Machine Learning Identifies Robust Matrisome Markers and Regulatory Mechanisms in Cancer

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
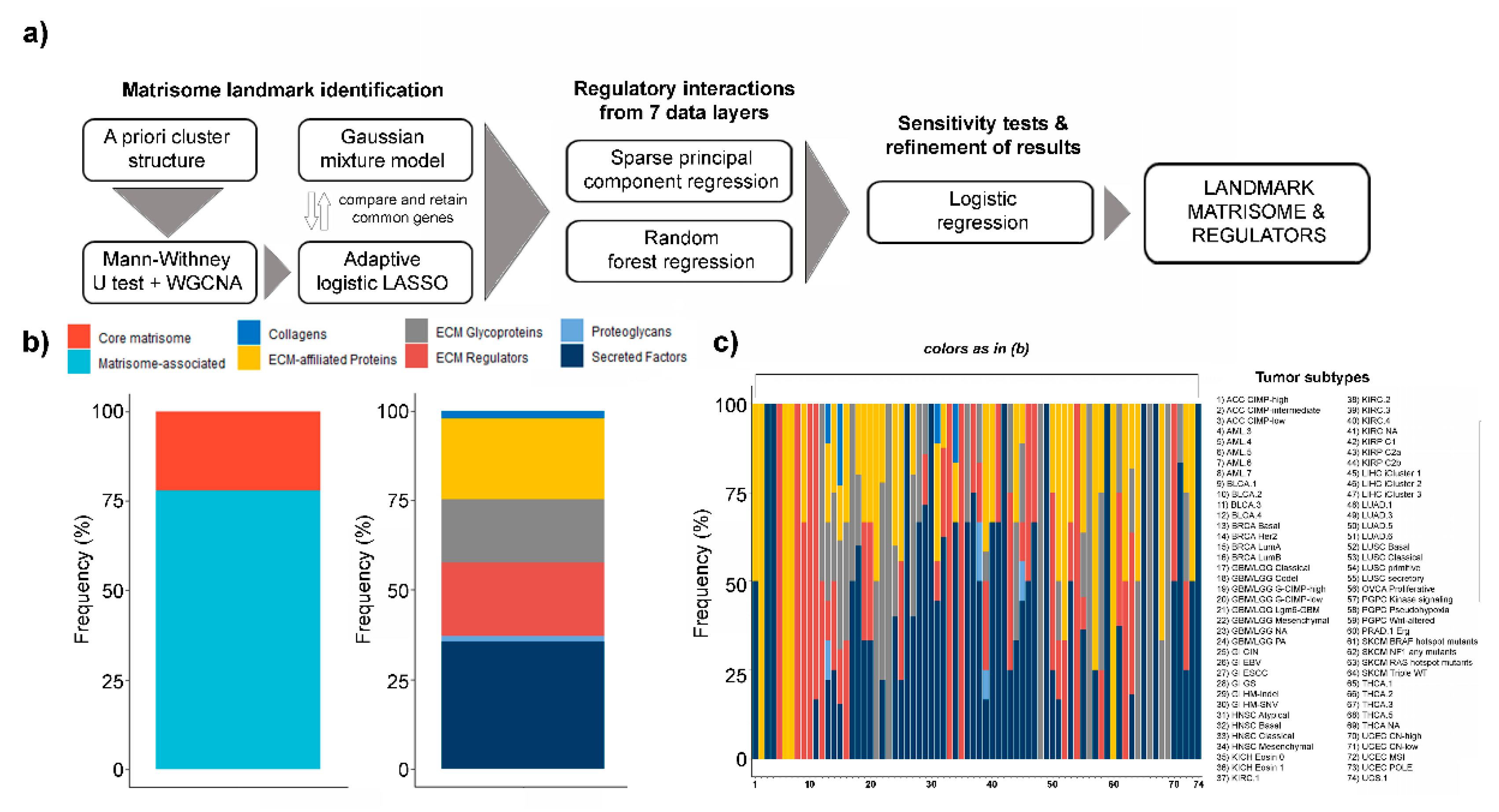
2. Results
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| WGCNA | Weighted gene coexpression analysis |
| LASSO | Least absolute shrinkage and selection operator |
| miRNA | Micro-RNAs |
| sPCR | Sparse principal component regression |
| RFR | Random forest regression |
| TCGA | The Cancer Genome Atlas |
| THPA | The Human Protein Atlas |
| TME | Tumor microenvironment |
| ECM | Extracellular matrix |
| CNA | Copy number alterations |
| GMM | Gaussian mixture model |
| EM | Expectation maximization |
| SD | Standard deviation |
Appendix A

References
- Ribeiro Franco, P.I.; Rodrigues, A.P.; de Menezes, L.B.; Pacheco Miguel, M. Tumor Microenvironment Components: Allies of Cancer Progression. Pathol. Res. Pract. 2020, 216, 152729. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The Tumor Microenvironment and its Role in Promoting Tumor Growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The Tumor Microenvironment at a Glance. J. Cell. Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rianna, C.; Kumar, P.; Radmacher, M. The Role of the Microenvironment in the Biophysics of Cancer. Semin. Cell Dev. Biol. 2018, 73, 107–114. [Google Scholar]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An Immeasurable Source of Knowledge. Contemp. Oncol. 2015, 19, 68. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in the Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Zhang, S. An Integrative and Comparative Study of Pan-Cancer Transcriptomes Reveals Distinct Cancer Common and Specific Signatures. Sci. Rep. 2016, 6, 33398. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic Cancer Genetics at High-Resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A Resource for Therapeutic Biomarker Discovery in Cancer Cells. Nucleic Acids Res. 2013, 41, 955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuzhalin, A.E.; Urbonas, T.; Silva, M.A.; Muschel, R.J.; Gordon-Weeks, A.N. A Core Matrisome Gene Signature Predicts Cancer Outcome. Br. J. Cancer 2018, 118, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzi, V.; Lakkala, J.; Devarajan, R.; Kääriäinen, A.; Koivunen, J.; Heljasvaara, R.; Pihlajaniemi, T. Pan-Cancer Analysis of the Expression and Regulation of Matrisome Genes Across 32 Tumor Types. Matrix Biol. Plus 2019, 1, 100004. [Google Scholar] [CrossRef]
- Izzi, V.; Davis, M.N.; Naba, A. Pan-Cancer Analysis of the Genomic Alterations and Mutations of the Matrisome. Cancers 2020, 12, 2046. [Google Scholar] [CrossRef]
- Lim, S.B.; Chua, M.L.K.; Yeong, J.P.S.; Tan, S.J.; Lim, W.; Lim, C.T. Pan-Cancer Analysis Connects Tumor Matrisome to Immune Response. NPJ Precis. Oncol. 2019, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The Extracellular Matrix: Tools and Insights for the “Omics” Era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The Extracellular Matrix Modulates the Hallmarks of Cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Hsu, J.B.; Chiu, C.; Hsu, S.; Huang, W.; Chien, C.; Lee, T.; Huang, H. miRTar: An Integrated System for Identifying miRNA-Target Interactions in Human. BMC Bioinform. 2011, 12, 300. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic Pan-Cancer Analysis of Tumour Purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Cho, J.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST V2: An Expanded Reference Database of Human and Mouse Transcriptional Regulatory Interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Marbach, D.; Lamparter, D.; Quon, G.; Kellis, M.; Kutalik, Z.; Bergmann, S. Tissue-Specific Regulatory Circuits Reveal Variable Modular Perturbations Across Complex Diseases. Nat. Methods 2016, 13, 366–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data Via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Akbani, R.; Ng, P.K.S.; Werner, H.M.J.; Shahmoradgoli, M.; Zhang, F.; Ju, Z.; Liu, W.; Yang, J.; Yoshihara, K.; Li, J.; et al. A Pan-Cancer Proteomic Perspective on the Cancer Genome Atlas. Nat. Commun. 2014, 5, 3887. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kaminska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-Cancer Analysis of Whole Genomes. Nature 2020, 578, 82–93. [Google Scholar]
- Law, J.W.S.; Lee, A.Y.W. The Role of Semaphorins and their Receptors in Gliomas. J. Signal Transduct. 2012, 2012, 902854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelucci, C.; Lama, G.; Sica, G. Multifaceted Functional Role of Semaphorins in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, Y.; Tsunematsu, T.; Takata, T. Oncogenic Role of RUNX3 in Head and Neck Cancer. J. Cell Biochem. 2011, 112, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Fu, J.; Yao, L.; Hou, A.; Xue, X. Runx3 is a Key Modulator during the Epithelial-Mesenchymal Transition of Alveolar Type II Cells in Animal Models of BPD. Int. J. Mol. Med. 2017, 40, 1466–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yemelyanova, A.; Gown, A.M.; Wu, L.; Holmes, B.J.; Ronnett, B.M.; Vang, R. PAX8 Expression in Uterine Adenocarcinomas and Mesonephric Proliferations. Int. J. Gynecol. Pathol. 2014, 33, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Xiu, X.; Wang, H.; Yun-Yi, L.; Fan-Dou, K.; Jin-Ping, H. Endometrial Stromal Sarcoma in Combination with Mixed Type Endometrial Carcinomas: A Case Report and Literature Review. Medicine 2017, 96, e8928. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.; Jin, W. The Updated Landscape of Tumor Microenvironment and Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 1–16. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally Portraying the Tissue Cellular Heterogeneity Landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
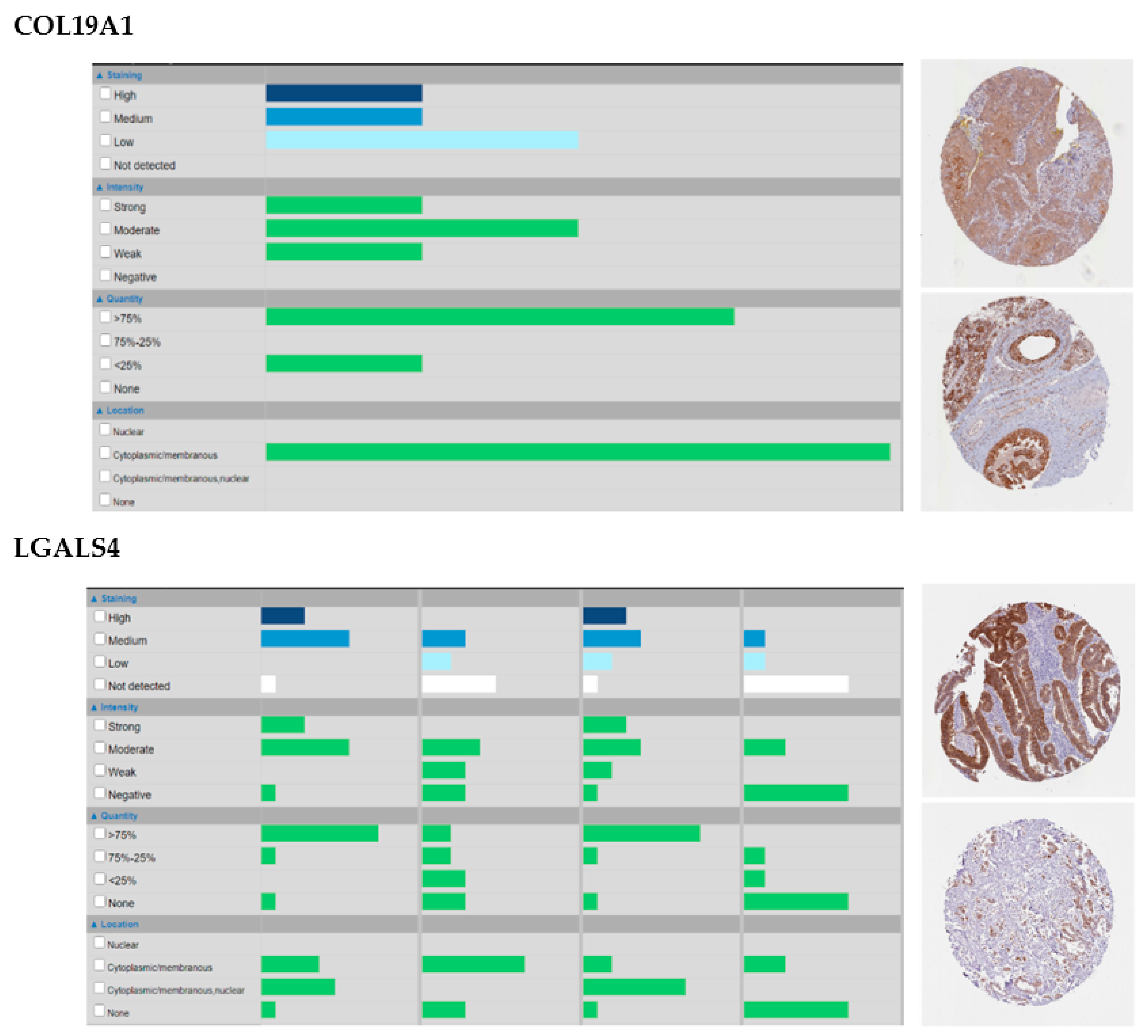
- Oudart, J.; Monboisse, J.; Maquart, F.; Brassart, B.; Brassart-Pasco, S.; Ramont, L. Type XIX Collagen: A New Partner in the Interactions between Tumor Cells and their Microenvironment. Matrix Biol. 2017, 57, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Amenta, P.S.; Hadad, S.; Lee, M.T.; Barnard, N.; Li, D.; Myers, J.C. Loss of Types XV and XIX Collagen Precedes Basement Membrane Invasion in Ductal Carcinoma of the Female Breast. J. Pathol. 2003, 199, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Oudart, J.; Doué, M.; Vautrin, A.; Brassart, B.; Sellier, C.; Dupont-Deshorgue, A.; Monboisse, J.; Maquart, F.; Brassart-Pasco, S.; Ramont, L. The Anti-Tumor NC1 Domain of Collagen XIX Inhibits the FAK/ PI3K/Akt/mTOR Signaling Pathway through Αvβ3 Integrin Interaction. Oncotarget 2015, 7, 1516–1528. [Google Scholar] [CrossRef] [Green Version]
- Suh, Y.; Lee, H.; Jung, E.; Kim, M.; Nam, K.T.; Goldenring, J.R.; Yang, H.; Kim, W.H. The Combined Expression of Metaplasia Biomarkers Predicts the Prognosis of Gastric Cancer. Ann Surg. Oncol. 2012, 19, 1240–1249. [Google Scholar] [CrossRef] [Green Version]
- Satelli, A.; Rao, P.S.; Thirumala, S.; Rao, U.S. Galectin-4 Functions as a Tumor Suppressor of Human Colorectal Cancer. Int. J. Cancer 2011, 129, 799–809. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Li, C.; Lin, L.; Wang, A.S.; Pu, Y.; Wang, H.; Mar, A.; Chen, C.; Lee, T. Promoter Hypermethylation of LGALS4 Correlates with Poor Prognosis in Patients with Urothelial Carcinoma. Oncotarget 2017, 8, 23787–23802. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Cyrta, J.; Murphy, D.A.; Walczak, E.G.; Ran, L.; Agrawal, P.; Xie, Y.; Chen, Y.; Wang, S.; Zhan, Y.; et al. Aberrant Activation of a Gastrointestinal Transcriptional Circuit in Prostate Cancer Mediates Castration Resistance. Cancer Cell 2017, 32, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Piperigkou, Z.; Karamanos, N.K. Dynamic Interplay between miRNAs and the Extracellular Matrix Influences the Tumor Microenvironment. Trends Biochem. Sci. 2019, 44, 1076–1088. [Google Scholar] [CrossRef]
- Kawano, S.; Fujisawa, H.; Takada, T.; Shiroishi, T. Sparse Principal Component Regression for Generalized Linear Models. Comput. Stat. Data Anal. 2016, 124, 180–196. [Google Scholar] [CrossRef]
- Auret, L.; Aldrich, C. Interpretation of Nonlinear Relationships between Process Variables by use of Random Forests. Miner. Eng. 2012, 35, 27–42. [Google Scholar] [CrossRef]
- Peeney, D.; Fan, Y.; Nguyen, T.; Meerzaman, D.; Stetler-Stevenson, W.G. Matrisome-Associated Gene Expression Patterns Correlating with TIMP2 in Cancer. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tomko, L.A.; Hill, R.C.; Barrett, A.; Szulczewski, J.M.; Conklin, M.W.; Eliceiri, K.W.; Keely, P.J.; Hansen, K.C.; Ponik, S.M. Targeted Matrisome Analysis Identifies Thrombospondin-2 and Tenascin-C in Aligned Collagen Stroma from Invasive Breast Carcinoma. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kobak, D.; Berens, P. The art of using t-SNE for single-cell transcriptomics. Nat. Commun. 2019, 10, 5416. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kääriäinen, A.; Pesola, V.; Dittmann, A.; Kontio, J.; Koivunen, J.; Pihlajaniemi, T.; Izzi, V. Machine Learning Identifies Robust Matrisome Markers and Regulatory Mechanisms in Cancer. Int. J. Mol. Sci. 2020, 21, 8837. https://doi.org/10.3390/ijms21228837
Kääriäinen A, Pesola V, Dittmann A, Kontio J, Koivunen J, Pihlajaniemi T, Izzi V. Machine Learning Identifies Robust Matrisome Markers and Regulatory Mechanisms in Cancer. International Journal of Molecular Sciences. 2020; 21(22):8837. https://doi.org/10.3390/ijms21228837
Chicago/Turabian StyleKääriäinen, Anni, Vilma Pesola, Annalena Dittmann, Juho Kontio, Jarkko Koivunen, Taina Pihlajaniemi, and Valerio Izzi. 2020. "Machine Learning Identifies Robust Matrisome Markers and Regulatory Mechanisms in Cancer" International Journal of Molecular Sciences 21, no. 22: 8837. https://doi.org/10.3390/ijms21228837
APA StyleKääriäinen, A., Pesola, V., Dittmann, A., Kontio, J., Koivunen, J., Pihlajaniemi, T., & Izzi, V. (2020). Machine Learning Identifies Robust Matrisome Markers and Regulatory Mechanisms in Cancer. International Journal of Molecular Sciences, 21(22), 8837. https://doi.org/10.3390/ijms21228837