Currently Applied Molecular Assays for Identifying ESR1 Mutations in Patients with Advanced Breast Cancer


Abstract
:1. Introduction
1.1. Epidemiology of Breast Cancer
1.2. Diagnosis and Molecular Heterogeneity of Breast Cancer
1.3. Endocrine Therapy and Resistance
1.3.1. Mechanisms of Resistance to Endocrine Therapy
1.3.2. ESR1 Mutation
ESR1 Amplification
ESR1 Rearrangements and Fusion
ESR1 Point Mutation
1.4. Aims of This Review for Molecular Assays
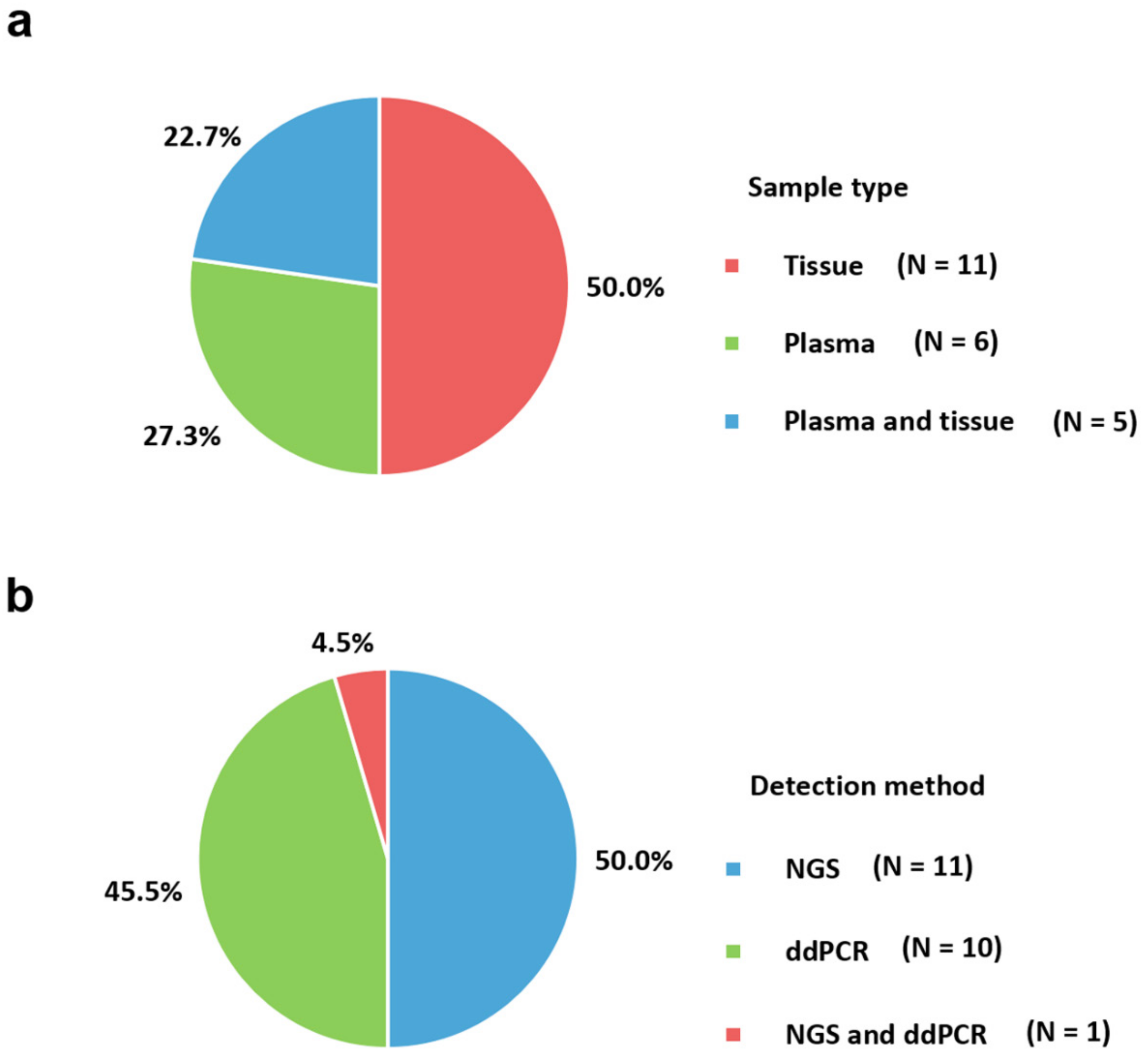
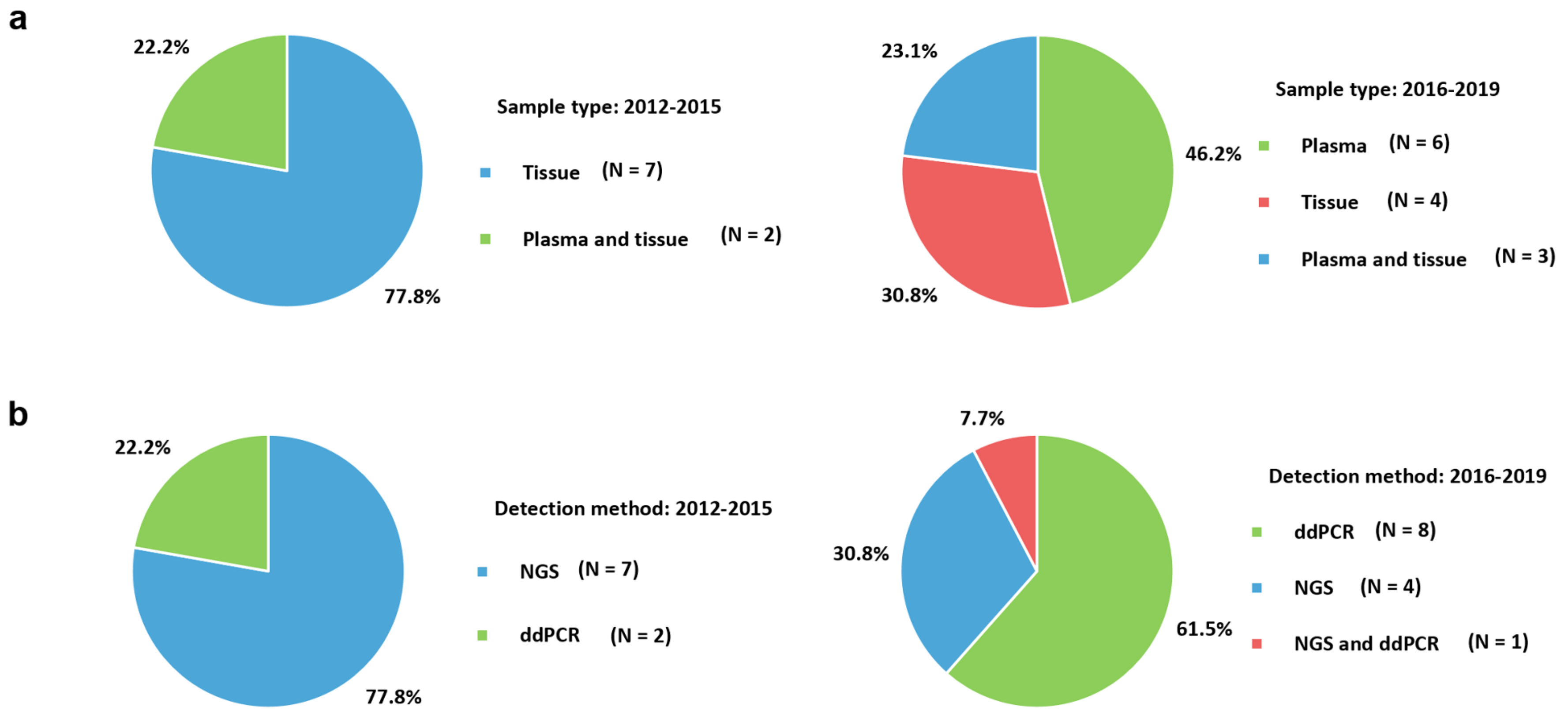
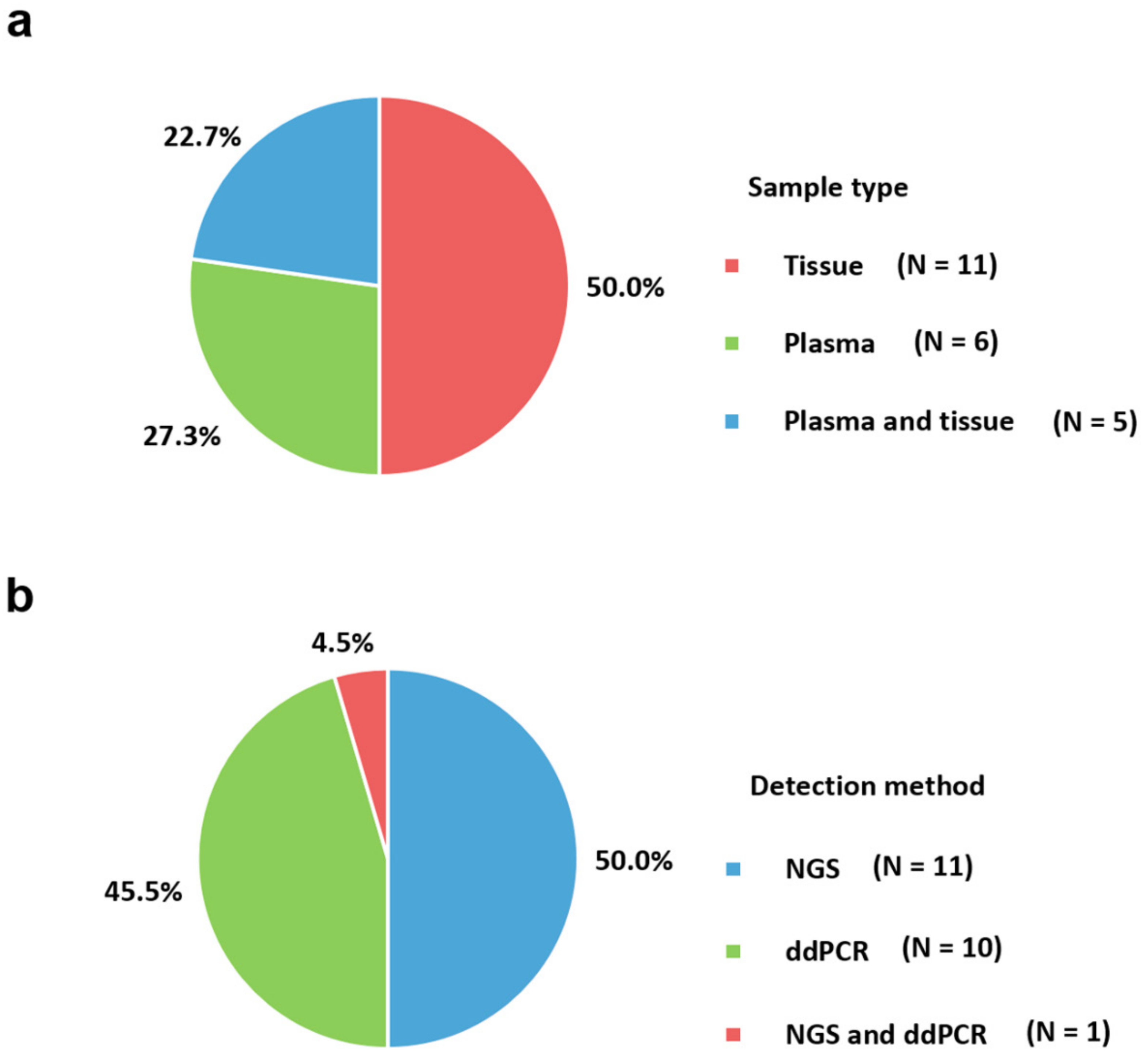
2. Sample Type
3. Trends in Molecular Assays

{kind=link}
{kind=link}
| First Author [Year] | Tumor Type (n) | Sample Type | Detection Method | Sequencing Equipment of Kit (Company) | Most Frequent ESR1 | Study Country |
|---|---|---|---|---|---|---|
| NGS based | ||||||
| Bartels et al., (2018), [78] | BC with bone metastases (231) | FFPE | NGS and ddPCR | Ion PGM Hi-Q Kit v2 using 318 v2 Chips and QuantStudio 3D Digital PCR System (Thermo Fisher Scientific, Germany) | D538G | Germany |
| Cancer Genome Atlas, (2012), [16] | Luminal BC (169) | Tissue | NGS and several methods | Illumina (Illumina, USA) | NA | Multi-national |
| Ellis et al., (2012), [17] | Luminal BC (46) | Snap-frozen tissue | NGS | Illumina (Illumina, USA) | NA | USA |
| Jeselsohn et al., (2014), [40] | Metastatic BC (76) | FFPE | NGS | HiSeq2000 (Illumina, USA) | D538G and Y537N | USA and Spain |
| Lefebvre et al., (2016), [29] | Metastatic BC (143) | Fresh frozen tumor biopsy | NGS | Illumina HiSeq2500, HiSeq4000, or NextSeq500 (Illumina, USA) | NA | France |
| Merenbakh-Lamin et al., (2013), [55] | Metastatic BC (13) | FFPE | NGS | Illumina HiSeq2000 (Illumina, USA) | D538G | Israel |
| Nik-Zainal et al., (2016) [83] | BC (560) | FFPE | NGS | Illumina GAIIx, Hiseq 2000 or Hiseq 2500 (Illumina, USA) | NA | Multi-national |
| Niu et al., (2015) [36] | Metastatic BC (222) | FFPE | NGS | Illumina HiSeq2000 platform (Illumina, USA) | Codon Y537 | USA |
| Robinson et al., (2013) [33] | Metastatic BC (11) | Frozen needle biopsy | NGS | Illumina HiSeq2000 platform (Illumina, USA) | NA | USA |
| Toy et al., (2013) [34] | Advanced BC and Metastatic BC (36) | Fresh frozen tissue and FFPE | NGS | Illumina Hiseq 2000 (Illumina, USA) | D538G | USA |
| Toy et al., (2017) [87] | Metastatic BC (265) | FFPE | NGS | Illumina HiSeq 2500 (Illumina, USA) | D538G | USA |
| Yanagawa et al., (2017) [89] | Primary BC (16) and recurrent BC (46) | FFPE and plasma | NGS | Torrent PGM instrument (Thermo Fisher Scientific, USA) | D538G | Japan |
| ddPCR based | ||||||
| Chandarlapaty et al., (2016) [79] | Metastatic BC (541) related to BOLERO-2 clinical trial | Plasma in EDTA | Single ddPCR | QX200 Droplet Digital PCR System (Bio-Rad Laboratories, USA) | D538G | USA |
| Chu et al., (2016) [80] | Metastatic BC (23) | Plasma in Streck BCT DNA tube or EDTA | ddPCR | QX200 Droplet Digital PCR System (Bio-Rad Laboratories, USA) | D538G | USA |
| Clatot et al., (2016) [81] | BC with progression (144) | Plasma in heparinized tube | Single ddPCR | QX200 Droplet Digital PCR System (Bio-Rad Laboratories, USA) | D538G | France |
| Gyanchandani et al., (2017) [90] | Relapsed or metastatic BC (16) | Plasma in Streck Cell-free DNA blood tubes | ddPCR | QX100 Droplet Digital PCR System (Bio-Rad Laboratories, USA) | D538G | USA |
| Fribbens et al., (2016) [82] | BC with relapse or progression (161) related to SoFEA and PALOMA-3 clinical trials | Plasma in EDTA | Multiplex and uniplex ddPCR | QX200 Droplet Digital PCR System (Bio-Rad Laboratories, USA) | D538G | USA |
| Schiavon et al., (2015] [84] | Advanced BC (171) | Plasma in EDTA or Streck Cell-Free DNA BCT tube, and FFPE | Multiplex ddPCR | QX200 Droplet Digital PCR System (Bio-Rad, USA), Ion AmpliSeq Breast Cancer Panel (Thermo Fisher Scientific, USA), and PI chip using the Ion PI OT2 200 Kit (Thermo Fisher Scientific, USA) | D538G | United Kingdom |
| Sefrioui et al., (2015) [35] | Metastatic BC (7) | Frozen pleural biopsy, FFPE for primary tumor sample, and plasma in heparinized tube | ddPCR | QuantStudio 3D Digital PCR System (Thermo Fisher Scientific, USA) | NA | France |
| Spoerke et al. (2016) [85] | Metastatic BC (153) related to FERGI clinical trial | Plasma and FFPE | ddPCR | QX200 Droplet Digital PCR System (Bio-Rad Laboratories, USA) | D538G | USA |
| Takeshita et al., (2017) [86] | Advanced BC (17) and Metastatic BC (69) | Plasma in EDTA | Single ddPCR | QX200 Droplet Digital PCR System (Bio-Rad Laboratories, USA) | Y537N | Japan |
| Wang et al., (2016) [88] | Primary or metastatic BC (29) | Frozen tissue and plasma in Streck tubes | ddPCR | QX100 Droplet Digital PCR System (Bio-Rad Laboratories, USA) | D538G | USA |
4. Molecular Assays
4.1. Next-Generation Sequencing
4.1.1. Library Preparation
4.1.2. Sequencing Platforms
Illumina
Ion Torrent
4.1.3. Bioinformatics
4.1.4. NGS strategies
Targeted Panel Sequencing
Whole Exome Sequencing
Whole Genome Sequencing
4.2. Droplet Digital PCR
4.3. Other Methods
4.3.1. Sanger Sequencing
4.3.2. Pyrosequencing
4.3.3. Real-Time PCR
4.3.4. Microarray
4.3.5. Methylation
5. Future of Molecular Assays
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BAM | Binary Alignment/map |
| CCDC170 | Coiled-Coil Domain Containing 170 |
| cfDNA | Cell-Free DNA |
| ddPCR | Droplet Digital Polymerase Chain Reaction |
| EDTA | Ethylenediaminetetraacetic Acid |
| EGFR | Epidermal Growth Factor Receptor |
| ER | Estrogen Receptor |
| FFPE | Formalin-Fixed Paraffin-Embedded |
| GATK | Genome Analysis Toolkit |
| HER2 | Human Epidermal Growth Factor Receptor2 |
| LBD | Ligand-Binding Domain |
| NGS | Next-Generation Sequencing |
| PGM | Personal Genome Machine |
| PR | Progesterone Receptor |
| SAM | Sequence Alignment/map |
| SMRT | Single Molecule Real-Time |
| VCF | Variant Calling Format |
References
- Global Cancer Observatory: Cancer Tomorrow. Available online: https://gco.iarc.fr/tomorrow/graphic-isotype (accessed on 10 November 2020).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Wen, W.; Zheng, Y.; Gao, Y.T.; Wu, C.; Bao, P.; Wang, C.; Gu, K.; Peng, P.; Gong, Y.; et al. Breast cancer incidence and mortality: Trends over 40 years among women in Shanghai, China. Ann. Oncol. 2016, 27, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romieu, I.; Biessy, C.; Carayol, M.; His, M.; Torres-Mejia, G.; Angeles-Llerenas, A.; Sanchez, G.I.; Jaramillo, R.; Navarro, E.; Porras, C.; et al. Reproductive factors and molecular subtypes of breast cancer among premenopausal women in Latin America: The PRECAMA study. Sci. Rep. 2018, 8, 13109. [Google Scholar] [CrossRef] [Green Version]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef]
- Hu, Z.; Fan, C.; Oh, D.S.; Marron, J.S.; He, X.; Qaqish, B.F.; Livasy, C.; Carey, L.A.; Reynolds, E.; Dressler, L.; et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genom. 2006, 7, 96. [Google Scholar] [CrossRef] [Green Version]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Sotiriou, C.; Neo, S.Y.; McShane, L.M.; Korn, E.L.; Long, P.M.; Jazaeri, A.; Martiat, P.; Fox, S.B.; Harris, A.L.; Liu, E.T. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc. Natl. Acad. Sci. USA 2003, 100, 10393–10398. [Google Scholar] [CrossRef] [Green Version]
- Veer, L.J.V.T.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [Green Version]
- Guarneri, V.; Conte, P. Metastatic breast cancer: Therapeutic options according to molecular subtypes and prior adjuvant therapy. Oncologist 2009, 14, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.; Osako, T.; Okumura, Y.; Hayashi, M.; Toyozumi, Y.; Arima, N. Ki-67 as a prognostic marker according to breast cancer subtype and a predictor of recurrence time in primary breast cancer. Exp. Ther. Med. 2010, 1, 747–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, M.J.; Tao, Y.; Luo, J.; A’Hern, R.; Evans, D.B.; Bhatnagar, A.S.; Chaudri Ross, H.A.; von Kameke, A.; Miller, W.R.; Smith, I.; et al. Outcome prediction for estrogen receptor-positive breast cancer based on postneoadjuvant endocrine therapy tumor characteristics. J. Natl. Cancer Inst. 2008, 100, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Ellis, M.J.; Ding, L.; Shen, D.; Luo, J.; Suman, V.J.; Wallis, J.W.; Van Tine, B.A.; Hoog, J.; Goiffon, R.J.; Goldstein, T.C.; et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 2012, 486, 353–360. [Google Scholar] [CrossRef]
- Nielsen, K.V.; Ejlertsen, B.; Muller, S.; Moller, S.; Rasmussen, B.B.; Balslev, E.; Laenkholm, A.V.; Christiansen, P.; Mouridsen, H.T. Amplification of ESR1 may predict resistance to adjuvant tamoxifen in postmenopausal patients with hormone receptor positive breast cancer. Breast Cancer Res. Treat. 2011, 127, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E. Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-updagger. Ann. Oncol. 2019, 30, 1194–1220. [Google Scholar] [CrossRef] [Green Version]
- Najim, O.; Seghers, S.; Sergoynne, L.; Van Gaver, H.; Papadimitriou, K.; Wouters, K.; Trinh, X.B.; Huizing, M.T.; Tjalma, W. The association between type of endocrine therapy and development of estrogen receptor-1 mutation(s) in patients with hormone-sensitive advanced breast cancer: A systematic review and meta-analysis of randomized and non-randomized trials. Biochim. Biophys. Acta. Rev. Cancer 2019, 1872, 188315. [Google Scholar] [CrossRef]
- Tabarestani, S.; Motallebi, M.; Akbari, M.E. Are Estrogen Receptor Genomic Aberrations Predictive of Hormone Therapy Response in Breast Cancer? Iran. J. Cancer Prev. 2016, 9, e6565. [Google Scholar] [CrossRef]
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG); Davies, C.; Godwin, J.; Gray, R.; Clarke, M.; Cutter, D.; Darby, S.; McGale, P.; Pan, H.C.; Taylor, C.; et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet 2011, 378, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Reinert, T.; Chmielewska, I.; Ellis, M.J. Mechanisms of aromatase inhibitor resistance. Nat. Rev. Cancer 2015, 15, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Bardou, V.; Hopp, T.A.; Chamness, G.C.; Hilsenbeck, S.G.; Fuqua, S.A.; Wong, J.; Allred, D.C.; Clark, G.M.; Schiff, R. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J. Natl. Cancer Inst. 2003, 95, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arpino, G.; Green, S.J.; Allred, D.C.; Lew, D.; Martino, S.; Osborne, C.K.; Elledge, R.M. HER-2 amplification, HER-1 expression, and tamoxifen response in estrogen receptor-positive metastatic breast cancer: A southwest oncology group study. Clin. Cancer Res. 2004, 10, 5670–5676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, C.K.; Neven, P.; Dirix, L.Y.; Mackey, J.R.; Robert, J.; Underhill, C.; Schiff, R.; Gutierrez, C.; Migliaccio, I.; Anagnostou, V.K.; et al. Gefitinib or placebo in combination with tamoxifen in patients with hormone receptor-positive metastatic breast cancer: A randomized phase II study. Clin. Cancer Res. 2011, 17, 1147–1159. [Google Scholar] [CrossRef] [Green Version]
- Thangavel, C.; Dean, J.L.; Ertel, A.; Knudsen, K.E.; Aldaz, C.M.; Witkiewicz, A.K.; Clarke, R.; Knudsen, E.S. Therapeutically activating RB: Reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr. Relat. Cancer 2011, 18, 333–345. [Google Scholar] [CrossRef]
- Ma, C.X.; Gao, F.; Luo, J.; Northfelt, D.W.; Goetz, M.; Forero, A.; Hoog, J.; Naughton, M.; Ademuyiwa, F.; Suresh, R.; et al. NeoPalAna: Neoadjuvant Palbociclib, a Cyclin-Dependent Kinase 4/6 Inhibitor, and Anastrozole for Clinical Stage 2 or 3 Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2017, 23, 4055–4065. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, C.; Bachelot, T.; Filleron, T.; Pedrero, M.; Campone, M.; Soria, J.C.; Massard, C.; Levy, C.; Arnedos, M.; Lacroix-Triki, M.; et al. Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 2016, 13, e1002201. [Google Scholar] [CrossRef]
- Dahlman-Wright, K.; Cavailles, V.; Fuqua, S.A.; Jordan, V.C.; Katzenellenbogen, J.A.; Korach, K.S.; Maggi, A.; Muramatsu, M.; Parker, M.G.; Gustafsson, J.A. International Union of Pharmacology. LXIV. Estrogen receptors. Pharmacol. Rev. 2006, 58, 773–781. [Google Scholar] [CrossRef] [Green Version]
- Tuteja, N. Signaling through G protein coupled receptors. Plant Signal. Behav. 2009, 4, 942–947. [Google Scholar] [CrossRef]
- Gelsomino, L.; Gu, G.; Rechoum, Y.; Beyer, A.R.; Pejerrey, S.M.; Tsimelzon, A.; Wang, T.; Huffman, K.; Ludlow, A.; Ando, S.; et al. ESR1 mutations affect anti-proliferative responses to tamoxifen through enhanced cross-talk with IGF signaling. Breast Cancer Res. Treat. 2016, 157, 253–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sefrioui, D.; Perdrix, A.; Sarafan-Vasseur, N.; Dolfus, C.; Dujon, A.; Picquenot, J.M.; Delacour, J.; Cornic, M.; Bohers, E.; Leheurteur, M.; et al. Short report: Monitoring ESR1 mutations by circulating tumor DNA in aromatase inhibitor resistant metastatic breast cancer. Int. J. Cancer 2015, 137, 2513–2519. [Google Scholar] [CrossRef] [Green Version]
- Niu, J.; Andres, G.; Kramer, K.; Kundranda, M.N.; Alvarez, R.H.; Klimant, E.; Parikh, A.R.; Tan, B.; Staren, E.D.; Markman, M. Incidence and clinical significance of ESR1 mutations in heavily pretreated metastatic breast cancer patients. Onco Targets Ther. 2015, 8, 3323–3328. [Google Scholar] [CrossRef] [Green Version]
- Pejerrey, S.M.; Dustin, D.; Kim, J.A.; Gu, G.; Rechoum, Y.; Fuqua, S.A.W. The Impact of ESR1 Mutations on the Treatment of Metastatic Breast Cancer. Horm. Cancer 2018, 9, 215–228. [Google Scholar] [CrossRef]
- Basudan, A.; Priedigkeit, N.; Hartmaier, R.J.; Sokol, E.S.; Bahreini, A.; Watters, R.J.; Boisen, M.M.; Bhargava, R.; Weiss, K.R.; Karsten, M.M.; et al. Frequent ESR1 and CDK Pathway Copy-Number Alterations in Metastatic Breast Cancer. Mol. Cancer Res. 2019, 17, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Brown, L.A.; Hoog, J.; Chin, S.F.; Tao, Y.; Zayed, A.A.; Chin, K.; Teschendorff, A.E.; Quackenbush, J.F.; Marioni, J.C.; Leung, S.; et al. ESR1 gene amplification in breast cancer: A common phenomenon? Nat. Genet. 2008, 40, 806–807. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gomez, H.; et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef] [Green Version]
- Nembrot, M.; Quintana, B.; Mordoh, J. Estrogen receptor gene amplification is found in some estrogen receptor-positive human breast tumors. Biochem. Biophys. Res. Commun. 1990, 166, 601–607. [Google Scholar] [CrossRef]
- Tomita, S.; Zhang, Z.; Nakano, M.; Ibusuki, M.; Kawazoe, T.; Yamamoto, Y.; Iwase, H. Estrogen receptor alpha gene ESR1 amplification may predict endocrine therapy responsiveness in breast cancer patients. Cancer Sci. 2009, 100, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Holst, F.; Stahl, P.R.; Ruiz, C.; Hellwinkel, O.; Jehan, Z.; Wendland, M.; Lebeau, A.; Terracciano, L.; Al-Kuraya, K.; Janicke, F.; et al. Estrogen receptor alpha (ESR1) gene amplification is frequent in breast cancer. Nat. Genet. 2007, 39, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, A.; Welnicka-Jaskiewicz, M.; Skokowski, J.; Jaskiewicz, J.; Szade, J.; Jassem, J.; Zaczek, A.J. Prognostic significance of ESR1 amplification and ESR1 PvuII, CYP2C19*2, UGT2B15*2 polymorphisms in breast cancer patients. PLoS ONE 2013, 8, e72219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holst, F.; Singer, C.F. ESR1-Amplification-Associated Estrogen Receptor alpha Activity in Breast Cancer. Trends Endocrinol. Metab. 2016, 27, 751–752. [Google Scholar] [CrossRef] [PubMed]
- Giltnane, J.M.; Hutchinson, K.E.; Stricker, T.P.; Formisano, L.; Young, C.D.; Estrada, M.V.; Nixon, M.J.; Du, L.; Sanchez, V.; Ericsson, P.G.; et al. Genomic profiling of ER(+) breast cancers after short-term estrogen suppression reveals alterations associated with endocrine resistance. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Veeraraghavan, J.; Tan, Y.; Cao, X.X.; Kim, J.A.; Wang, X.; Chamness, G.C.; Maiti, S.N.; Cooper, L.J.; Edwards, D.P.; Contreras, A.; et al. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat. Commun. 2014, 5, 4577. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.T.; Gou, X.; Seker, S.; Ellis, M.J. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. J. Cancer Metastasis Treat. 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Lin, L.; Veeraraghavan, J.; Hu, Y.; Wang, X.; Lee, S.; Tan, Y.; Schiff, R.; Wang, X.S. Therapeutic role of recurrent ESR1-CCDC170 gene fusions in breast cancer endocrine resistance. Breast Cancer Res. 2020, 22, 84. [Google Scholar] [CrossRef]
- Ross, D.S.; Liu, B.; Schram, A.M.; Razavi, P.; Lagana, S.M.; Zhang, Y.; Scaltriti, M.; Bromberg, J.F.; Ladanyi, M.; Hyman, D.M.; et al. Enrichment of kinase fusions in ESR1 wild-type, metastatic breast cancer revealed by a systematic analysis of 4854 patients. Ann. Oncol. 2020, 31, 991–1000. [Google Scholar] [CrossRef]
- Weis, K.E.; Ekena, K.; Thomas, J.A.; Lazennec, G.; Katzenellenbogen, B.S. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol. Endocrinol. 1996, 10, 1388–1398. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.X.; Borg, A.; Wolf, D.M.; Oesterreich, S.; Fuqua, S.A. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res. 1997, 57, 1244–1249. [Google Scholar] [PubMed]
- Jeselsohn, R.; Bergholz, J.S.; Pun, M.; Cornwell, M.; Liu, W.; Nardone, A.; Xiao, T.; Li, W.; Qiu, X.; Buchwalter, G.; et al. Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer Cell 2018, 33, 173–186.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yeheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Yelensky, R.; Brown, M.; Miller, V.A.; Sarid, D.; et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013, 73, 6856–6864. [Google Scholar] [CrossRef] [Green Version]
- Pavlin, M.; Spinello, A.; Pennati, M.; Zaffaroni, N.; Gobbi, S.; Bisi, A.; Colombo, G.; Magistrato, A. A Computational Assay of Estrogen Receptor alpha Antagonists Reveals the Key Common Structural Traits of Drugs Effectively Fighting Refractory Breast Cancers. Sci. Rep. 2018, 8, 649. [Google Scholar] [CrossRef] [PubMed]
- Gates, L.A.; Gu, G.; Chen, Y.; Rohira, A.D.; Lei, J.T.; Hamilton, R.A.; Yu, Y.; Lonard, D.M.; Wang, J.; Wang, S.P.; et al. Proteomic profiling identifies key coactivators utilized by mutant ERalpha proteins as potential new therapeutic targets. Oncogene 2018, 37, 4581–4598. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e6. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Levine, K.M.; Bahreini, A.; Wang, P.; Chu, D.; Park, B.H.; Oesterreich, S.; Lee, A.V. Upregulation of IRS1 Enhances IGF1 Response in Y537S and D538G ESR1 Mutant Breast Cancer Cells. Endocrinology 2018, 159, 285–296. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Bahreini, A.; Li, Z.; Wang, P.; Levine, K.M.; Tasdemir, N.; Cao, L.; Weir, H.M.; Puhalla, S.L.; Davidson, N.E.; Stern, A.M.; et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res. 2017, 19, 60. [Google Scholar] [CrossRef] [Green Version]
- Clatot, F.; Perdrix, A.; Beaussire, L.; Lequesne, J.; Levy, C.; Emile, G.; Bubenheim, M.; Lacaille, S.; Calbrix, C.; Augusto, L.; et al. Risk of early progression according to circulating ESR1 mutation, CA-15.3 and cfDNA increases under first-line anti-aromatase treatment in metastatic breast cancer. Breast Cancer Res. 2020, 22, 56. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.; Qiao, N.; Gross, B.; Meric-Bernstam, F.; Guerriero, J.; Chen, K.; Philips, A.V.; Peoples, G.E.; Alatrash, G.; Mittendorf, E.A. ESR1 mutations provide novel targets for breast cancer immunotherapy. J. Clin. Oncol. 2020, 38, 3135. [Google Scholar] [CrossRef]
- Lee, J.H.; Jeong, H.; Choi, J.W.; Oh, H.E.; Kim, Y.S. Liquid biopsy prediction of axillary lymph node metastasis, cancer recurrence, and patient survival in breast cancer: A meta-analysis. Med. Baltim. 2018, 97, e12862. [Google Scholar] [CrossRef] [PubMed]
- Cornen, S.; Guille, A.; Adelaide, J.; Addou-Klouche, L.; Finetti, P.; Saade, M.R.; Manai, M.; Carbuccia, N.; Bekhouche, I.; Letessier, A.; et al. Candidate luminal B breast cancer genes identified by genome, gene expression and DNA methylation profiling. PLoS ONE 2014, 9, e81843. [Google Scholar] [CrossRef] [PubMed]
- McDonough, S.J.; Bhagwate, A.; Sun, Z.; Wang, C.; Zschunke, M.; Gorman, J.A.; Kopp, K.J.; Cunningham, J.M. Use of FFPE-derived DNA in next generation sequencing: DNA extraction methods. PLoS ONE 2019, 14, e0211400. [Google Scholar] [CrossRef] [Green Version]
- Oh, E.; Choi, Y.L.; Kwon, M.J.; Kim, R.N.; Kim, Y.J.; Song, J.Y.; Jung, K.S.; Shin, Y.K. Comparison of Accuracy of Whole-Exome Sequencing with Formalin-Fixed Paraffin-Embedded and Fresh Frozen Tissue Samples. PLoS ONE 2015, 10, e0144162. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Q.; Li, J.; Tan, A.Y.; Vedururu, R.; Pang, J.M.; Do, H.; Ellul, J.; Doig, K.; Bell, A.; MacArthur, G.A.; et al. Sequence artefacts in a prospective series of formalin-fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med. Genom. 2014, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Greytak, S.R.; Engel, K.B.; Zmuda, E.; Casas-Silva, E.; Guan, P.; Hoadley, K.A.; Mungall, A.J.; Wheeler, D.A.; Doddapaneni, H.V.; Moore, H.M. National Cancer Institute Biospecimen Evidence-Based Practices: Harmonizing Procedures for Nucleic Acid Extraction from Formalin-Fixed, Paraffin-Embedded Tissue. Biopreserv. Biobank. 2018, 16, 247–250. [Google Scholar] [CrossRef]
- Kofanova, O.; Bellora, C.; Garcia Frasquilho, S.; Antunes, L.; Hamot, G.; Mathay, C.; Mommaerts, K.; Muller, A.; DeWitt, B.; Betsou, F. Standardization of the preanalytical phase of DNA extraction from fixed tissue for next-generation sequencing analyses. N. Biotechnol. 2020, 54, 52–61. [Google Scholar] [CrossRef]
- Ivanov, M.; Laktionov, K.; Breder, V.; Chernenko, P.; Novikova, E.; Telysheva, E.; Musienko, S.; Baranova, A.; Mileyko, V. Towards standardization of next-generation sequencing of FFPE samples for clinical oncology: Intrinsic obstacles and possible solutions. J. Transl. Med. 2017, 15, 22. [Google Scholar] [CrossRef] [Green Version]
- Tozaki, T.; Ohnuma, A.; Takasu, M.; Kikuchi, M.; Kakoi, H.; Hirota, K.I.; Kusano, K.; Nagata, S.I. Droplet Digital PCR Detection of the Erythropoietin Transgene from Horse Plasma and Urine for Gene-Doping Control. Genes 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ginkel, J.H.; van den Broek, D.A.; van Kuik, J.; Linders, D.; de Weger, R.; Willems, S.M.; Huibers, M.M.H. Preanalytical blood sample workup for cell-free DNA analysis using Droplet Digital PCR for future molecular cancer diagnostics. Cancer Med. 2017, 6, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Grolz, D.; Hauch, S.; Schlumpberger, M.; Guenther, K.; Voss, T.; Sprenger-Haussels, M.; Oelmuller, U. Liquid Biopsy Preservation Solutions for Standardized Pre-Analytical Workflows-Venous Whole Blood and Plasma. Curr. Pathobiol. Rep. 2018, 6, 275–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, T.; Taschner-Mandl, S.; Saloberger-Sindhoringer, L.; Popitsch, N.; Heitzer, E.; Witt, V.; Geyeregger, R.; Hutter, C.; Schwentner, R.; Ambros, I.M.; et al. Assessment of Pre-Analytical Sample Handling Conditions for Comprehensive Liquid Biopsy Analysis. J. Mol. Diagn. 2020, 22, 1070–1086. [Google Scholar] [CrossRef]
- Kang, Q.; Henry, N.L.; Paoletti, C.; Jiang, H.; Vats, P.; Chinnaiyan, A.M.; Hayes, D.F.; Merajver, S.D.; Rae, J.M.; Tewari, M. Comparative analysis of circulating tumor DNA stability In K3EDTA, Streck, and CellSave blood collection tubes. Clin. Biochem. 2016, 49, 1354–1360. [Google Scholar] [CrossRef]
- Medina Diaz, I.; Nocon, A.; Mehnert, D.H.; Fredebohm, J.; Diehl, F.; Holtrup, F. Performance of Streck cfDNA Blood Collection Tubes for Liquid Biopsy Testing. PLoS ONE 2016, 11, e0166354. [Google Scholar] [CrossRef]
- Bartels, S.; Christgen, M.; Luft, A.; Persing, S.; Jodecke, K.; Lehmann, U.; Kreipe, H. Estrogen receptor (ESR1) mutation in bone metastases from breast cancer. Mod. Pathol. 2018, 31, 56–61. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Voi, M.; Gnant, M.; et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncol. 2016, 2, 1310–1315. [Google Scholar] [CrossRef] [Green Version]
- Chu, D.; Paoletti, C.; Gersch, C.; VanDenBerg, D.A.; Zabransky, D.J.; Cochran, R.L.; Wong, H.Y.; Toro, P.V.; Cidado, J.; Croessmann, S.; et al. ESR1 Mutations in Circulating Plasma Tumor DNA from Metastatic Breast Cancer Patients. Clin. Cancer Res. 2016, 22, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Clatot, F.; Perdrix, A.; Augusto, L.; Beaussire, L.; Delacour, J.; Calbrix, C.; Sefrioui, D.; Viailly, P.J.; Bubenheim, M.; Moldovan, C.; et al. Kinetics, prognostic and predictive values of ESR1 circulating mutations in metastatic breast cancer patients progressing on aromatase inhibitor. Oncotarget 2016, 7, 74448–74459. [Google Scholar] [CrossRef] [Green Version]
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. J. Clin. Oncol. 2016, 34, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Schiavon, G.; Hrebien, S.; Garcia-Murillas, I.; Cutts, R.J.; Pearson, A.; Tarazona, N.; Fenwick, K.; Kozarewa, I.; Lopez-Knowles, E.; Ribas, R.; et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci. Transl. Med. 2015, 7, 313ra182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spoerke, J.M.; Gendreau, S.; Walter, K.; Qiu, J.; Wilson, T.R.; Savage, H.; Aimi, J.; Derynck, M.K.; Chen, M.; Chan, I.T.; et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat. Commun. 2016, 7, 11579. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, T.; Yamamoto, Y.; Yamamoto-Ibusuki, M.; Tomiguchi, M.; Sueta, A.; Murakami, K.; Omoto, Y.; Iwase, H. Analysis of ESR1 and PIK3CA mutations in plasma cell-free DNA from ER-positive breast cancer patients. Oncotarget 2017, 8, 52142–52155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Bahreini, A.; Gyanchandani, R.; Lucas, P.C.; Hartmaier, R.J.; Watters, R.J.; Jonnalagadda, A.R.; Trejo Bittar, H.E.; Berg, A.; Hamilton, R.L.; et al. Sensitive Detection of Mono- and Polyclonal ESR1 Mutations in Primary Tumors, Metastatic Lesions, and Cell-Free DNA of Breast Cancer Patients. Clin. Cancer Res. 2016, 22, 1130–1137. [Google Scholar] [CrossRef] [Green Version]
- Yanagawa, T.; Kagara, N.; Miyake, T.; Tanei, T.; Naoi, Y.; Shimoda, M.; Shimazu, K.; Kim, S.J.; Noguchi, S. Detection of ESR1 mutations in plasma and tumors from metastatic breast cancer patients using next-generation sequencing. Breast Cancer Res. Treat. 2017, 163, 231–240. [Google Scholar] [CrossRef]
- Gyanchandani, R.; Kota, K.J.; Jonnalagadda, A.R.; Minteer, T.; Knapick, B.A.; Oesterreich, S.; Brufsky, A.M.; Lee, A.V.; Puhalla, S.L. Detection of ESR1 mutations in circulating cell-free DNA from patients with metastatic breast cancer treated with palbociclib and letrozole. Oncotarget 2017, 8, 66901–66911. [Google Scholar] [CrossRef] [Green Version]
- Laig, M.; Fekete, C.; Majumdar, N. Digital PCR and the QuantStudio 3D Digital PCR System. Methods Mol. Biol. 2020, 2065, 209–231. [Google Scholar] [CrossRef]
- Head, S.R.; Komori, H.K.; LaMere, S.A.; Whisenant, T.; Van Nieuwerburgh, F.; Salomon, D.R.; Ordoukhanian, P. Library construction for next-generation sequencing: Overviews and challenges. Biotechniques 2014, 56, 61–64, 66, 68, passim. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Thermes, C. Library preparation methods for next-generation sequencing: Tone down the bias. Exp. Cell Res. 2014, 322, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.; Oliveira, J.; Sousa, M. Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. J. Clin. Med. 2020, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakharkar, M.K.; Chow, V.T.; Kangueane, P. Distributions of exons and introns in the human genome. Silico Biol. 2004, 4, 387–393. [Google Scholar]
- Hung, S.S.; Meissner, B.; Chavez, E.A.; Ben-Neriah, S.; Ennishi, D.; Jones, M.R.; Shulha, H.P.; Chan, F.C.; Boyle, M.; Kridel, R.; et al. Assessment of Capture and Amplicon-Based Approaches for the Development of a Targeted Next-Generation Sequencing Pipeline to Personalize Lymphoma Management. J. Mol. Diagn. 2018, 20, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Samorodnitsky, E.; Jewell, B.M.; Hagopian, R.; Miya, J.; Wing, M.R.; Lyon, E.; Damodaran, S.; Bhatt, D.; Reeser, J.W.; Datta, J.; et al. Evaluation of Hybridization Capture Versus Amplicon-Based Methods for Whole-Exome Sequencing. Hum. Mutat. 2015, 36, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Wu, P.H.; Beane, T.; Zamore, P.D.; Weng, Z. Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers. BMC Genom. 2018, 19, 531. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Gresham, D. Incorporation of unique molecular identifiers in TruSeq adapters improves the accuracy of quantitative sequencing. Biotechniques 2017, 63, 221–226. [Google Scholar] [CrossRef] [Green Version]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef]
- Fuller, C.W.; Middendorf, L.R.; Benner, S.A.; Church, G.M.; Harris, T.; Huang, X.; Jovanovich, S.B.; Nelson, J.R.; Schloss, J.A.; Schwartz, D.C.; et al. The challenges of sequencing by synthesis. Nat. Biotechnol. 2009, 27, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Heyn, P.; Kelso, J. Addressing challenges in the production and analysis of illumina sequencing data. BMC Genom. 2011, 12, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Xu, F.; Wu, J.; Schubert, J.; Li, M.M. Application of Next Generation Sequencing in Laboratory Medicine. Ann. Lab. Med. 2021, 41, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Desmedt, C.; Voet, T.; Sotiriou, C.; Campbell, P.J. Next-generation sequencing in breast cancer: First take home messages. Curr. Opin. Oncol. 2012, 24, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Dressman, D.; Yan, H.; Traverso, G.; Kinzler, K.W.; Vogelstein, B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl. Acad. Sci. USA 2003, 100, 8817–8822. [Google Scholar] [CrossRef] [Green Version]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef]
- Merriman, B.; Ion Torrent, R.; Team, D.; Rothberg, J.M. Progress in ion torrent semiconductor chip based sequencing. Electrophoresis 2012, 33, 3397–3417. [Google Scholar] [CrossRef]
- Boland, J.F.; Chung, C.C.; Roberson, D.; Mitchell, J.; Zhang, X.; Im, K.M.; He, J.; Chanock, S.J.; Yeager, M.; Dean, M. The new sequencer on the block: Comparison of Life Technology’s Proton sequencer to an Illumina HiSeq for whole-exome sequencing. Hum. Genet. 2013, 132, 1153–1163. [Google Scholar] [CrossRef] [Green Version]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.11–11.10.33. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Su, X.; Wang, A.; Xu, J.; Ning, K. QC-Chain: Fast and holistic quality control method for next-generation sequencing data. PLoS ONE 2013, 8, e60234. [Google Scholar] [CrossRef] [PubMed]
- Del Fabbro, C.; Scalabrin, S.; Morgante, M.; Giorgi, F.M. An extensive evaluation of read trimming effects on Illumina NGS data analysis. PLoS ONE 2013, 8, e85024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flicek, P.; Birney, E. Sense from sequence reads: Methods for alignment and assembly. Nat. Methods 2009, 6, S6–S12. [Google Scholar] [CrossRef]
- Fonseca, N.A.; Rung, J.; Brazma, A.; Marioni, J.C. Tools for mapping high-throughput sequencing data. Bioinformatics 2012, 28, 3169–3177. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.R.; Koren, S.; Sutton, G. Assembly algorithms for next-generation sequencing data. Genomics 2010, 95, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Sedlazeck, F.J.; Lee, H.; Darby, C.A.; Schatz, M.C. Piercing the dark matter: Bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet. 2018, 19, 329–346. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.; Kim, E.; Lee, I.; Marcotte, E.M. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci. Rep. 2015, 5, 17875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Perez, A.; Lopez-Bigas, N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 2011, 88, 440–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International HapMap, C.; Altshuler, D.M.; Gibbs, R.A.; Peltonen, L.; Altshuler, D.M.; Gibbs, R.A.; Peltonen, L.; Dermitzakis, E.; Schaffner, S.F.; Yu, F.; et al. Integrating common and rare genetic variation in diverse human populations. Nature 2010, 467, 52–58. [Google Scholar] [CrossRef]
- Siva, N. 1000 Genomes project. Nat. Biotechnol. 2008, 26, 256. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Harper, P.S. The European Society of Human Genetics: Beginnings, early history and development over its first 25 years. Eur. J. Hum. Genet. 2017. [Google Scholar] [CrossRef] [Green Version]
- Schwarze, K.; Buchanan, J.; Taylor, J.C.; Wordsworth, S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet. Med. 2018, 20, 1122–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, E.; Dear, P.H.; McCaughan, F. Digital PCR strategies in the development and analysis of molecular biomarkers for personalized medicine. Methods 2013, 59, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Shen, S.; Jiang, H.; Chen, Z. Application of Digital PCR in Detecting Human Diseases Associated Gene Mutation. Cell. Physiol. Biochem. 2017, 43, 1718–1730. [Google Scholar] [CrossRef] [PubMed]
- Kanagal-Shamanna, R. Digital PCR: Principles and Applications. Methods Mol. Biol. 2016, 1392, 43–50. [Google Scholar] [CrossRef]
- Mao, X.; Liu, C.; Tong, H.; Chen, Y.; Liu, K. Principles of digital PCR and its applications in current obstetrical and gynecological diseases. Am. J. Transl. Res. 2019, 11, 7209–7222. [Google Scholar]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [Green Version]
- Besser, J.; Carleton, H.A.; Gerner-Smidt, P.; Lindsey, R.L.; Trees, E. Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clin. Microbiol. Infect. 2018, 24, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [Green Version]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef] [Green Version]
- Andergassen, U.; Kolbl, A.C.; Mahner, S.; Jeschke, U. Real-time RT-PCR systems for CTC detection from blood samples of breast cancer and gynaecological tumour patients (Review). Oncol. Rep. 2016, 35, 1905–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.S.; Tsai, C.L.; Chang, J.; Hsu, T.C.; Lin, S.; Lee, C.C. Multiplex PCR system for the rapid diagnosis of respiratory virus infection: Systematic review and meta-analysis. Clin. Microbiol. Infect. 2018, 24, 1055–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeselsohn, R.; Buchwalter, G.; De Angelis, C.; Brown, M.; Schiff, R. ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapino, A.; Roepman, P.; Linn, S.C.; Snel, M.H.; Delahaye, L.J.; van den Akker, J.; Glas, A.M.; Simon, I.M.; Barth, N.; de Snoo, F.A.; et al. MammaPrint molecular diagnostics on formalin-fixed, paraffin-embedded tissue. J. Mol. Diagn. 2014, 16, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.; Edmiston, S.N.; May, R.; Kuan, P.F.; Chu, H.; Bryant, C.; Tse, C.K.; Swift-Scanlan, T.; Geradts, J.; Troester, M.A.; et al. DNA methylation profiling in the Carolina Breast Cancer Study defines cancer subclasses differing in clinicopathologic characteristics and survival. Breast Cancer Res. 2014, 16, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, X.; Guo, Y.; Lu, Y. Prognostic role of methylated GSTP1, p16, ESR1 and PITX2 in patients with breast cancer: A systematic meta-analysis under the guideline of PRISMA. Med. Baltim. 2017, 96, e7476. [Google Scholar] [CrossRef] [PubMed]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Danenberg, P.V.; Laird, P.W. MethyLight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28, E32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widschwendter, M.; Siegmund, K.D.; Muller, H.M.; Fiegl, H.; Marth, C.; Muller-Holzner, E.; Jones, P.A.; Laird, P.W. Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res. 2004, 64, 3807–3813. [Google Scholar] [CrossRef] [Green Version]
- Ramos, E.A.; Camargo, A.A.; Braun, K.; Slowik, R.; Cavalli, I.J.; Ribeiro, E.M.; Pedrosa Fde, O.; de Souza, E.M.; Costa, F.F.; Klassen, G. Simultaneous CXCL12 and ESR1 CpG island hypermethylation correlates with poor prognosis in sporadic breast cancer. BMC Cancer 2010, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Sharma, G.; Mirza, S.; Yang, Y.H.; Parshad, R.; Hazrah, P.; Datta Gupta, S.; Ralhan, R. Prognostic relevance of promoter hypermethylation of multiple genes in breast cancer patients. Cell. Oncol. 2009, 31, 487–500. [Google Scholar] [CrossRef]
- Berlin, K.; Koren, S.; Chin, C.S.; Drake, J.P.; Landolin, J.M.; Phillippy, A.M. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 2015, 33, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Ferrarini, M.; Moretto, M.; Ward, J.A.; Surbanovski, N.; Stevanovic, V.; Giongo, L.; Viola, R.; Cavalieri, D.; Velasco, R.; Cestaro, A.; et al. An evaluation of the PacBio RS platform for sequencing and de novo assembly of a chloroplast genome. BMC Genom. 2013, 14, 670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Deamer, D.; Akeson, M.; Branton, D. Three decades of nanopore sequencing. Nat. Biotechnol. 2016, 34, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.L.; Afshar, P.T.; Clark, T.A.; Tseng, E.; Powers, L.S.; Underwood, J.G.; Zabner, J.; Korlach, J.; Wong, W.H.; Au, K.F. Characterization of fusion genes and the significantly expressed fusion isoforms in breast cancer by hybrid sequencing. Nucleic Acids Res. 2015, 43, e116. [Google Scholar] [CrossRef] [Green Version]
- Elazezy, M.; Joosse, S.A. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- O’Leary, B.; Hrebien, S.; Beaney, M.; Fribbens, C.; Garcia-Murillas, I.; Jiang, J.; Li, Y.; Huang Bartlett, C.; Andre, F.; Loibl, S.; et al. Comparison of BEAMing and Droplet Digital PCR for Circulating Tumor DNA Analysis. Clin. Chem. 2019, 65, 1405–1413. [Google Scholar] [CrossRef]
- Cristofanilli, M.; DeMichele, A.; Giorgetti, C.; Turner, N.C.; Slamon, D.J.; Im, S.A.; Masuda, N.; Verma, S.; Loi, S.; Colleoni, M.; et al. Predictors of prolonged benefit from palbociclib plus fulvestrant in women with endocrine-resistant hormone receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer in PALOMA-3. Eur. J. Cancer 2018, 104, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Lupini, L.; Moretti, A.; Bassi, C.; Schirone, A.; Pedriali, M.; Querzoli, P.; Roncarati, R.; Frassoldati, A.; Negrini, M. High-sensitivity assay for monitoring ESR1 mutations in circulating cell-free DNA of breast cancer patients receiving endocrine therapy. Sci. Rep. 2018, 8, 4371. [Google Scholar] [CrossRef]
- Shatsky, R.; Parker, B.A.; Bui, N.Q.; Helsten, T.; Schwab, R.B.; Boles, S.G.; Kurzrock, R. Next-Generation Sequencing of Tissue and Circulating Tumor DNA: The UC San Diego Moores Center for Personalized Cancer Therapy Experience with Breast Malignancies. Mol. Cancer Ther. 2019, 18, 1001–1011. [Google Scholar] [CrossRef] [Green Version]

| Detection Method | Principle | Advantage | Limitation |
|---|---|---|---|
| NGS platforms |
|
|
|
| Illumina |
|
|
|
| Ion Torrent |
|
|
|
| ddPCR |
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, N.; Park, M.-J.; Song, W.; Jeon, K.; Jeong, S. Currently Applied Molecular Assays for Identifying ESR1 Mutations in Patients with Advanced Breast Cancer. Int. J. Mol. Sci. 2020, 21, 8807. https://doi.org/10.3390/ijms21228807
Lee N, Park M-J, Song W, Jeon K, Jeong S. Currently Applied Molecular Assays for Identifying ESR1 Mutations in Patients with Advanced Breast Cancer. International Journal of Molecular Sciences. 2020; 21(22):8807. https://doi.org/10.3390/ijms21228807
Chicago/Turabian StyleLee, Nuri, Min-Jeong Park, Wonkeun Song, Kibum Jeon, and Seri Jeong. 2020. "Currently Applied Molecular Assays for Identifying ESR1 Mutations in Patients with Advanced Breast Cancer" International Journal of Molecular Sciences 21, no. 22: 8807. https://doi.org/10.3390/ijms21228807
APA StyleLee, N., Park, M.-J., Song, W., Jeon, K., & Jeong, S. (2020). Currently Applied Molecular Assays for Identifying ESR1 Mutations in Patients with Advanced Breast Cancer. International Journal of Molecular Sciences, 21(22), 8807. https://doi.org/10.3390/ijms21228807