Mechanistic Insights into the Allosteric Regulation of the Clr4 Protein Lysine Methyltransferase by Autoinhibition and Automethylation



Abstract
1. Introduction
2. Results
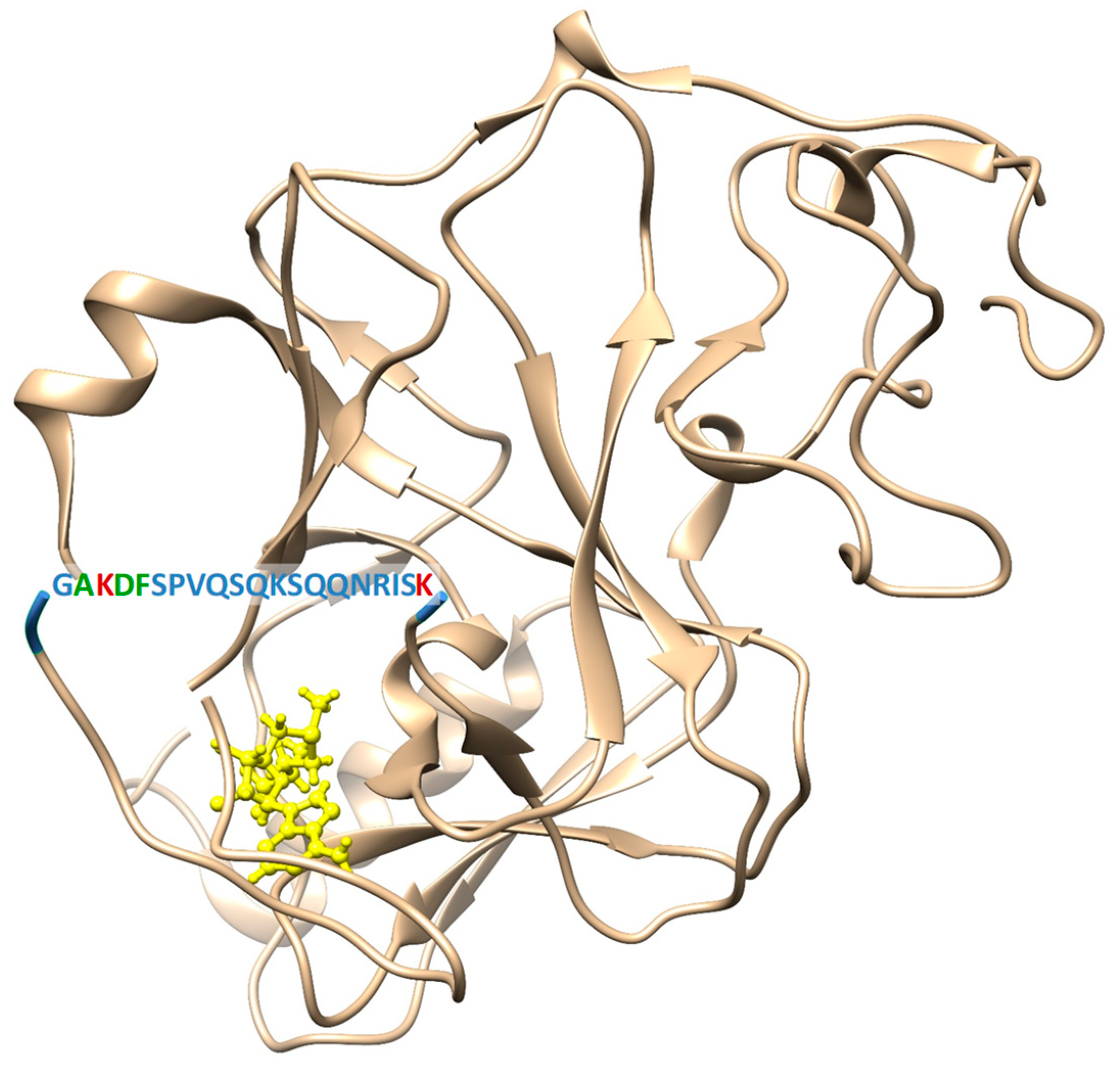
2.1. Site-Directed Mutagenesis, Clr4 Expression and Purification
2.2. Effect of the Clr4 Mutations on Automethylation and Methyltransferase Activity
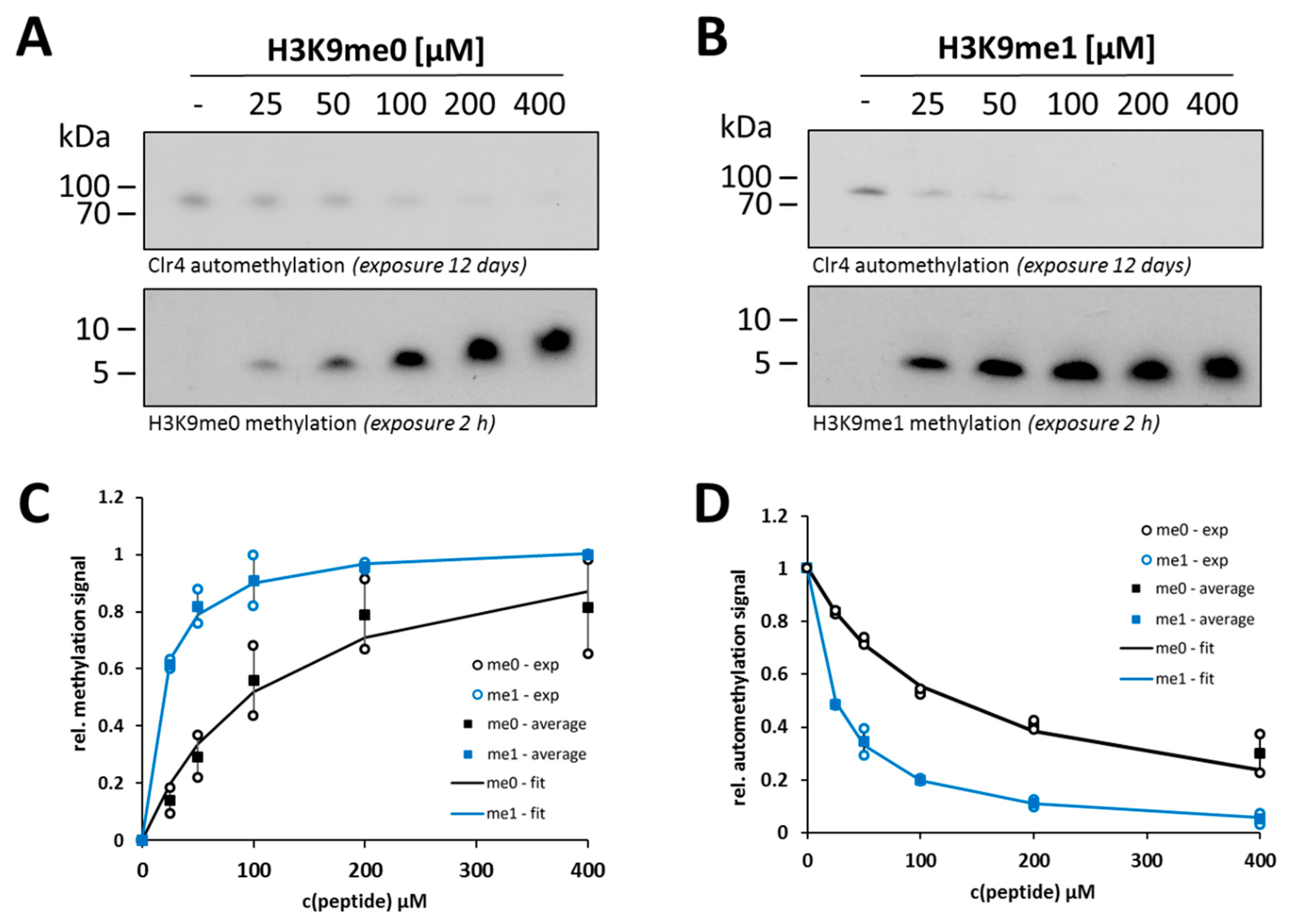
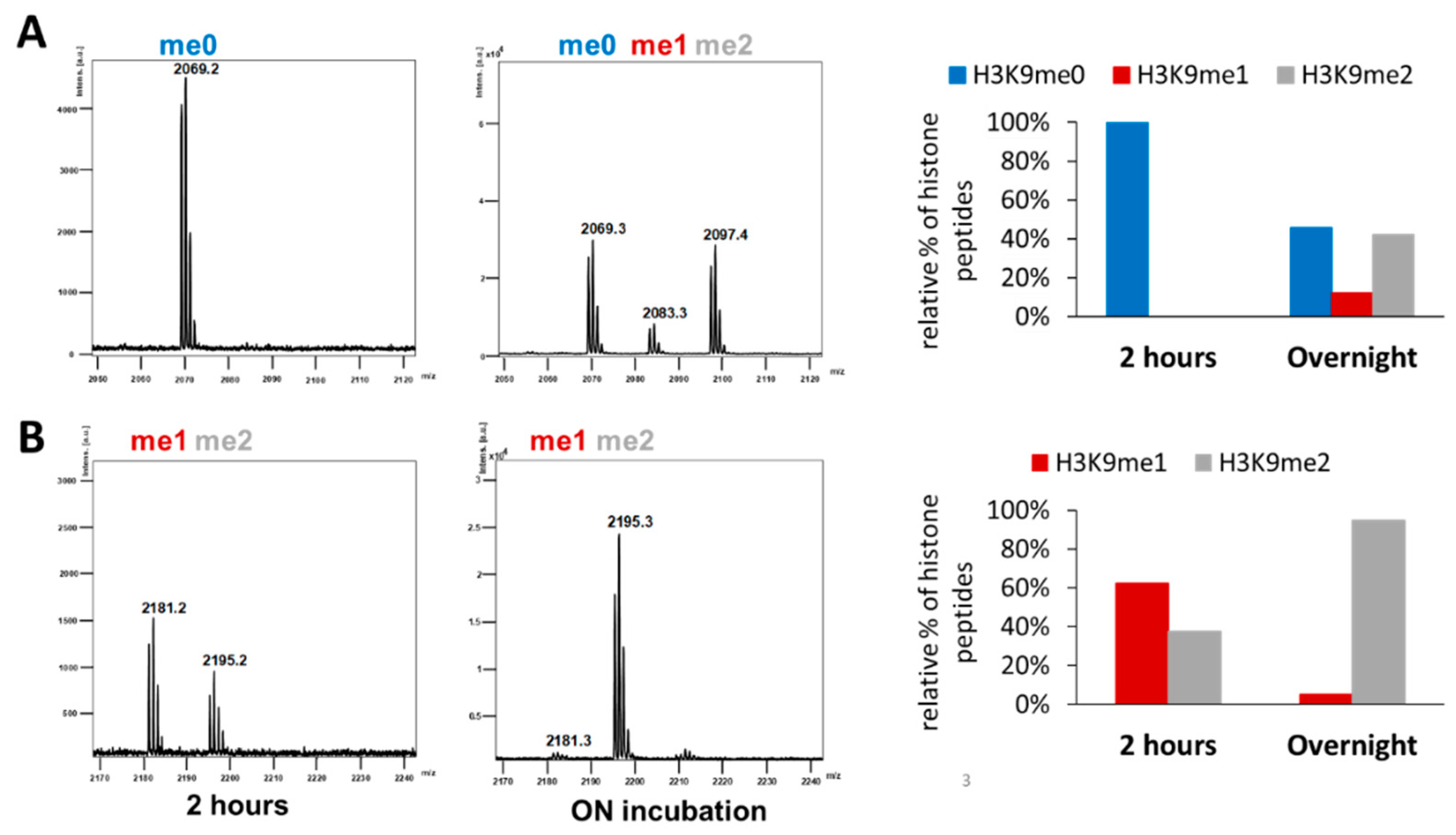
2.3. Clr4 Prefers H3K9me1 over H3K9me0 as Substrate and Its Automethylation Is More Strongly Inhibited by H3K9me1 than by H3K9me0
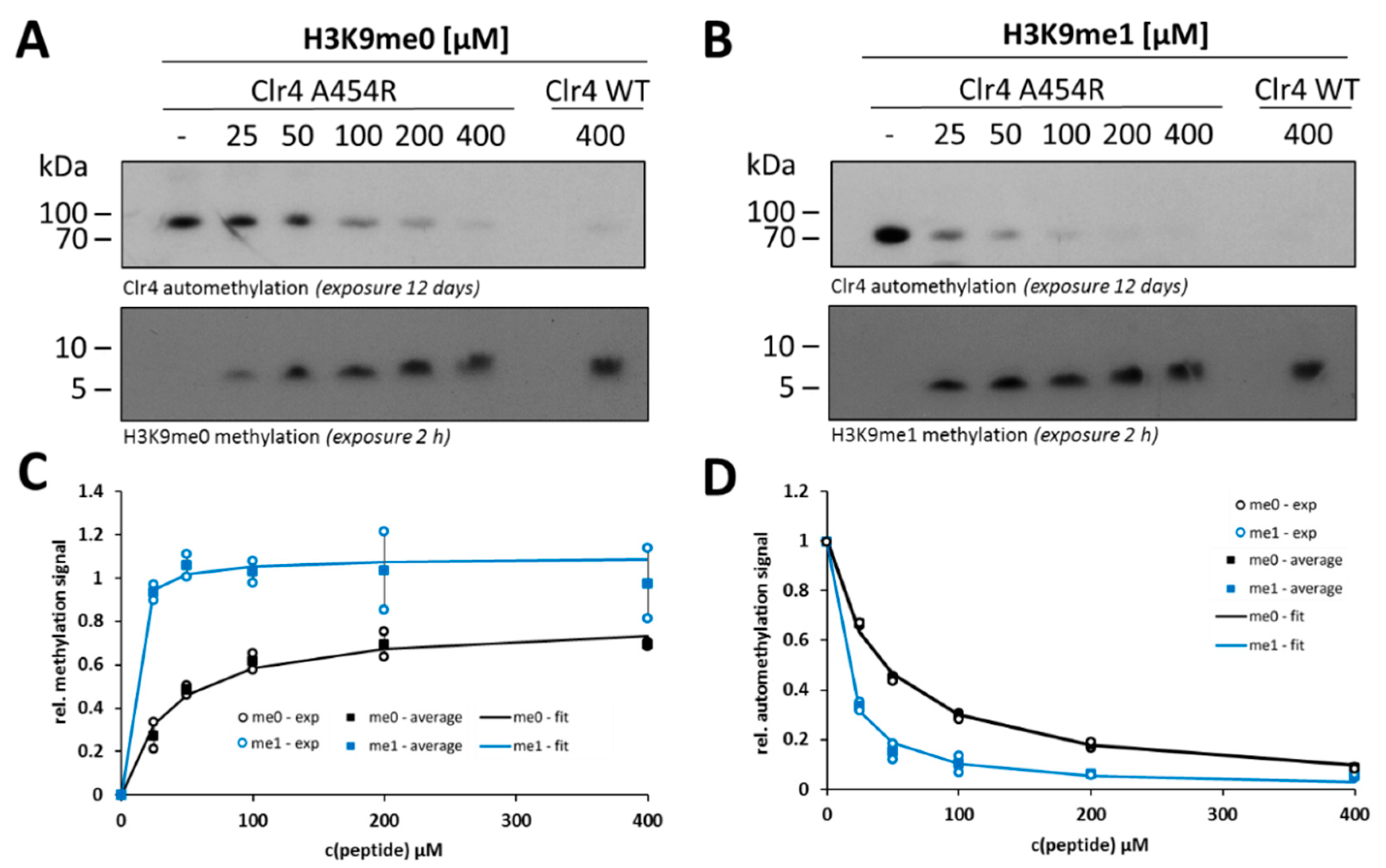
2.4. Steady-State Kinetics with the Clr4 A454R Mutant Show Increased Automethylation and Less Autoinhibition
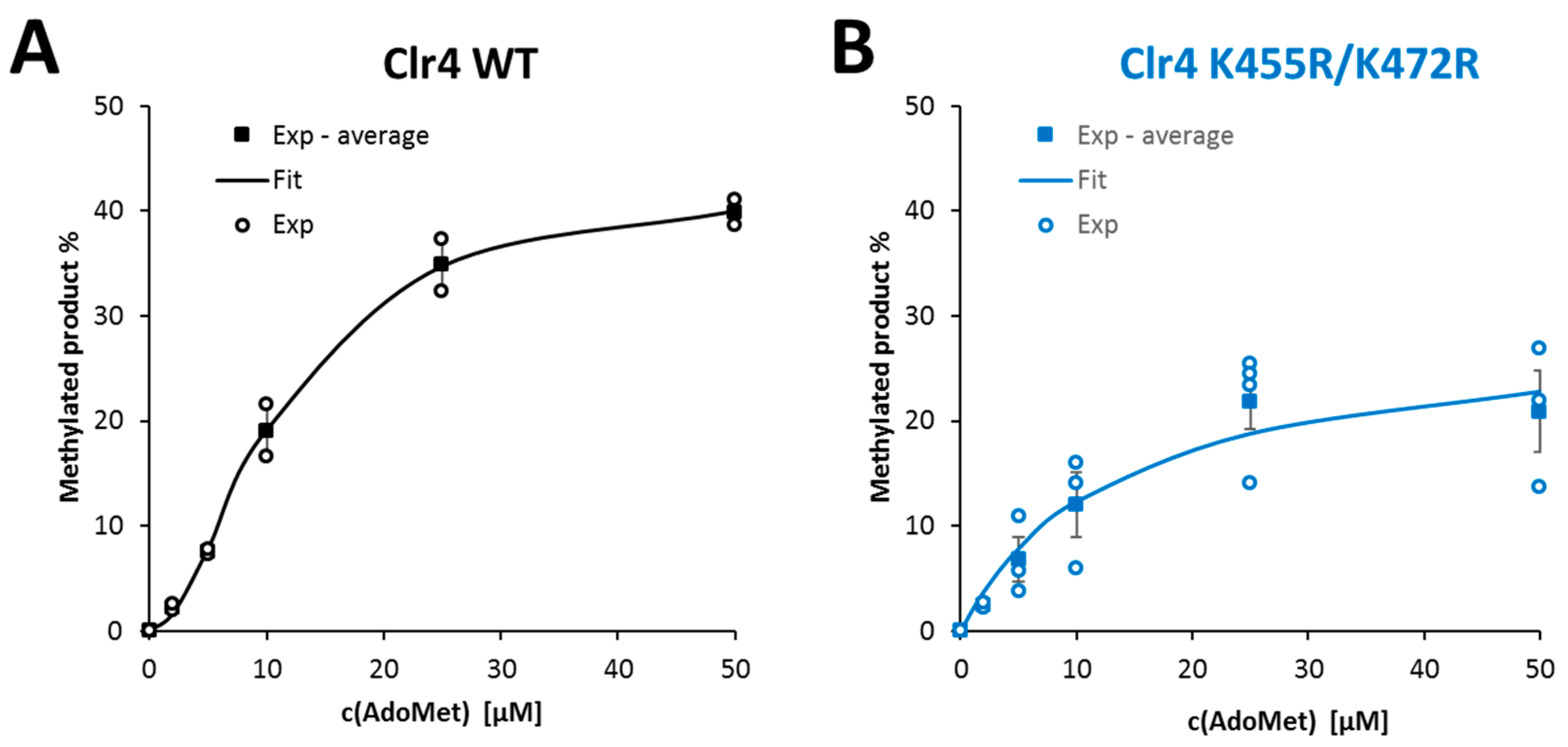
2.5. Clr4 WT Reaction Rate Exhibited Sigmoidal Response towards Increasing AdoMet Concentrations
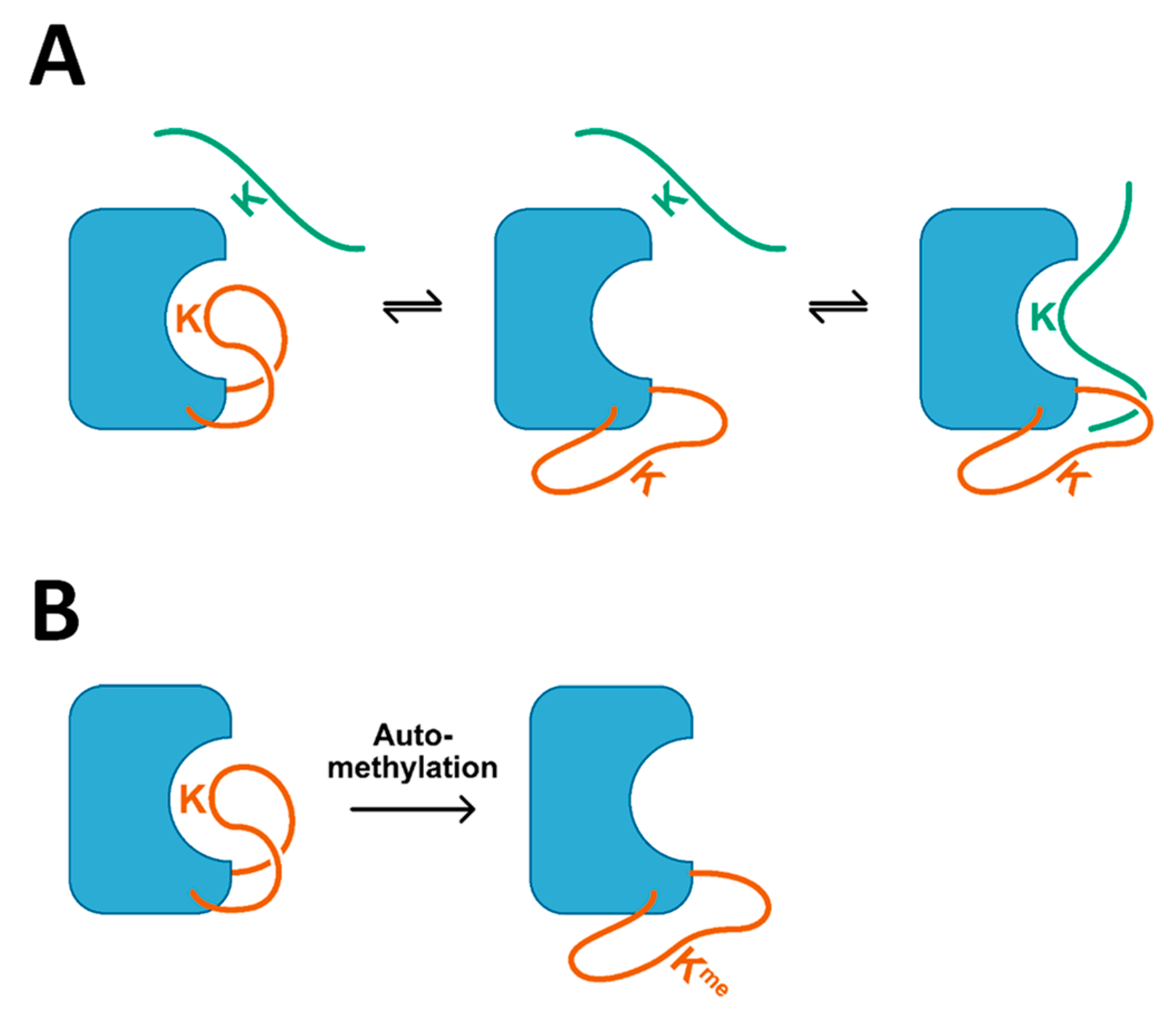
3. Discussion
4. Materials and Methods
4.1. Site-Directed Mutagenesis, Protein Expression and Purification
4.2. Peptide In Vitro Methylation Activity Assay
4.3. Km Determination of H3K9me0 and H3K9me1 Substrates
4.4. Km Determination of AdoMet Using MALDI-TOF Mass Spectrometry
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J., Jr. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. 2005, 44, 7342–7372. [Google Scholar] [CrossRef]
- Murn, J.; Shi, Y. The winding path of protein methylation research: Milestones and new frontiers. Nat. Rev. Mol. Cell Biol. 2017, 18, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Lin, S.; Garcia, B.A.; Zhao, Y. Quantitative proteomic analysis of histone modifications. Chem. Rev. 2015, 115, 2376–2418. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.C.; Maze, I. Nothing Is yet Set in (Hi)stone: Novel Post-Translational Modifications Regulating Chromatin Function. Trends Biochem. Sci. 2020. [Google Scholar] [CrossRef]
- Biggar, K.K.; Li, S.S. Non-histone protein methylation as a regulator of cellular signalling and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications—Miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Becker, J.S.; Nicetto, D.; Zaret, K.S. H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet. TIG 2016, 32, 29–41. [Google Scholar] [CrossRef]
- Janssen, A.; Colmenares, S.U.; Karpen, G.H. Heterochromatin: Guardian of the Genome. Annu. Rev. Cell Dev. Biol. 2018, 34, 265–288. [Google Scholar] [CrossRef]
- Nakayama, J.; Rice, J.C.; Strahl, B.D.; Allis, C.D.; Grewal, S.I. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 2001, 292, 110–113. [Google Scholar] [CrossRef]
- Min, J.; Zhang, X.; Cheng, X.; Grewal, S.I.; Xu, R.M. Structure of the SET domain histone lysine methyltransferase Clr4. Nat. Struct. Biol. 2002, 9, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Wang, X.; Moazed, D. Epigenetic inheritance mediated by coupling of RNAi and histone H3K9 methylation. Nature 2018, 558, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Jih, G.; Iglesias, N.; Currie, M.A.; Bhanu, N.V.; Paulo, J.A.; Gygi, S.P.; Garcia, B.A.; Moazed, D. Unique roles for histone H3K9me states in RNAi and heritable silencing of transcription. Nature 2017, 547, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Mosch, K.; Fischle, W.; Grewal, S.I. Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat. Struct. Mol. Biol. 2008, 15, 381–388. [Google Scholar] [CrossRef]
- Cheng, X.; Collins, R.E.; Zhang, X. Structural and sequence motifs of protein (histone) methylation enzymes. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 267–294. [Google Scholar] [CrossRef]
- Akoury, E.; Ma, G.; Demolin, S.; Bronner, C.; Zocco, M.; Cirilo, A.; Ivic, N.; Halic, M. Disordered region of H3K9 methyltransferase Clr4 binds the nucleosome and contributes to its activity. Nucleic Acids Res. 2019, 47, 6726–6736. [Google Scholar] [CrossRef]
- Al-Sady, B.; Madhani, H.D.; Narlikar, G.J. Division of labor between the chromodomains of HP1 and Suv39 methylase enables coordination of heterochromatin spread. Mol. Cell 2013, 51, 80–91. [Google Scholar] [CrossRef]
- Haldar, S.; Saini, A.; Nanda, J.S.; Saini, S.; Singh, J. Role of Swi6/HP1 self-association-mediated recruitment of Clr4/Suv39 in establishment and maintenance of heterochromatin in fission yeast. J. Biol. Chem. 2011, 286, 9308–9320. [Google Scholar] [CrossRef]
- Lewis, P.W.; Muller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef]
- Chan, K.M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef]
- Justin, N.; Zhang, Y.; Tarricone, C.; Martin, S.R.; Chen, S.; Underwood, E.; De Marco, V.; Haire, L.F.; Walker, P.A.; Reinberg, D.; et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 2016, 7, 11316. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, H.; Hoelper, D.; Jain, S.U.; Cantone, N.; Lundgren, S.M.; Poy, F.; Allis, C.D.; Cummings, R.; Bellon, S.; Lewis, P.W. S-adenosyl methionine is necessary for inhibition of the methyltransferase G9a by the lysine 9 to methionine mutation on histone H3. Proc. Natl. Acad. Sci. USA 2016, 113, 6182–6187. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R.; et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016, 352, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zheng, X.; Lu, C.; Li, G.M.; Allis, C.D.; Li, H. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev. 2016, 30, 1611–1616. [Google Scholar] [CrossRef]
- Schuhmacher, M.; Kusevic, D.; Kudithipudi, S.; Jeltsch, A. Kinetic Analysis of the Inhibition of the NSD1, NSD2 and SETD2 Protein Lysine Methyltransferases by a K36M Oncohistone Peptide. Chemistryselect 2017, 2, 9532–9536. [Google Scholar] [CrossRef]
- Shan, C.M.; Wang, J.; Xu, K.; Chen, H.; Yue, J.X.; Andrews, S.; Moresco, J.J.; Yates, J.R.; Nagy, P.L.; Tong, L.; et al. A histone H3K9M mutation traps histone methyltransferase Clr4 to prevent heterochromatin spreading. eLife 2016, 5. [Google Scholar] [CrossRef]
- Bayne, E.H.; White, S.A.; Kagansky, A.; Bijos, D.A.; Sanchez-Pulido, L.; Hoe, K.L.; Kim, D.U.; Park, H.O.; Ponting, C.P.; Rappsilber, J.; et al. Stc1: A critical link between RNAi and chromatin modification required for heterochromatin integrity. Cell 2010, 140, 666–677. [Google Scholar] [CrossRef]
- Rathert, P.; Dhayalan, A.; Murakami, M.; Zhang, X.; Tamas, R.; Jurkowska, R.; Komatsu, Y.; Shinkai, Y.; Cheng, X.; Jeltsch, A. Protein lysine methyltransferase G9a acts on non-histone targets. Nat. Chem. Biol. 2008, 4, 344–346. [Google Scholar] [CrossRef]
- Dhayalan, A.; Kudithipudi, S.; Rathert, P.; Jeltsch, A. Specificity analysis-based identification of new methylation targets of the SET7/9 protein lysine methyltransferase. Chem. Biol. 2011, 18, 111–120. [Google Scholar] [CrossRef]
- Levy, D.; Liu, C.L.; Yang, Z.; Newman, A.M.; Alizadeh, A.A.; Utz, P.J.; Gozani, O. A proteomic approach for the identification of novel lysine methyltransferase substrates. Epigenet. Chromatin 2011, 4, 19. [Google Scholar] [CrossRef]
- Kudithipudi, S.; Dhayalan, A.; Kebede, A.F.; Jeltsch, A. The SET8 H4K20 protein lysine methyltransferase has a long recognition sequence covering seven amino acid residues. Biochimie 2012, 94, 2212–2218. [Google Scholar] [CrossRef] [PubMed]
- Kudithipudi, S.; Kusevic, D.; Weirich, S.; Jeltsch, A. Specificity analysis of protein lysine methyltransferases using SPOT peptide arrays. J. Vis. Exp. 2014, e52203. [Google Scholar] [CrossRef] [PubMed]
- Schuhmacher, M.K.; Kudithipudi, S.; Kusevic, D.; Weirich, S.; Jeltsch, A. Activity and specificity of the human SUV39H2 protein lysine methyltransferase. Biochim. Biophys. Acta 2015, 1849, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Duan, S.; Arrowsmith, C.H.; Barsyte-Lovejoy, D.; Schapira, M. An Integrative Proteomic Approach Identifies Novel Cellular SMYD2 Substrates. J. Proteome Res. 2016, 15, 2052–2059. [Google Scholar] [CrossRef] [PubMed]
- Weirich, S.; Kudithipudi, S.; Jeltsch, A. Specificity of the SUV4-20H1 and SUV4-20H2 protein lysine methyltransferases and methylation of novel substrates. J. Mol. Biol. 2016, 428, 2344–2358. [Google Scholar] [CrossRef] [PubMed]
- Kudithipudi, S.; Schuhmacher, M.K.; Kebede, A.F.; Jeltsch, A. The SUV39H1 Protein Lysine Methyltransferase Methylates Chromatin Proteins Involved in Heterochromatin Formation and VDJ Recombination. ACS Chem. Biol. 2017, 12, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Cornett, E.M.; Dickson, B.M.; Krajewski, K.; Spellmon, N.; Umstead, A.; Vaughan, R.M.; Shaw, K.M.; Versluis, P.P.; Cowles, M.W.; Brunzelle, J.; et al. A functional proteomics platform to reveal the sequence determinants of lysine methyltransferase substrate selectivity. Sci. Adv. 2018, 4, eaav2623. [Google Scholar] [CrossRef]
- Brohm, A.; Elsawy, H.; Rathert, P.; Kudithipudi, S.; Schoch, T.; Schuhmacher, M.K.; Weirich, S.; Jeltsch, A. Somatic Cancer Mutations in the SUV420H1 Protein Lysine Methyltransferase Modulate Its Catalytic Activity. J. Mol. Biol. 2019, 431, 3068–3080. [Google Scholar] [CrossRef]
- Kusevic, D.; Kudithipudi, S.; Iglesias, N.; Moazed, D.; Jeltsch, A. Clr4 specificity and catalytic activity beyond H3K9 methylation. Biochimie 2017, 135, 83–88. [Google Scholar] [CrossRef]
- Iglesias, N.; Currie, M.A.; Jih, G.; Paulo, J.A.; Siuti, N.; Kalocsay, M.; Gygi, S.P.; Moazed, D. Automethylation-induced conformational switch in Clr4 (Suv39h) maintains epigenetic stability. Nature 2018, 560, 504–508. [Google Scholar] [CrossRef]
- Pinheiro, I.; Margueron, R.; Shukeir, N.; Eisold, M.; Fritzsch, C.; Richter, F.M.; Mittler, G.; Genoud, C.; Goyama, S.; Kurokawa, M.; et al. Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell 2012, 150, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Yu, J.R.; Granat, J.; Saldana-Meyer, R.; Andrade, J.; LeRoy, G.; Jin, Y.; Lund, P.; Stafford, J.M.; Garcia, B.A.; et al. Automethylation of PRC2 promotes H3K27 methylation and is impaired in H3K27M pediatric glioma. Genes Dev. 2019, 33, 1428–1440. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Long, Y.; Paucek, R.D.; Gooding, A.R.; Lee, T.; Burdorf, R.M.; Cech, T.R. Regulation of histone methylation by automethylation of PRC2. Genes Dev. 2019, 33, 1416–1427. [Google Scholar] [CrossRef] [PubMed]
- Collazo, E.; Couture, J.F.; Bulfer, S.; Trievel, R.C. A coupled fluorescent assay for histone methyltransferases. Anal. Biochem. 2005, 342, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Thompson, C.B. Metabolic regulation of epigenetics. Cell Metab. 2012, 16, 9–17. [Google Scholar] [CrossRef]
- Fan, J.; Krautkramer, K.A.; Feldman, J.L.; Denu, J.M. Metabolic regulation of histone post-translational modifications. ACS Chem. Biol. 2015, 10, 95–108. [Google Scholar] [CrossRef]
- Etchegaray, J.P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef]
- Chin, H.G.; Esteve, P.O.; Pradhan, M.; Benner, J.; Patnaik, D.; Carey, M.F.; Pradhan, S. Automethylation of G9a and its implication in wider substrate specificity and HP1 binding. Nucleic Acids Res. 2007, 35, 7313–7323. [Google Scholar] [CrossRef]
- Piao, L.; Nakakido, M.; Suzuki, T.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. Automethylation of SUV39H2, an oncogenic histone lysine methyltransferase, regulates its binding affinity to substrate proteins. Oncotarget 2016, 7, 22846–22856. [Google Scholar] [CrossRef]
- Weirich, S.; Kudithipudi, S.; Jeltsch, A. Somatic cancer mutations in the MLL1 histone methyltransferase modulate its enzymatic activity and dependence on the WDR5/RBBP5/ASH2L complex. Mol. Oncol. 2017, 11, 373–387. [Google Scholar] [CrossRef]
- Koh-Stenta, X.; Poulsen, A.; Li, R.; Wee, J.L.; Kwek, P.Z.; Chew, S.Y.; Peng, J.; Wu, L.; Guccione, E.; Joy, J.; et al. Discovery and characterisation of the automethylation properties of PRDM9. Biochem. J. 2017, 474, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Weil, L.E.; Shmidov, Y.; Kublanovsky, M.; Morgenstern, D.; Feldman, M.; Bitton, R.; Levy, D. Oligomerization and Auto-methylation of the Human Lysine Methyltransferase SETD6. J. Mol. Biol. 2018, 430, 4359–4368. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.; Chumanov, R.; Wang, Y.; Ge, Y.; Burgess, R.R.; Xu, W. Automethylation of CARM1 allows coupling of transcription and mRNA splicing. Nucleic Acids Res. 2011, 39, 2717–2726. [Google Scholar] [CrossRef] [PubMed]
- Dillon, M.B.; Rust, H.L.; Thompson, P.R.; Mowen, K.A. Automethylation of protein arginine methyltransferase 8 (PRMT8) regulates activity by impeding S-adenosylmethionine sensitivity. J. Biol. Chem. 2013, 288, 27872–27880. [Google Scholar] [CrossRef]
- Singhroy, D.N.; Mesplede, T.; Sabbah, A.; Quashie, P.K.; Falgueyret, J.P.; Wainberg, M.A. Automethylation of protein arginine methyltransferase 6 (PRMT6) regulates its stability and its anti-HIV-1 activity. Retrovirology 2013, 10, 73. [Google Scholar] [CrossRef]
- Geng, P.; Zhang, Y.; Liu, X.; Zhang, N.; Liu, Y.; Lin, C.; Yan, X.; Li, Z.; Wang, G.; Li, Y.; et al. Automethylation of protein arginine methyltransferase 7 and its impact on breast cancer progression. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 2287–2300. [Google Scholar] [CrossRef]
- Vinci, C.R.; Clarke, S.G. Homocysteine methyltransferases Mht1 and Sam4 prevent the accumulation of age-damaged (R,S)-AdoMet in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 2010, 285, 20526–20531. [Google Scholar] [CrossRef]
- Kanai, M.; Mizunuma, M.; Fujii, T.; Iefuji, H. A genetic method to enhance the accumulation of S-adenosylmethionine in yeast. Appl. Microbiol. Biotechnol. 2017, 101, 1351–1357. [Google Scholar] [CrossRef]
- Jeltsch, A.; Lanio, T. Site-directed mutagenesis by polymerase chain reaction. Methods Mol. Biol. 2002, 182, 85–94. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Clr4 WT | Clr4 A454R | ||||
|---|---|---|---|---|---|---|
| Km (µM) | rel. vmax | Ki (µM) | Km (µM) | rel. vmax | Ki (µM) | |
| H3K9me0 | 118.9 ± 9.4 | 1.13 ± 0.18 | 126.8 ± 7.9 | 37.2 ± 9.1 | 0.8 ± 0.05 | 43.3 ± 0.6 |
| H3K9me1 | 16.38 ± 2.2 | 1.05 ± 0.02 | 24.6 ± 1.82 | 5.5 ± 1.6 | 1.14 ± 0.03 | 11.5 ± 1.6 |
| Substrate | Clr4 WT | Clr4 K455R/K472R | ||||
|---|---|---|---|---|---|---|
| Km (µM) | rel. vmax | N | Km (µM) | rel. vmax | n | |
| AdoMet | 11.4 ± 1.5 | 1.0 ± 0.01 | 1.87 ± 0.19 | 13.6 ± 1.0 | 0.71 ± 0.08 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khella, M.S.; Bröhm, A.; Weirich, S.; Jeltsch, A. Mechanistic Insights into the Allosteric Regulation of the Clr4 Protein Lysine Methyltransferase by Autoinhibition and Automethylation. Int. J. Mol. Sci. 2020, 21, 8832. https://doi.org/10.3390/ijms21228832
Khella MS, Bröhm A, Weirich S, Jeltsch A. Mechanistic Insights into the Allosteric Regulation of the Clr4 Protein Lysine Methyltransferase by Autoinhibition and Automethylation. International Journal of Molecular Sciences. 2020; 21(22):8832. https://doi.org/10.3390/ijms21228832
Chicago/Turabian StyleKhella, Mina S., Alexander Bröhm, Sara Weirich, and Albert Jeltsch. 2020. "Mechanistic Insights into the Allosteric Regulation of the Clr4 Protein Lysine Methyltransferase by Autoinhibition and Automethylation" International Journal of Molecular Sciences 21, no. 22: 8832. https://doi.org/10.3390/ijms21228832
APA StyleKhella, M. S., Bröhm, A., Weirich, S., & Jeltsch, A. (2020). Mechanistic Insights into the Allosteric Regulation of the Clr4 Protein Lysine Methyltransferase by Autoinhibition and Automethylation. International Journal of Molecular Sciences, 21(22), 8832. https://doi.org/10.3390/ijms21228832