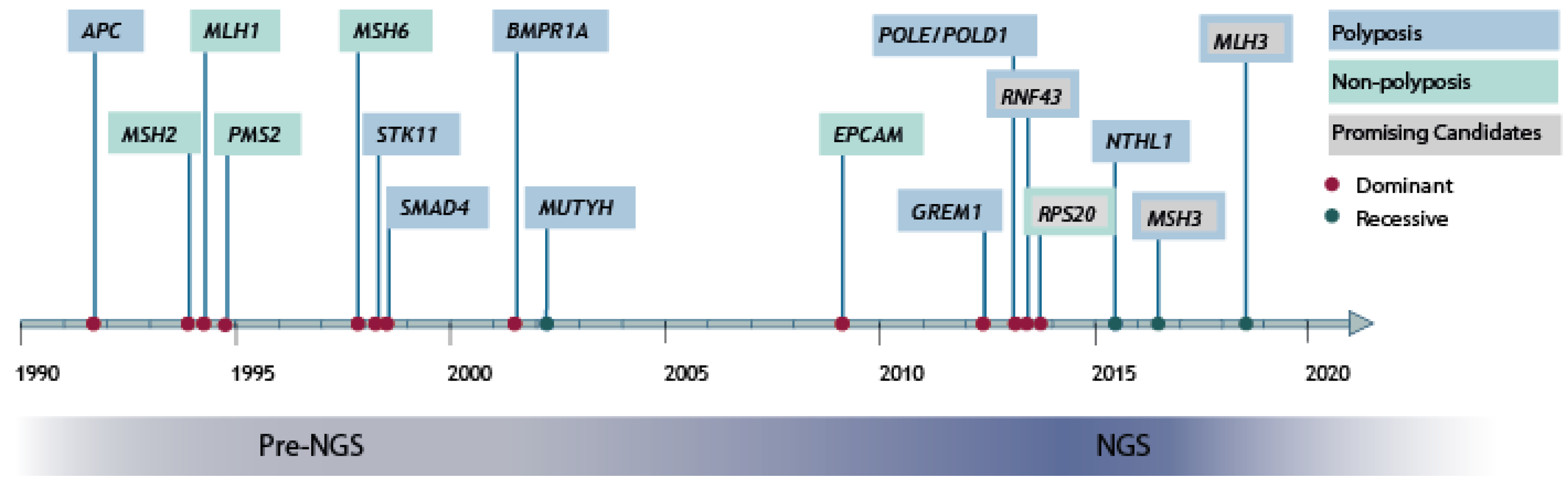
2. High-Penetrant Risk Genes Discovery in hCRC and Polyposis
3. Strategies for Identification of Rare High-Penetrant Risk Genes
3.1. Discovery Cohort
3.2. Variant Prioritization
3.2.1. Locus Prioritization
3.2.2. Allele Frequency Cut-Offs
3.2.3. In Silico Pathogenicity
3.2.4. Co-Segregation
3.3. Variant Validation
3.3.1. Molecular Tumor Analysis
3.3.2. Functional Characterization of the Variant
3.3.3. Case-Control Validation
4. Missing Heritability Explained by Known or Common Risk Genes
4.1. Identification of Variants in Known hCRC and Polyposis Risk Genes by Whole-Genome Sequencing
4.2. Mosaic and De Novo Variant in Known hCRC and Polyposis Syndrome Genes
4.3. Polygenic Risk Scores
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AFAP | Attenuated familial adenomatous polyposis syndrome |
| BER | Base-excision repair pathway |
| CRC | Colorectal cancer |
| FAP | Familial adenomatous polyposis |
| FH | Family history |
| hCRC | Hereditary colorectal cancer |
| MAF | Minor allele frequency |
| MAP | MutyH-associated polyposis |
| MMR | Mismatch repair |
| NATS | NTHL1-associated tumor syndrome |
| NGS | Next-generation sequencing |
| PPAP | Polymerase proofreading associated polyposis |
| PRS | Polygenic risk score |
| WES | Whole exome sequencing |
| WGS | Whole genome sequencing |
References
- Cancer Today; International Agency for Research on Cancer: Lyon, France, 2020.
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer—Analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Czene, K.; Lichtenstein, P.; Hemminki, K. Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish Family-Cancer Database. Int. J. Cancer 2002, 99, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Mucci, L.A.; Hjelmborg, J.B.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike, T.; Graff, R.E.; Holst, K.; Moller, S.; Unger, R.H.; et al. Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 2016, 315, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Graff, R.E.; Moller, S.; Passarelli, M.N.; Witte, J.S.; Skytthe, A.; Christensen, K.; Tan, Q.; Adami, H.O.; Czene, K.; Harris, J.R.; et al. Familial Risk and Heritability of Colorectal Cancer in the Nordic Twin Study of Cancer. Clin. Gastroenterol. Hepatol. 2017, 15, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.L.; Ukaegbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer Susceptibility Gene Mutations in Individuals With Colorectal Cancer. J. Clin. Oncol. 2017, 35, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [CrossRef]
- Huyghe, J.R.; Bien, S.A.; Harrison, T.A.; Kang, H.M.; Chen, S.; Schmit, S.L.; Conti, D.V.; Qu, C.; Jeon, J.; Edlund, C.K.; et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat. Genet. 2019, 51, 76–87. [Google Scholar] [CrossRef]
- Weigl, K.; Chang-Claude, J.; Hsu, L.; Hoffmeister, M.; Brenner, H. Establishing a valid approach for estimating familial risk of cancer explained by common genetic variants. Int. J. Cancer 2019. [Google Scholar] [CrossRef]
- Schmit, S.L.; Edlund, C.K.; Schumacher, F.R.; Gong, J.; Harrison, T.A.; Huyghe, J.R.; Qu, C.; Melas, M.; Van Den Berg, D.J.; Wang, H.; et al. Novel Common Genetic Susceptibility Loci for Colorectal Cancer. J. Natl. Cancer Inst. 2019, 111, 146–157. [Google Scholar] [CrossRef]
- Law, P.J.; Timofeeva, M.; Fernandez-Rozadilla, C.; Broderick, P.; Studd, J.; Fernandez-Tajes, J.; Farrington, S.; Svinti, V.; Palles, C.; Orlando, G.; et al. Association analyses identify 31 new risk loci for colorectal cancer susceptibility. Nat. Commun. 2019, 10, 2154. [Google Scholar] [CrossRef]
- De Voer, R.M.; Hahn, M.M.; Weren, R.D.; Mensenkamp, A.R.; Gilissen, C.; van Zelst-Stams, W.A.; Spruijt, L.; Kets, C.M.; Zhang, J.; Venselaar, H.; et al. Identification of Novel Candidate Genes for Early-Onset Colorectal Cancer Susceptibility. PLoS Genet. 2016, 12, e1005880. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.M.; de Voer, R.M.; Hoogerbrugge, N.; Ligtenberg, M.J.; Kuiper, R.P.; van Kessel, A.G. The genetic heterogeneity of colorectal cancer predisposition—Guidelines for gene discovery. Cell. Oncol. 2016, 39, 491–510. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.A.; Morreau, H.; de Miranda, N.; van Wezel, T. The missing heritability of familial colorectal cancer. Mutagenesis 2019. [Google Scholar] [CrossRef]
- Risch, N. The genetic epidemiology of cancer: Interpreting family and twin studies and their implications for molecular genetic approaches. Cancer Epidemiol. Biomark. Prev. 2001, 10, 733–741. [Google Scholar]
- Bodmer, W.F.; Bailey, C.J.; Bodmer, J.; Bussey, H.J.; Ellis, A.; Gorman, P.; Lucibello, F.C.; Murday, V.A.; Rider, S.H.; Scambler, P.; et al. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature 1987, 328, 614–616. [Google Scholar] [CrossRef]
- Solomon, E.; Voss, R.; Hall, V.; Bodmer, W.F.; Jass, J.R.; Jeffreys, A.J.; Lucibello, F.C.; Patel, I.; Rider, S.H. Chromosome 5 allele loss in human colorectal carcinomas. Nature 1987, 328, 616–619. [Google Scholar] [CrossRef]
- Leppert, M.; Burt, R.; Hughes, J.P.; Samowitz, W.; Nakamura, Y.; Woodward, S.; Gardner, E.; Lalouel, J.M.; White, R. Genetic analysis of an inherited predisposition to colon cancer in a family with a variable number of adenomatous polyps. N. Engl. J. Med. 1990, 322, 904–908. [Google Scholar] [CrossRef]
- Nishisho, I.; Nakamura, Y.; Miyoshi, Y.; Miki, Y.; Ando, H.; Horii, A.; Koyama, K.; Utsunomiya, J.; Baba, S.; Hedge, P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991, 253, 665–669. [Google Scholar] [CrossRef]
- Peltomaki, P.; Aaltonen, L.A.; Sistonen, P.; Pylkkanen, L.; Mecklin, J.P.; Jarvinen, H.; Green, J.S.; Jass, J.R.; Weber, J.L.; Leach, F.S.; et al. Genetic mapping of a locus predisposing to human colorectal cancer. Science 1993, 260, 810–812. [Google Scholar] [CrossRef]
- Lindblom, A.; Tannergard, P.; Werelius, B.; Nordenskjold, M. Genetic mapping of a second locus predisposing to hereditary non-polyposis colon cancer. Nat. Genet. 1993, 5, 279–282. [Google Scholar] [CrossRef]
- Leach, F.S.; Nicolaides, N.C.; Papadopoulos, N.; Liu, B.; Jen, J.; Parsons, R.; Peltomaki, P.; Sistonen, P.; Aaltonen, L.A.; Nystrom-Lahti, M.; et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993, 75, 1215–1225. [Google Scholar] [CrossRef]
- Bronner, C.E.; Baker, S.M.; Morrison, P.T.; Warren, G.; Smith, L.G.; Lescoe, M.K.; Kane, M.; Earabino, C.; Lipford, J.; Lindblom, A.; et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994, 368, 258–261. [Google Scholar] [CrossRef]
- Nicolaides, N.C.; Papadopoulos, N.; Liu, B.; Wei, Y.F.; Carter, K.C.; Ruben, S.M.; Rosen, C.A.; Haseltine, W.A.; Fleischmann, R.D.; Fraser, C.M.; et al. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 1994, 371, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, N.; Nicolaides, N.C.; Liu, B.; Parsons, R.; Lengauer, C.; Palombo, F.; D’Arrigo, A.; Markowitz, S.; Willson, J.K.; Kinzler, K.W.; et al. Mutations of GTBP in genetically unstable cells. Science 1995, 268, 1915–1917. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, M.E.; Papp, J.; Szentirmay, Z.; Otto, S.; Olah, E. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum. Mutat. 2009, 30, 197–203. [Google Scholar] [CrossRef]
- Ligtenberg, M.J.; Kuiper, R.P.; Chan, T.L.; Goossens, M.; Hebeda, K.M.; Voorendt, M.; Lee, T.Y.; Bodmer, D.; Hoenselaar, E.; Hendriks-Cornelissen, S.J.; et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat. Genet. 2009, 41, 112–117. [Google Scholar] [CrossRef]
- Nakamura, Y.; Nishisho, I.; Kinzler, K.W.; Vogelstein, B.; Miyoshi, Y.; Miki, Y.; Ando, H.; Horii, A.; Nagase, H. Mutations of the adenomatous polyposis coli gene in familial polyposis coli patients and sporadic colorectal tumors. Princess Takamatsu Symp. 1991, 22, 285–292. [Google Scholar] [CrossRef]
- Lynch, H.T.; Smyrk, T.C.; Lanspa, S.J.; Lynch, P.M.; Watson, P.; Strayhorn, P.C.; Bronson, E.K.; Lynch, J.F.; Priluck, I.A.; Appelman, H.D. Phenotypic variation in colorectal adenoma/cancer expression in two families. Hereditary flat adenoma syndrome. Cancer 1990, 66, 909–915. [Google Scholar] [CrossRef]
- Knudsen, A.L.; Bisgaard, M.L.; Bulow, S. Attenuated familial adenomatous polyposis (AFAP). A review of the literature. Fam. Cancer 2003, 2, 43–55. [Google Scholar] [CrossRef]
- Spirio, L.; Olschwang, S.; Groden, J.; Robertson, M.; Samowitz, W.; Joslyn, G.; Gelbert, L.; Thliveris, A.; Carlson, M.; Otterud, B.; et al. Alleles of the APC gene: An attenuated form of familial polyposis. Cell 1993, 75, 951–957. [Google Scholar] [CrossRef]
- Al-Tassan, N.; Chmiel, N.H.; Maynard, J.; Fleming, N.; Livingston, A.L.; Williams, G.T.; Hodges, A.K.; Davies, D.R.; David, S.S.; Sampson, J.R.; et al. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat. Genet. 2002, 30, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Franken, P.F.; Reinards, T.H.C.M.; Weiss, M.M.; Wagner, A.; van der Klift, H.; Kloosterman, S.; Houwing-Duistermaat, J.J.; Aalfs, C.M.; Ausems, M.G.E.M.; et al. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP). J. Med. Genet. 2005, 42, e54. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Baudhuin, L.M.; Boardman, L.A.; Steenblock, K.J.; Petersen, G.M.; Halling, K.C.; French, A.J.; Johnson, R.A.; Burgart, L.J.; Rabe, K.; et al. MYH mutations in patients with attenuated and classic polyposis and with young-onset colorectal cancer without polyps. Gastroenterology 2004, 127, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Jenne, D.E.; Reimann, H.; Nezu, J.; Friedel, W.; Loff, S.; Jeschke, R.; Muller, O.; Back, W.; Zimmer, M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat. Genet. 1998, 18, 38–43. [Google Scholar] [CrossRef]
- Howe, J.R.; Roth, S.; Ringold, J.C.; Summers, R.W.; Jarvinen, H.J.; Sistonen, P.; Tomlinson, I.P.; Houlston, R.S.; Bevan, S.; Mitros, F.A.; et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998, 280, 1086–1088. [Google Scholar] [CrossRef]
- Howe, J.R.; Bair, J.L.; Sayed, M.G.; Anderson, M.E.; Mitros, F.A.; Petersen, G.M.; Velculescu, V.E.; Traverso, G.; Vogelstein, B. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat. Genet. 2001, 28, 184–187. [Google Scholar] [CrossRef]
- Zbuk, K.M.; Eng, C. Hamartomatous polyposis syndromes. Nat. Clin. Pract. Gastroenterol. Hepatol. 2007, 4, 492–502. [Google Scholar] [CrossRef]
- Olkinuora, A.; Nieminen, T.T.; Martensson, E.; Rohlin, A.; Ristimaki, A.; Koskenvuo, L.; Lepisto, A.; Swedish Extended Genetic Analysis of Colorectal Neoplasia Study Group; Gebre-Medhin, S.; Nordling, M.; et al. Biallelic germline nonsense variant of MLH3 underlies polyposis predisposition. Genet. Med. 2018. [Google Scholar] [CrossRef]
- Adam, R.; Spier, I.; Zhao, B.; Kloth, M.; Marquez, J.; Hinrichsen, I.; Kirfel, J.; Tafazzoli, A.; Horpaopan, S.; Uhlhaas, S.; et al. Exome Sequencing Identifies Biallelic MSH3 Germline Mutations as a Recessive Subtype of Colorectal Adenomatous Polyposis. Am. J. Hum. Genet. 2016, 99, 337–351. [Google Scholar] [CrossRef]
- Nieminen, T.T.; O’Donohue, M.F.; Wu, Y.; Lohi, H.; Scherer, S.W.; Paterson, A.D.; Ellonen, P.; Abdel-Rahman, W.M.; Valo, S.; Mecklin, J.P.; et al. Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology 2014, 147, 595–598.e5. [Google Scholar] [CrossRef]
- Gala, M.K.; Mizukami, Y.; Le, L.P.; Moriichi, K.; Austin, T.; Yamamoto, M.; Lauwers, G.Y.; Bardeesy, N.; Chung, D.C. Germline mutations in oncogene-induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology 2014, 146, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Weren, R.D.; Ligtenberg, M.J.; Kets, C.M.; de Voer, R.M.; Verwiel, E.T.; Spruijt, L.; van Zelst-Stams, W.A.; Jongmans, M.C.; Gilissen, C.; Hehir-Kwa, J.Y.; et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet. 2015, 47, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Valle, L. Recent Discoveries in the Genetics of Familial Colorectal Cancer and Polyposis. Clin. Gastroenterol. Hepatol. 2017, 15, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Valle, L.; de Voer, R.M.; Goldberg, Y.; Sjursen, W.; Forsti, A.; Ruiz-Ponte, C.; Caldes, T.; Garre, P.; Olsen, M.F.; Nordling, M.; et al. Update on genetic predisposition to colorectal cancer and polyposis. Mol. Aspects Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.G.; Naven, M.; Harris, R.; Colley, J.; West, H.; Li, N.; Liu, Y.; Adams, R.; Maughan, T.S.; Nichols, L.; et al. Exome resequencing identifies potential tumor-suppressor genes that predispose to colorectal cancer. Hum. Mutat. 2013, 34, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- DeRycke, M.S.; Gunawardena, S.R.; Middha, S.; Asmann, Y.W.; Schaid, D.J.; McDonnell, S.K.; Riska, S.M.; Eckloff, B.W.; Cunningham, J.M.; Fridley, B.L.; et al. Identification of novel variants in colorectal cancer families by high-throughput exome sequencing. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1239–1251. [Google Scholar] [CrossRef]
- De Voer, R.M.; Geurts van Kessel, A.; Weren, R.D.; Ligtenberg, M.J.; Smeets, D.; Fu, L.; Vreede, L.; Kamping, E.J.; Verwiel, E.T.; Hahn, M.M.; et al. Germline mutations in the spindle assembly checkpoint genes BUB1 and BUB3 are risk factors for colorectal cancer. Gastroenterology 2013, 145, 544–547. [Google Scholar] [CrossRef]
- Gylfe, A.E.; Katainen, R.; Kondelin, J.; Tanskanen, T.; Cajuso, T.; Hanninen, U.; Taipale, J.; Taipale, M.; Renkonen-Sinisalo, L.; Jarvinen, H.; et al. Eleven candidate susceptibility genes for common familial colorectal cancer. PLoS Genet. 2013, 9, e1003876. [Google Scholar] [CrossRef]
- Rohlin, A.; Zagoras, T.; Nilsson, S.; Lundstam, U.; Wahlstrom, J.; Hulten, L.; Martinsson, T.; Karlsson, G.B.; Nordling, M. A mutation in POLE predisposing to a multi-tumour phenotype. Int. J. Oncol. 2014, 45, 77–81. [Google Scholar] [CrossRef]
- Schulz, E.; Klampfl, P.; Holzapfel, S.; Janecke, A.R.; Ulz, P.; Renner, W.; Kashofer, K.; Nojima, S.; Leitner, A.; Zebisch, A.; et al. Germline variants in the SEMA4A gene predispose to familial colorectal cancer type X. Nat. Commun. 2014, 5, 5191. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Jurado, C.; Vila-Casadesus, M.; Garre, P.; Lozano, J.J.; Pristoupilova, A.; Beltran, S.; Munoz, J.; Ocana, T.; Balaguer, F.; Lopez-Ceron, M.; et al. Whole-exome sequencing identifies rare pathogenic variants in new predisposition genes for familial colorectal cancer. Genet. Med. 2015, 17, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Tanskanen, T.; Gylfe, A.E.; Katainen, R.; Taipale, M.; Renkonen-Sinisalo, L.; Jarvinen, H.; Mecklin, J.P.; Bohm, J.; Kilpivaara, O.; Pitkanen, E.; et al. Systematic search for rare variants in Finnish early-onset colorectal cancer patients. Cancer Genet. 2015, 208, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Peng, B.; Han, Y.; Chen, W.V.; Rother, J.; Tomlinson, G.E.; Boland, C.R.; Chaussabel, D.; Frazier, M.L.; Amos, C.I. Mutations of HNRNPA0 and WIF1 predispose members of a large family to multiple cancers. Fam. Cancer 2015, 14, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Fu, L.; de Voer, R.M.; Hahn, M.M.; Jin, P.; Lv, C.X.; Verwiel, E.T.; Ligtenberg, M.J.; Hoogerbrugge, N.; Kuiper, R.P.; et al. Candidate colorectal cancer predisposing gene variants in Chinese early-onset and familial cases. World J. Gastroenterol. 2015, 21, 4136–4149. [Google Scholar] [CrossRef] [PubMed]
- Segui, N.; Mina, L.B.; Lazaro, C.; Sanz-Pamplona, R.; Pons, T.; Navarro, M.; Bellido, F.; Lopez-Doriga, A.; Valdes-Mas, R.; Pineda, M.; et al. Germline Mutations in FAN1 Cause Hereditary Colorectal Cancer by Impairing DNA Repair. Gastroenterology 2015, 149, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Ngeow, J.; Yu, W.; Yehia, L.; Niazi, F.; Chen, J.; Tang, X.; Heald, B.; Lei, J.; Romigh, T.; Tucker-Kellogg, L.; et al. Exome Sequencing Reveals Germline SMAD9 Mutation That Reduces Phosphatase and Tensin Homolog Expression and Is Associated With Hamartomatous Polyposis and Gastrointestinal Ganglioneuromas. Gastroenterology 2015, 149, 886–889.e5. [Google Scholar] [CrossRef]
- Arora, S.; Yan, H.; Cho, I.; Fan, H.Y.; Luo, B.; Gai, X.; Bodian, D.L.; Vockley, J.G.; Zhou, Y.; Handorf, E.A.; et al. Genetic Variants That Predispose to DNA Double-Strand Breaks in Lymphocytes From a Subset of Patients With Familial Colorectal Carcinomas. Gastroenterology 2015, 149, 1872–1883.e9. [Google Scholar] [CrossRef]
- Goldberg, Y.; Halpern, N.; Hubert, A.; Adler, S.N.; Cohen, S.; Plesser-Duvdevani, M.; Pappo, O.; Shaag, A.; Meiner, V. Mutated MCM9 is associated with predisposition to hereditary mixed polyposis and colorectal cancer in addition to primary ovarian failure. Cancer Genet. 2015, 208, 621–624. [Google Scholar] [CrossRef]
- Rohlin, A.; Eiengard, F.; Lundstam, U.; Zagoras, T.; Nilsson, S.; Edsjo, A.; Pedersen, J.; Svensson, J.; Skullman, S.; Karlsson, B.G.; et al. GREM1 and POLE variants in hereditary colorectal cancer syndromes. Genes, chromosomes & cancer 2016, 55, 95–106. [Google Scholar] [CrossRef]
- Spier, I.; Kerick, M.; Drichel, D.; Horpaopan, S.; Altmuller, J.; Laner, A.; Holzapfel, S.; Peters, S.; Adam, R.; Zhao, B.; et al. Exome sequencing identifies potential novel candidate genes in patients with unexplained colorectal adenomatous polyposis. Fam. Cancer 2016, 15, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Thutkawkorapin, J.; Picelli, S.; Kontham, V.; Liu, T.; Nilsson, D.; Lindblom, A. Exome sequencing in one family with gastric- and rectal cancer. BMC Genet. 2016, 17, 41. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Jurado, C.; Franch-Exposito, S.; Munoz, J.; Ocana, T.; Carballal, S.; Lopez-Ceron, M.; Cuatrecasas, M.; Vila-Casadesus, M.; Lozano, J.J.; Serra, E.; et al. The Fanconi anemia DNA damage repair pathway in the spotlight for germline predisposition to colorectal cancer. Eur. J. Hum. Genet. 2016, 24, 1501–1505. [Google Scholar] [CrossRef] [PubMed]
- Chubb, D.; Broderick, P.; Dobbins, S.E.; Frampton, M.; Kinnersley, B.; Penegar, S.; Price, A.; Ma, Y.P.; Sherborne, A.L.; Palles, C.; et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat. Commun. 2016, 7, 11883. [Google Scholar] [CrossRef]
- Schubert, S.A.; Ruano, D.; Elsayed, F.A.; Boot, A.; Crobach, S.; Sarasqueta, A.F.; Wolffenbuttel, B.; van der Klauw, M.M.; Oosting, J.; Tops, C.M.; et al. Evidence for genetic association between chromosome 1q loci and predisposition to colorectal neoplasia. Br. J. Cancer 2017, 117, 1215–1223. [Google Scholar] [CrossRef]
- Martin-Morales, L.; Feldman, M.; Vershinin, Z.; Garre, P.; Caldes, T.; Levy, D. SETD6 dominant negative mutation in familial colorectal cancer type X. Hum. Mol. Genet. 2017, 26, 4481–4493. [Google Scholar] [CrossRef]
- Bellido, F.; Sowada, N.; Mur, P.; Lazaro, C.; Pons, T.; Valdes-Mas, R.; Pineda, M.; Aiza, G.; Iglesias, S.; Soto, J.L.; et al. Association Between Germline Mutations in BRF1, a Subunit of the RNA Polymerase III Transcription Complex, and Hereditary Colorectal Cancer. Gastroenterology 2018, 154, 181–194.e20. [Google Scholar] [CrossRef]
- Franch-Exposito, S.; Esteban-Jurado, C.; Garre, P.; Quintanilla, I.; Duran-Sanchon, S.; Diaz-Gay, M.; Bonjoch, L.; Cuatrecasas, M.; Samper, E.; Munoz, J.; et al. Rare germline copy number variants in colorectal cancer predisposition characterized by exome sequencing analysis. J. Genet. Genom. 2018, 45, 41–45. [Google Scholar] [CrossRef]
- Yu, L.; Yin, B.; Qu, K.; Li, J.; Jin, Q.; Liu, L.; Liu, C.; Zhu, Y.; Wang, Q.; Peng, X.; et al. Screening for susceptibility genes in hereditary non-polyposis colorectal cancer. Oncol. Lett. 2018, 15, 9413–9419. [Google Scholar] [CrossRef]
- Thutkawkorapin, J.; Lindblom, A.; Tham, E. Exome sequencing in 51 early onset non-familial CRC cases. Mol. Genet. Genom. Med. 2019, 7, e605. [Google Scholar] [CrossRef]
- Diaz-Gay, M.; Franch-Exposito, S.; Arnau-Collell, C.; Park, S.; Supek, F.; Munoz, J.; Bonjoch, L.; Gratacos-Mulleras, A.; Sanchez-Rojas, P.A.; Esteban-Jurado, C.; et al. Integrated Analysis of Germline and Tumor DNA Identifies New Candidate Genes Involved in Familial Colorectal Cancer. Cancers 2019, 11, 362. [Google Scholar] [CrossRef] [PubMed]
- Toma, C.; Diaz-Gay, M.; Soares de Lima, Y.; Arnau-Collell, C.; Franch-Exposito, S.; Munoz, J.; Overs, B.; Bonjoch, L.; Carballal, S.; Ocana, T.; et al. Identification of a Novel Candidate Gene for Serrated Polyposis Syndrome Germline Predisposition by Performing Linkage Analysis Combined With Whole-Exome Sequencing. Clin. Transl. Gastroenterol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.M.L.; Ghosh, P.; Dakal, T.C.; Slavin, T.P.; Boland, C.R.; Goel, A. Novel candidates in early-onset familial colorectal cancer. Fam. Cancer 2020, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Toma, C.; Diaz-Gay, M.; Franch-Exposito, S.; Arnau-Collell, C.; Overs, B.; Munoz, J.; Bonjoch, L.; Soares de Lima, Y.; Ocana, T.; Cuatrecasas, M.; et al. Using linkage studies combined with whole-exome sequencing to identify novel candidate genes for familial colorectal cancer. Int. J. Cancer 2020, 146, 1568–1577. [Google Scholar] [CrossRef] [PubMed]
- Bonjoch, L.; Franch-Exposito, S.; Garre, P.; Belhadj, S.; Munoz, J.; Arnau-Collell, C.; Diaz-Gay, M.; Gratacos-Mulleras, A.; Raimondi, G.; Esteban-Jurado, C.; et al. Germline mutations in FAF1 are associated with hereditary colorectal cancer. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Chubb, D.; Broderick, P.; Frampton, M.; Kinnersley, B.; Sherborne, A.; Penegar, S.; Lloyd, A.; Ma, Y.P.; Dobbins, S.E.; Houlston, R.S. Genetic diagnosis of high-penetrance susceptibility for colorectal cancer (CRC) is achievable for a high proportion of familial CRC by exome sequencing. J. Clin. Oncol. 2015, 33, 426–432. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Weren, R.D.; Ligtenberg, M.J.; Geurts van Kessel, A.; De Voer, R.M.; Hoogerbrugge, N.; Kuiper, R.P. NTHL1 and MUTYH polyposis syndromes: Two sides of the same coin? J. Pathol. 2018, 244, 135–142. [Google Scholar] [CrossRef]
- Ghosh, R.; Oak, N.; Plon, S.E. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017, 18, 225. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- J. Craig Venter Institute—PROVEAN. Available online: http://provean.jcvi.org/about.php (accessed on 12 August 2020).
- Mahmood, K.; Jung, C.H.; Philip, G.; Georgeson, P.; Chung, J.; Pope, B.J.; Park, D.J. Variant effect prediction tools assessed using independent, functional assay-based datasets: Implications for discovery and diagnostics. Hum. Genom. 2017, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Spier, I.; Drichel, D.; Kerick, M.; Kirfel, J.; Horpaopan, S.; Laner, A.; Holzapfel, S.; Peters, S.; Adam, R.; Zhao, B.; et al. Low-level APC mutational mosaicism is the underlying cause in a substantial fraction of unexplained colorectal adenomatous polyposis cases. J. Med. Genet. 2016, 53, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.T.; Buchanan, D.D.; Thompson, B.; Young, J.P.; Spurdle, A.B. Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: A literature review assessing utility of tumour features for MMR variant classification. J. Med. Genet. 2012, 49, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Viel, A.; Bruselles, A.; Meccia, E.; Fornasarig, M.; Quaia, M.; Canzonieri, V.; Policicchio, E.; Urso, E.D.; Agostini, M.; Genuardi, M.; et al. A Specific Mutational Signature Associated with DNA 8-Oxoguanine Persistence in MUTYH-defective Colorectal Cancer. EBioMedicine 2017, 20, 39–49. [Google Scholar] [CrossRef]
- Pilati, C.; Shinde, J.; Alexandrov, L.B.; Assie, G.; Andre, T.; Helias-Rodzewicz, Z.; Ducoudray, R.; Le Corre, D.; Zucman-Rossi, J.; Emile, J.F.; et al. Mutational signature analysis identifies MUTYH deficiency in colorectal cancers and adrenocortical carcinomas. J. Pathol. 2017, 242, 10–15. [Google Scholar] [CrossRef]
- Grolleman, J.E.; de Voer, R.M.; Elsayed, F.A.; Nielsen, M.; Weren, R.D.A.; Palles, C.; Ligtenberg, M.J.L.; Vos, J.R.; Ten Broeke, S.W.; de Miranda, N.; et al. Mutational Signature Analysis Reveals NTHL1 Deficiency to Cause a Multi-tumor Phenotype. Cancer Cell 2019, 35, 256–266.e5. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Grolleman, J.E.; Diaz-Gay, M.; Franch-Exposito, S.; Castellvi-Bel, S.; de Voer, R.M. Somatic mutational signatures in polyposis and colorectal cancer. Mol. Aspects Med. 2019, 69, 62–72. [Google Scholar] [CrossRef]
- Drost, J.; van Boxtel, R.; Blokzijl, F.; Mizutani, T.; Sasaki, N.; Sasselli, V.; de Ligt, J.; Behjati, S.; Grolleman, J.E.; van Wezel, T.; et al. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 2017, 358, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Mur, P.; Sanchez-Cuartielles, E.; Ausso, S.; Aiza, G.; Valdes-Mas, R.; Pineda, M.; Navarro, M.; Brunet, J.; Urioste, M.; Lazaro, C.; et al. Scarce evidence of the causal role of germline mutations in UNC5C in hereditary colorectal cancer and polyposis. Sci. Rep. 2016, 6, 20697. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, S.H.; Spielmann, M.; Mundlos, S.; Veltman, J.A.; Gilissen, C. Comparison of Exome and Genome Sequencing Technologies for the Complete Capture of Protein-Coding Regions. Hum. Mutat. 2015, 36, 815–822. [Google Scholar] [CrossRef]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.L.; Abel, L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef]
- Lin, Y.; Lin, S.; Baxter, M.D.; Lin, L.; Kennedy, S.M.; Zhang, Z.; Goodfellow, P.J.; Chapman, W.C.; Davidson, N.O. Novel APC promoter and exon 1B deletion and allelic silencing in three mutation-negative classic familial adenomatous polyposis families. Genome Med. 2015, 7, 42. [Google Scholar] [CrossRef]
- Teresi, R.E.; Zbuk, K.M.; Pezzolesi, M.G.; Waite, K.A.; Eng, C. Cowden syndrome-affected patients with PTEN promoter mutations demonstrate abnormal protein translation. Am. J. Hum. Genet. 2007, 81, 756–767. [Google Scholar] [CrossRef]
- Snow, A.K.; Tuohy, T.M.; Sargent, N.R.; Smith, L.J.; Burt, R.W.; Neklason, D.W. APC promoter 1B deletion in seven American families with familial adenomatous polyposis. Clin. Genet. 2015, 88, 360–365. [Google Scholar] [CrossRef]
- Pavicic, W.; Nieminen, T.T.; Gylling, A.; Pursiheimo, J.P.; Laiho, A.; Gyenesei, A.; Jarvinen, H.J.; Peltomaki, P. Promoter-specific alterations of APC are a rare cause for mutation-negative familial adenomatous polyposis. Genes Chromosomes Cancer 2014, 53, 857–864. [Google Scholar] [CrossRef]
- Nieminen, T.T.; Pavicic, W.; Porkka, N.; Kankainen, M.; Jarvinen, H.J.; Lepisto, A.; Peltomaki, P. Pseudoexons provide a mechanism for allele-specific expression of APC in familial adenomatous polyposis. Oncotarget 2016, 7, 70685–70698. [Google Scholar] [CrossRef]
- Spier, I.; Horpaopan, S.; Vogt, S.; Uhlhaas, S.; Morak, M.; Stienen, D.; Draaken, M.; Ludwig, M.; Holinski-Feder, E.; Nothen, M.M.; et al. Deep intronic APC mutations explain a substantial proportion of patients with familial or early-onset adenomatous polyposis. Hum. Mutat. 2012, 33, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Lam, E.; Saghbini, M.; Bocklandt, S.; Hastie, A.; Cao, H.; Holmlin, E.; Borodkin, M. Structural Variation Detection and Analysis Using Bionano Optical Mapping. Methods Mol. Biol. 2018, 1833, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Aretz, S.; Stienen, D.; Friedrichs, N.; Stemmler, S.; Uhlhaas, S.; Rahner, N.; Propping, P.; Friedl, W. Somatic APC mosaicism: A frequent cause of familial adenomatous polyposis (FAP). Hum. Mutat. 2007, 28, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Hes, F.J.; Nielsen, M.; Bik, E.C.; Konvalinka, D.; Wijnen, J.T.; Bakker, E.; Vasen, H.F.; Breuning, M.H.; Tops, C.M. Somatic APC mosaicism: An underestimated cause of polyposis coli. Gut 2008, 57, 71–76. [Google Scholar] [CrossRef]
- Ciavarella, M.; Miccoli, S.; Prossomariti, A.; Pippucci, T.; Bonora, E.; Buscherini, F.; Palombo, F.; Zuntini, R.; Balbi, T.; Ceccarelli, C.; et al. Somatic APC mosaicism and oligogenic inheritance in genetically unsolved colorectal adenomatous polyposis patients. Eur. J. Hum. Genet. 2018, 26, 387–395. [Google Scholar] [CrossRef]
- Kim, B.; Won, D.; Jang, M.; Kim, H.; Choi, J.R.; Kim, T.I.; Lee, S.T. Next-generation sequencing with comprehensive bioinformatics analysis facilitates somatic mosaic APC gene mutation detection in patients with familial adenomatous polyposis. BMC Med. Genom. 2019, 12, 103. [Google Scholar] [CrossRef]
- Acuna-Hidalgo, R.; Veltman, J.A.; Hoischen, A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 2016, 17, 241. [Google Scholar] [CrossRef]
- Mavaddat, N.; Michailidou, K.; Dennis, J.; Lush, M.; Fachal, L.; Lee, A.; Tyrer, J.P.; Chen, T.H.; Wang, Q.; Bolla, M.K.; et al. Polygenic Risk Scores for Prediction of Breast Cancer and Breast Cancer Subtypes. Am. J. Hum. Genet. 2019, 104, 21–34. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Gruber, S.B.; Wu, Z.; Schmidt, E.M.; Zawistowski, M.; Moser, S.E.; Blanc, V.M.; Brummett, C.M.; Kheterpal, S.; Abecasis, G.R.; et al. Association of Polygenic Risk Scores for Multiple Cancers in a Phenome-wide Study: Results from The Michigan Genomics Initiative. Am. J. Hum. Genet. 2018, 102, 1048–1061. [Google Scholar] [CrossRef]
- Weigl, K.; Chang-Claude, J.; Knebel, P.; Hsu, L.; Hoffmeister, M.; Brenner, H. Strongly enhanced colorectal cancer risk stratification by combining family history and genetic risk score. Clin. Epidemiol. 2018, 10, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Fahed, A.C.; Wang, M.; Homburger, J.R.; Patel, A.P.; Bick, A.G.; Neben, C.L.; Lai, C.; Brockman, D.; Philippakis, A.; Ellinor, P.T.; et al. Polygenic background modifies penetrance of monogenic variants conferring risk for coronary artery disease, breast cancer, or colorectal cancer. medRxiv 2019. [Google Scholar] [CrossRef]
- Schlafly, A.; Pfeiffer, R.M.; Nagore, E.; Puig, S.; Calista, D.; Ghiorzo, P.; Menin, C.; Fargnoli, M.C.; Peris, K.; Song, L.; et al. Contribution of Common Genetic Variants to Familial Aggregation of Disease and Implications for Sequencing Studies. PLoS Genet. 2019, 15, e1008490. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Author | Key Gene(s) | Inclusion Criteria Index Phenotype | Inclusion Criteria Age | Inclusion Criteria FH | Size Discovery Cohort |
|---|---|---|---|---|---|
| Palles et al., 2013 [43] | POLD1, POLE | ≥10 colorectal tumors | <60 years | FDR or SDR with CRC | 15 families (20 cases) |
| Smith et al., 2013 [47] | FANCM, LAMB4, PTCHD3, LAMC3, REX2 | Advanced CRC | ≤35 years (18 cases) | No, sporadic | 50 cases |
| DeRycke et al., 2013 [48] | CENPE, KIF23 | Familial CRC | NS | ≥2 members affected | 16 families (40 cases) |
| de Voer et al., 2013 [49] | BUB1, BUB3 | Non-polyposis MMR-proficient CRC | ≤40 years | NS | 33 cases |
| Gylfe et al., 2013 [50] | UACA, SFXN4, TWSG1, PSPH, NUDT7, ZNF490, PRSS37, CCDC18, PRADC1, MRPL3, ARK1C4 | Familial CRC | NS | ≥1 FDR with CRC | 96 cases |
| Gala et al., 2014 [42] | RNF43 | Sessile serrated adenomas | NS | If <5 SSAs, ≥1 FDR with SSAs or CRC | 20 cases |
| Rohlin et al., 2014 [51] | No novel genes found, POLE found | Hereditary CRC | n/a | n/a | 1 family (3 affected, 1 unaffected) |
| Nieminen et al., 2014 [41] | RPS20 | Amsterdam/Bethesda FCCTX | n/a | n/a | 1 family (4 cases) |
| Schulz et al., 2014 [52] | SEMA4A | Amsterdam I FCCTX | n/a | n/a | 1 family (4 cases) |
| Esteban-Jurado et al., 2015 [53] | CDKN1B, XRCC4, EPHX1, NFKBIZ, SMARCA4, BARD1 | Familial CRC 1 | ≥1 relative diagnosed <60 1 | ≥3 affected, ≥2 in consecutive generations | 29 families (43 cases) 1 |
| Tanskanen et al., 2015 [54] | ADAMTS4, CYTL1, SYNE1, MCTP2, ARHGAP12, ATM, DONSON, ROS1 | Non-syndromic early-onset CRC | <40 years | NS | 22 cases |
| Wei et al., 2015 [55] | HNRNPA0 and WIF1 | Multiple early-onset cancer | n/a | n/a | 1 family (4 affected, 4 unaffected) |
| Zhang et al., 2015 [56] | EIF2AK4, LRP5, BUB1 | Familial CRC without polyposis | ≤55 years | If ≥40 years, ≥1 FDR with CRC | 21 families (23 cases) |
| Weren et al., 2015 [44] | NTHL1 | Multiple adenomatous polyps | NS | NS | 51 cases (48 families) |
| Segui et al., 2015 [57] | FAN1 | Amsterdam I MMR-proficient CRC | n/a | n/a | 1 family (3 cases) |
| Ngeow et al., 2015 [58] | SMAD9 | HPS | n/a | n/a | 1 family (1 case) |
| Arora et al., 2015 [59] | ERCC6, WRN | CRC or polyposis (≥10 polyps) | <50 years | ≥1 relative with CRC | 25 cases |
| Goldberg et al., 2015 [60] | MCM9 | Multiple mixed polyposis and metastatic CRC | n/a | n/a | 1 family (1 cases, 1 unaffected) |
| Rohlin et al., 2016 [61] | No novel genes found, GREM1 and POLE found | AFAP/atypical polyposis | n/a | n/a | 1 family (4 affected, 4 unaffected cases) |
| Spier et al., 2016 [62] | DSC2, PIEZO1 | Colorectal adenomatous polyposis | NS | NS | 7 cases |
| Thutkawkorapin et al., 2016 [63] | DZIP1L, IGSF10, NOTCH1, SF3A1, GAL3ST1 | Familial rectal- and gastric cancer | n/a | n/a | 1 family (3 cases) |
| de Voer et al., 2016 [13] | PTPN12, LRP6 | non-polyposis MMR-proficient CRC | ≤45 years | NS | 55 cases |
| Esteban-Jurado et al., 2016 [64] | BRCA2/FANCD1, BRIP1/FANCJ, FANCC, FANCE, REV3L/POLZ | Familial CRC 1 | ≥1 relative diagnosed <60 1 | ≥3 affected, ≥2 in consecutive generations | 40 families (74 cases) 1 |
| Chubb et al., 2016 [65] | POT1, POLE2, MRE11 | CRC | ≤55 years | ≥1 FDR with CRC | 1006 cases |
| Adam et al., 2016 [40] | MSH3 | ≥20 synchronous or ≥40 metachronous colorectal adenomas | NS | NS | 102 cases |
| Schubert et al., 2017, 2018 [66] | MIA3 | Amsterdam I MMR stable familial CRC | n/a | n/a | 1 family (3 cases WES, 2 cases WES/WGS, 1 cases WGS) |
| Martín-Morales et al., 2017 [67] | SETD6 | Amsterdam I FCCTX | ≥1 relative diagnosed <50 | ≥3 affected (≥1 FDR), ≥2 in consecutive generations | 1 family (2 cases, 1 unaffected) |
| Bellido et al., 2018 [68] | BRF1 | Amsterdam I hereditary CRC | n/a | n/a | 1 family (3 CRC cases, 1 BC case) |
| Franch-Expósito et al., 2018 [69] | TTF2, TMEM158 | Familial CRC 1 | ≥1 relative diagnosed <601 | ≥3 affected, ≥2 in consecutive generations | WES: 38 families (71 cases), WGS: 1 case 1 |
| Yu et al., 2018 [70] | DDX20, ZFYVE26, PIK3R3, SLC26A8, ZEB2, TP53INP1, SLC11A1, LRBA, CEBPZ, ETAA1, SEMA3G, IFRD2 and FAT1 | Amsterdam I/II non-polyposis hereditary CRC | ≥1 relative diagnosed <50 | ≥1 FDR & 2 generations affected | 1 family (3 cases) |
| Olkinuora et al., 2018 [39] | MLH3 | Adenomatous polyposis | NS | NS | 40 cases |
| Thutkawkorapin et al., 2019 [71] | BMPR1A, BRIP1, SRC, CLSPN, SEC24B, SSH2, ACACA, NR2C2, INPP4A, DIDO1, ATP10B, PKHD1, UGGT2, MYH13, TFF3 | Simplex early-onset CRC | <40 years | NS | 51 cases |
| Diaz-Gay et al., 2019 [72] | BRCA2, BLM, ERCC2, RECQL(=WRN), REV3L and RIF1 | Familial CRC 1 | ≥1 relative diagnosed <60 1 | ≥3 affected, ≥2 in consecutive generations | 18 cases 1 |
| Toma et al., 2019 [73] | FBLN2 | Familial CRC/SPS | NS | ≥2 affected in consecutive generations | 16 families (39 cases) |
| Jansen et al., 2020 [74] | NOTCH2, RAB25 | Familial CRC | NS | NS | 5 families (9 cases) |
| Toma et al., 2020 [75] | SMO | Familial CRC 1 | ≥1 relative diagnosed <60 1 | ≥3 affected, ≥2 in consecutive generations | 18 families (47 cases) 1 |
| Bonjoch et al., 2020 [76] | FAF1 | Familial CRC 1 | ≥1 relative diagnosed <60 1 | ≥3 affected, ≥2 in consecutive generations | 40 families (75 cases) 1 |
| Discovery Cohort Selection |
|
| Variant Prioritization |
|
|
|
|
| Variant Validation |
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
te Paske, I.B.A.W.; Ligtenberg, M.J.L.; Hoogerbrugge, N.; de Voer, R.M. Candidate Gene Discovery in Hereditary Colorectal Cancer and Polyposis Syndromes–Considerations for Future Studies. Int. J. Mol. Sci. 2020, 21, 8757. https://doi.org/10.3390/ijms21228757
te Paske IBAW, Ligtenberg MJL, Hoogerbrugge N, de Voer RM. Candidate Gene Discovery in Hereditary Colorectal Cancer and Polyposis Syndromes–Considerations for Future Studies. International Journal of Molecular Sciences. 2020; 21(22):8757. https://doi.org/10.3390/ijms21228757
Chicago/Turabian Stylete Paske, Iris B. A. W., Marjolijn J. L. Ligtenberg, Nicoline Hoogerbrugge, and Richarda M. de Voer. 2020. "Candidate Gene Discovery in Hereditary Colorectal Cancer and Polyposis Syndromes–Considerations for Future Studies" International Journal of Molecular Sciences 21, no. 22: 8757. https://doi.org/10.3390/ijms21228757
APA Stylete Paske, I. B. A. W., Ligtenberg, M. J. L., Hoogerbrugge, N., & de Voer, R. M. (2020). Candidate Gene Discovery in Hereditary Colorectal Cancer and Polyposis Syndromes–Considerations for Future Studies. International Journal of Molecular Sciences, 21(22), 8757. https://doi.org/10.3390/ijms21228757