Abstract
Despite being discovered and isolated more than one hundred years ago, tuberculosis (TB) remains a global public health concern arch. Our inability to eradicate this bacillus is strongly related with the growing resistance, low compliance to current drugs, and the capacity of the bacteria to coexist in a state of asymptomatic latency. This last state can be sustained for years or even decades, waiting for a breach in the immune system to become active again. Furthermore, most current therapies are not efficacious against this state, failing to completely clear the infection. Over the years, a series of experimental methods have been developed to mimic the latent state, currently used in drug discovery, both in vitro and in vivo. Most of these methods focus in one specific latency inducing factor, with only a few taking into consideration the complexity of the granuloma and the genomic and proteomic consequences of each physiological factor. A series of targets specifically involved in latency have been studied over the years with promising scaffolds being discovered and explored. Taking in account that solving the latency problem is one of the keys to eradicate the disease, herein we compile current therapies and diagnosis techniques, methods to mimic latency and new targets and compounds in the pipeline of drug discovery.
1. Introduction
The bacillus Mycobacterium tuberculosis (M.tb), causative agent for tuberculosis, was first identified in 1882 by Robert Koch. Koch was also responsible for the first extraction of tuberculin, a combination of proteins that would later become one of the basis of tuberculosis diagnosis [1]. Nowadays, more than 130 years after its discovery, M. tuberculosis remains a hot research topic and TB stands as one of the top 10 causes of death worldwide [2]. According to the latest reports, in 2018, 10 million new cases emerged, with 1.5 million people dying from the disease. Furthermore, 66% of the new cases occurred in specific countries: India, China, Indonesia, the Philippines, Pakistan, Nigeria, Bangladesh, and South Africa [3]. Besides the increasing resistance of the bacillus towards current therapies [4], one of the reasons for the success of this pathogen is its ability to coexist within the host in an asymptomatic latent state [5]. The phagocytosis of the bacillus by alveolar macrophages and dendritic cells along with the formation of the granuloma creates an avascular, inflammatory, and necrotic environment, in which the bacteria faces low oxygen levels and oxidative stress [6,7]. As a strictly aerobic microorganism, any decrease in the available oxygen leads to a significant decrease in bacillus growth, until it ceases completely [8]. The bacillus then activates a series of pathways leading to protein stability and homeostatic regulation, with maintenance of a basal metabolic level. M.tb adjusts energy sources, reduces energy expenditure, preparing itself for the dormancy state [9,10]. When this nonreplication is reached, the disease enters a state of latent infection. This dormancy state can be maintained for months, years, or even decades. It is estimated that 5 to 10% of individuals with this subclinical infection may develop clinical manifestations 2 to 5 years after the initial infection [11,12,13].
The current antitubercular therapy is a combination of four antibiotics: isoniazid, rifampicin, ethambutol, and pyrazinamide. The treatment regimen consists of a 2-month initial treatment, followed by a 4- or a 7-month continuation therapy. The initial phase uses the four drugs, with different mechanisms of action, each performing an essential role. Isoniazid and rifampicin show high cure rates in short-course regimens. Pyrazinamide is active against the latent bacillus, showing a potent sterilizing activity. Ethambutol prevents the emergence of resistance against rifampicin when resistance to isoniazid is present. The most common continuation phase is the 4-month therapy and it generally includes only isoniazid and rifampicin [14,15]. This long-term therapy is essential for a complete eradication of M.tb and to prevent dormant bacilli from remaining in the host [16]. However, and despite all these efforts, a study performed in 2014 estimated the global burden of latent tuberculosis as 23%, approximately 1.7 billion people, with the regions South-East Asia, Western-Pacific, and Africa accounting for 80% of this value [17]. The End TB Strategy sets 2035 as the year in which the number of deaths caused by the disease would have dropped by 95%. Not only to increase monitoring and therapy compliance, to reach this goal, it is essential to understand and find permanent and highly efficient solutions for latent tuberculosis [18].
2. Latent Tuberculosis Diagnosis and Current Therapies
The most common therapy for the latent tuberculosis infection (LTBI) is a 6-months’ daily monotherapy with isoniazid [14,19]. This was initially compared with the placebo, showing a significant decrease in TB incidence [20]. The efficacy of this therapy was also compared with twelve- and thirty-six-months treatments, with no significant differences [14,21]. Another analysis, however, suggests that 9 months is the optimal period for the treatment of LTBI using isoniazid monotherapy [22]. Therefore, the recommended duration for this therapy is considered 6 to 9 months [19]. Isoniazid advantages rely on its considerable clinical experience and low cost, however, the suboptimal compliance of such a long treatment and its hepatotoxicity led to alternative therapies [23,24,25]. Despite being used to treat LTBI, isoniazid is not active against nonreplicant bacillus, being used only as a preventive drug and requiring long therapeutic periods to prevent the progression into the active disease [26,27]. Furthermore, isoniazid has been shown to induce its own resistance when used in nonreplicant M.tb. After 1-week incubation in liquid media at concentrations of 0.1 and 0.4 µg/mL, subpopulations of bacilli were no longer susceptible to isoniazid. A similar experiment was performed with rifampicin and induction of resistance was not observed [28].
The development of techniques that mimic latency allowed insights on more efficacious drugs and shorter therapies. Overall, the most active drugs against dormant M.tb were the rifamycins (rifampicin and rifapentine), followed by pyrazinamide [29]. Three new treatment regimens are used as shorter and equally efficacious therapeutic alternatives for LTBI: 3-, 4-months’ daily rifampicin monotherapy; 3-, 4-months’ daily rifampicin plus isoniazid; 3-months’ weekly rifapentine plus isoniazid [14,19]. The use of rifampicin has been associated with lower hepatotoxicity and higher completion rates when compared with isoniazid in 6-months’ monotherapy [30,31]. Rifapentine displays longer half-life and increased potency than rifampicin, allowing weekly treatments with similar efficacy. Despite the efficacy of pyrazinamide against dormant bacilli, this drug is not in any of the recommended therapies due to its high hepatotoxicity [32,33].
The recommended methods to diagnose LTBI are the tuberculin skin test (TST) and the interferon-γ release assay (IGRA) [34]. For nearly one hundred years, TST was the only test available. It involves the injection of the tuberculin purified protein derivative (PPD) intradermally on the forearm and this area is then examined 48 to 72 h later. A delayed-type hypersensitivity reaction is observed in all individuals that were already sensitized for the antigens in PPD [35]. The major limitation of this test is the false-positive obtained from people who have been administered with the Bacillus Calmette–Guérin (BCG) vaccine [36]. IGRA is an in vitro test that measures the interferon-γ produced by lymphocytes, after incubation with the antigens early secretory antigen target-6 (ESAT-6) and culture filtrate protein-10 (CFP-10). The main advantage of ESAT-6 and CFP-10 is their absence from the BCG vaccine, overcoming the main problem of TST [35,37]. However, false positives may occur from infection with other mycobacteria species [37,38]. The sensitivity of both tests can be compromised in HIV-infected individuals with low CD4 T-cells count and patients on immunosuppressive drug therapy [39,40]. Furthermore, and since they are both indirect methods to detect an M.tb infection, it is not possible to distinguish individuals who are latently infected from those who have cleared the infection but still have a persistent anti-mycobacterial immune response [39]. This major drawback can favor the administration of anti-tubercular therapy on healthy individuals, increasing the disease-related costs and the possibility of unnecessary adverse effects [41].
3. Methods to Mimic Latency
In order to reach a nonreplicative state of M.tb for experimental purposes, it is essential to understand the conditions within the granuloma and mimic them as faithfully as possible [42]. The granuloma is characterized by a variety of stress conditions, being the most studied hypoxia, nutrient deprivation and limited carbon sources, and high concentration of nitric oxide [43,44,45]. The combination of all these factors would ideally replicate the phenotype found in the latent interaction between pathogen and host. However, and despite a few cases where several of these conditions are met, the majority of in vitro models is based on only one of the factors involved in latency [42].
3.1. Hypoxia
Hypoxia is the best characterized factor that triggers the latency state [46]. The first and most famous model was developed by Lawrence Wayne, upon the observation of arrested growth in the event of discontinued aeration of an M.tb culture [47]. This state, similar to latency, but with subtle differences, was then named nonreplicating persistence (NRP) [8,48]. The Wayne model, introduced in 1996, aims to simulate the depletion of oxygen within the granuloma and reach that NRP state. The M.tb growth occurs in a sealed container, with a controlled air ratio. As the available oxygen is consumed, the organisms slowly shift to anaerobic conditions. Two different NRP stages can be observed. Stage I is attained when the oxygen saturation achieves 1% and, even though the bacilli are not replicating or synthesizing DNA, there is still high production of ATP and some mechanisms of DNA repair remain functional [48,49,50]. When the oxygen saturation reaches 0.06%, the culture enters Stage II, reaching full anaerobic conditions. Even though NRP Stage II is often described as simply NRP, it is not possible for an organism to survive by being placed directly in Stage II. A gradual decrease in oxygen levels is essential [8,48,49,50]. When screened under this model, M.tb is resistant to isoniazid but susceptible to metronidazole, a drug with no inhibitory effect in aerobic conditions [51,52]. This bactericidal effect was also observed in non-human primate and rabbit models but was not reflected in mice, putting into question the activity of compounds identified using the latter model [7,53,54,55]. The reason for this difference lies on the methodologies itself. Being based only on hypoxia, the Wayne model generates bacilli with physiological and metabolic profiles different from other methods used for in vivo studies [53]. Over the years, adaptations of the Wayne model have been developed, in order to facilitate the screening of new drugs [42].
The hypoxic resazurin reduction assay (HyRRA) is a conjugation of the Wayne model with the resazurin microtiter assay (REMA) [56]. This colorimetric assay uses alamar blue for the identification of the minimum inhibitory concentration (MIC) of compounds against the NRP M.tb. After cultures reach the hypoxic and NRP state, the drugs are added and the system is incubated for 96 h. The resulting cultures are then dispensed into microtiter plates and the alamar blue is added to the wells. The viability of these cells is evaluated, and the MIC is determined by the colorimetric result [57]. Despite being a promising conjugation of methods, it may fail to identify potential compounds since 96 h may not be enough for some drugs to display activity [42].
Another adaptation of the Wayne model is the low oxygen recovery assay (LORA) [58,59]. This assay measures the metabolic activity levels of the culture by taking advantage of a luciferase reporter (luxAB gene) [60,61]. When entering the NRP state, the luminescence of the culture decreases, allowing the method to be used in high-throughput screening (HTS). The hypoxia is reached as previously described and a chemostat can be used to accurately control the oxygen levels [62].
In 2008, Yeware and Sarkar developed another method derived from the Wayne model, using red fluorescent protein (RFP) to measure the culture growth [63]. RFP appears to give a stronger and more stable signal in dormant mycobacteria than other reporter genes, such as green fluorescent protein (GFP) and the luciferase reporter (used in LORA) [62,64,65]. Furthermore, hypoxia was achieved by adding a layer of paraffin oil, impermeable to oxygen. The compounds for the HTS were inserted directly through the paraffin layer. Contrary to other hypoxic models, metronidazole showed no activity against the NRP bacilli using this method [63].
3.2. Nutrient Deprivation
Nutrient deprivation was early identified as a way to reach the NRP state. In 1933, Loebel et al. observed that it was possible to transfer a M.tb culture from a rich media to PBS and leave it for many years in solution [44]. The bacilli remained viable, but their respiration levels were slowly decreased as the culture entered a stationary state [66]. When reintroduced to a rich media, the bacteria resumed normal growth [44]. These findings were the basis of the first NRP state model based on nutrient deprivation, the nutrient deprivation model. Developed in 2002 by Betts et al., this method predicts the growth of mycobacteria in a rich media for 7 days, followed by centrifugation and resuspension in PBS. The viability of the culture was determined by colony-forming unit (CFU) counts in specific timepoints. Despite being kept in a sealed container, like in the Wayne model, methylene blue analysis reflected the presence of oxygen in the media [67]. Being deprived from ‘foodstuff’, the bacteria reduced their respiration rates and entered the NRP state, regardless of oxygen availability [66,67]. When screened against antibiotics, the new culture gained resistance to isoniazid without acquiring susceptibility to metronidazole [67]. This difference to hypoxia can be explained by the different transcriptomes derived from the two different conditions [68].
In 2004, Hampshire et al. developed a method of progressive nutrient depletion, measured by a chemostat. Apart from reporting the carbon sources, the device allowed a stable control over pH, temperature, and oxygen levels. After a quick depletion in glucose and a gradual depletion in glycerol, the culture reached a stationary state by day 15. The exhaustion of resources led to a progressive drop in viability until day 78. On day 78, a small population of fully adapted bacilli restarted to grow, reaching 4-fold of the previous measurement by day 111. Analysis of these 3 phases showed notorious differences, with the second phase, from day 15 to 78, matching the expected transcriptomic alterations for an NRP state [69].
3.3. Nitric Oxide
Even though hypoxia and nutrient starvation are the most studied environmental conditions of the granuloma, they are not the only parameters that can induce the NRP state [42]. Nitric oxide (NO) was identified as capable of inducting the expression of M.tb dormancy regulon genes at nanomolar concentrations, causing the bacillus to enter dormancy in vitro [70,71,72]. By inhibiting mycobacterial respiration and halting replication, NO generates an NRP state similar to the Wayne model and hypoxic conditions [71]. No functional model has yet been developed based on nitric oxide, however it would be expected a phenotype comparable to the one observed for hypoxia [42].
3.4. Streptomycin-Dependent Model
In 1955, Hashimoto et al. isolated the M.tb strain 18 b from a patient with tuberculosis. This strain was discovered to be not only resistant to streptomycin, but also streptomycin-growth dependent. Despite these findings, this strain remains viable in streptomycin-free media for several weeks, without multiplying, resuming growth upon the adding of the antibiotic [73,74]. Later, in 2010, Sala et al. exploited the M.tb strain 18 b for its potential as an NRP model. They observed that the growth rate was directly correlated with the concentration of streptomycin in the media. Looking into the phenotype, this strain entered the NRP state after 10 days without antibiotic, maintaining it until streptomycin was reinstated in the culture [75]. When analyzed for drug susceptibility, isoniazid showed no activity but rifampicin displayed an increased efficacy. New compounds in clinical trials were also tested with positive results. For HTS purposes, this strain was screened using the REMA technique already described [75,76]. This model has been used in drug discovery, not only allied with REMA, but also with the luciferase assay [77,78].
3.5. Multiple Stress Models
Despite the success of single factor stress models, more complex and multifactorial methods replicate more accurately the environment within the granuloma. In 2009, Deb et al. reached the NRP state of M.tb with a low-nutrient and acidic medium (pH 5), using an atmosphere with 10% CO2 and 5% O2 for 18 days. By screening the colony at day 18, the bacilli were resistant to rifampicin by 310-fold and to isoniazid by 2800-fold, when compared to day 0 [79]. When the results are compared to the Wayne model, the bacteria show a much higher resistance to rifampicin in this multi-stress method [8,79]. Another multi-stress model was purposed by Gold et al. in 2015. To reach the dormant state, the culture was exposed to a mildly acidic environment (pH 5), a flux of nitric oxide, and other reactive nitrogen intermediates, hypoxia with 1% oxygen levels and fatty acids as the carbon source. The assay was divided into two phases: a first phase with nonreplicant bacilli exposed to the drugs for 3 to 7 days; a second phase of outgrowth, that would facilitate the reading of viable cells. In order to prevent false positives in compounds that were only active in the outgrowth phase, a second assay was performed in parallel with replicating M.tb and the results were compared [80]. Despite these models being the most complex and granuloma-like available, still relevant host factors are not being taken into consideration. Examples of these conditions are deprivation of iron, magnesium, amino acids, and vitamins; copper intoxication; exposure to reactive oxygen intermediates and carbon monoxide [81,82,83,84,85,86,87,88,89,90].
3.6. In Vivo Models
Several animal models have been used to replicate the latency of M.tb [91]. Even though mice would be the most common choice due to its convenience, low-cost, availability, and technical benefits, the bacterial burden observed in these animals is a lot higher, with crucial differences in the structure of the granuloma [92,93]. Other models such as guinea pigs, rabbits, and non-human primates have also been studied, with advantageous similarities to humans [94]. Zebrafish and medaka models have been developed, recurring to Mycobacterium marinum. Analyzing these last two methods, zebrafish appears to be more useful when representing the active infection, while medaka is more representative of the latency and chronic tuberculosis [95,96,97]. Since our main goal is to analyze latency models that can be used in drug development for M.tb and as zebrafish and medaka models use M. marinum, these will not be further explored in this paper.
The first model for in vivo latent tuberculosis was developed at Cornel University, by McCune and McDermott in 1966. For this method, the mice were infected with a high burden of bacilli for a long period and then treated with isoniazid and pyrazinamide until the levels of mycobacteria were undetectable. Once this state was achieved, they observed that the remaining ‘persisters’ could cause relapses by cessation of therapy or by injection of corticosteroids [98,99]. More accurately, the Cornel model is described as a model of M.tb persistence rather than latency, since the undetectable state is achieved by drug treatment and not by defense mechanisms of the host immune system [91,98,99]. However, this low bacterial burden is still similar to latent tuberculosis in humans and this method has been used in drug discovery [100]. Another disadvantage of this method is the premature death of several mice due to the high bacterial burden imposed [91,98,99]. A different approach to obtain an NRP state within the mice was demonstrated by directly applying the Wayne model in vivo. The animals are infected with NRP bacilli and it was observed that this state is maintained for slightly less than 10 days. This variant of the Wayne model was used to measure the impact of vaccination in the reactivation of M.tb, but could be applied to antimycobacterial drug discovery following the same rational [8,101]. The streptomycin-dependent model has also been applied in vivo. The mice were infected with the streptomycin-dependent mutant strain and injected with streptomycin for 3 weeks. After this period, the drug administration was interrupted, and the bacilli entered the NRP state [78]. Despite all these advances, none of these methods was able to replicate the immune response that generated the latency state. An immune-based murine model was firstly developed in 2004. The mice were immunized using a recombinant BCG strain and infected with the bacteria 6 weeks later. The immunization overcame the problem of the premature death of mice and allowed a state of bacterial burden very similar to latent tuberculosis in humans [102,103]. After its development, the method was enhanced in 2014, by using C3Heb/FeJ mice, a mutant variation that allows the development of necrotic lung granulomas and reactivation with TNF-α [104]. Finally, a novel hypoxic granuloma method was also proposed, with the encapsulation of the bacteria in semi-diffusible hollow fibers and subsequent subcutaneous implantation. Once encapsulated in these fibers, M.tb upregulates the expression of several genes also expressed in the mouse model of chronic tuberculosis. Furthermore, genes involved in metabolism and DNA replication were downregulated, matching the expected physiological profile of persistent bacilli. The main drawback of this method is the site of infection, different from the physiological environment of the lungs [105].
Guinea pigs display a granuloma similar to humans, with traits of necrosis and hypoxia [7,91]. However, they still show some similarities with mice, exhibiting high bacterial burden infections and premature death, with low tendency to latency and chronicity [91]. A similar rational to Cornell model was developed, with guinea pigs being subcutaneously infected with M.tb and receiving drug treatment until the bacterial load was undetected [106]. In this particular model, the authors stated that there is no significant difference between subcutaneous and aerosol administration when it comes to demonstrating the efficacy of vaccines, which was their goal [107]. The streptomycin-dependent model has also been used in guinea pigs in a procedure identical to mice [108].
Rabbits are considered the most resistant animal model to M.tb, despite developing granulomas similar to humans and guinea pigs [7,109]. In 2008, Manabe et al. showed that the aerosol infection of rabbits with M.tb naturally evolved from an active infection state to latency in 72% of the hosts, with undetectable bacterial levels but reactivation with immunosuppressant therapy [109]. Later, in 2012, a similar study was conducted. Rabbit infection with M.tb led to an exponential bacterial growth and active infection for 4 weeks, after which the bacterial load gradually decreased. By week 20, all subjects showed undetectable levels. After 4 weeks of immunosuppressant treatment, M.tb resumed an active growth state, showing that the apparent bacterial clearance was not a result of sterilization but of latency and chronic infection. Rabbits can then be characterized as natural and spontaneous models for latent tuberculosis [110,111].
The efficacy of vaccines and drugs has been tested in both rhesus and cynomolgus macaques, however, rhesus macaques are more susceptible to the infection than cynomolgus, being more appropriate for the study of the active disease [112]. Cynomolgus macaques are the model that better represents latency and the chronic tuberculosis [113]. Studies have shown that a low-bacterial dose infection of cynomolgus macaques may lead to active or latent tuberculosis. The chronic bacilli were naturally reactivated in a low percentage of hosts [114,115,116,117,118,119]. The non-human primate has been described as the model that most accurately reproduces the traits of the latent M.tb infection in humans [120].
4. Targeting Nonreplicating M.tb
The growing understanding of the latent phase of M.tb and its implications in the reactivation of the disease led to the search for compounds that can block latent form activation or target active enzymes during the dormant phase. The combination of bioinformatic prediction and genomic studies allowed the identification of several potential targets, crucial for survival during this phase [121,122]. Furthermore, the screening of new compounds against dormant bacilli increased in the past few years, contributing to the identification of new potent scaffolds with sterilizing effects on M.tb.
4.1. Alanine Dehydrogenase and Isocitrate Lyase
When entering the latent state, M.tb adapts to hypoxia by remodeling its metabolic routes for anaerobic pathways, with alternative electron acceptors being activated [123,124,125]. One of the enzymes upregulated in this process is the alanine dehydrogenase (Ald), whose function is the conversion of pyruvate into alanine and glyoxalate into glycine, with oxidation of NADH to NAD (Figure 1). Besides playing a critical role in maintaining the redox balance NADH/NAD, alanine is then used as a nitrogen source for new electron acceptors [126]. Glyoxalate and pyruvate are supplied by the glyoxalate and methylcitrate cycles, respectively, with the two isoforms of isocitrate lyase (Icl1 and Icl2) being responsible for the catalysis of its formation [127]. Expression of both enzymes has been analyzed under hypoxia, using the Wayne model [8]. It was observed that the levels of Ald were upregulated 5-fold in NRP-1 and 16-fold in NRP-2, while Icl1 was increased 39-fold in NRP-1 and downregulated in NRP-2 and Icl2 was similar to Ald. All these values are calculated in comparison with its expression in an aerobic state. Ald and Icl have therefore been considered as latency specific targets over the past few years [128,129,130].
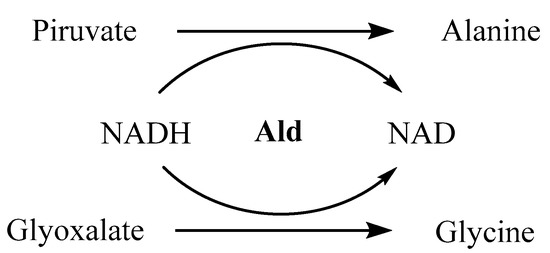
Figure 1.
Catalytic function of alanine dehydrogenase.
In 2014 and 2015, Saxena et al. successfully identified several Ald inhibitors using molecular docking studies. Some of these compounds were synthesized and tested against both active and dormant bacilli, showing modest to potent activities in active bacteria and effectively inhibiting dormant cells. This last screening was performed using the Betts model [67] and two compounds (1 and 2, Figure 2) were identified as causing a bacterial reduction of 3.2-log, showing also an IC50 for the M.tb Ald below 1 µM [131,132]. In 2016, the same group synthesized another library of inhibitors, with the most promising compound (3, Figure 2) displaying a 2-log bacterial reduction and an IC50 of 3.83 µM [133]. Following a whole-cell screen reported by GSK, Samala et al. were also able to identify Ald inhibitors, with one compound (4, Figure 2) providing a bacterial reduction of 2.7-log using the Betts method [67]. The interaction with the enzyme was confirmed using differential scanning fluorimetry, with the referred compound showing a thermal shift of 1.4 °C and an IC50 of 1.82 µM. Despite the activity against dormant strains, the MIC for active bacilli was weak, with values of 37.04 µM, suggesting that the scaffold still needs further optimization [134].
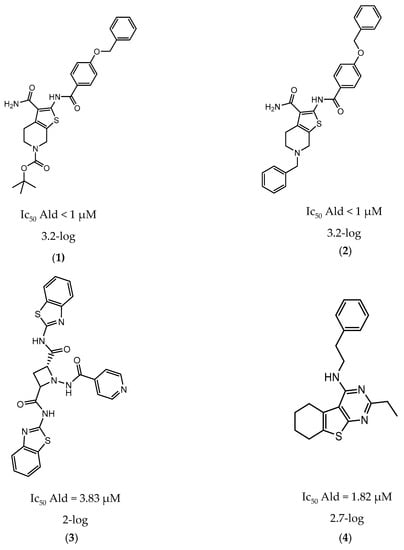
Figure 2.
Selected Alanine Dehydrogenase (Ald) inhibitors (1, 2, 3, 4), with respective Ald IC50 and bacterial reduction values.
Icl inhibitors were reported by Sriram et al. in 2011, when a full library of 3-nitroproprionamides was synthesized [135]. 3-Nitroproprionate was already a known inhibitor of Icl [136]. The newly synthesized compounds were shown to inhibit the enzyme, with IC50 below 1 µM, and displayed high selectivity as no cytotoxicity against mammalian cells was observed. They were screened against bacilli in the active and dormant phase (achieved through starvation) with promising MIC. The most potent compound (5, Figure 3) exhibited an MIC of 0.16 µM against active bacilli and 0.04 µM against starved M.tb [135]. Other Icl inhibitors have been successfully identified, however none of them were tested against dormant M.tb forms [137,138].
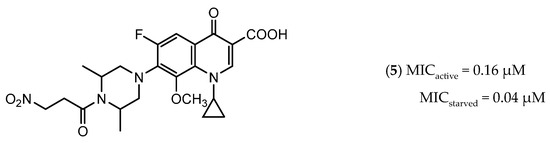
Figure 3.
Isocitrate Lyase (Icl) inhibitor with described Minimum Inhibitory Concentration (MIC) against active and starved bacilli.
4.2. Sulfur-Mediated Redox Homeostasis
After phagocytosis, the macrophages kill the bacteria engulfed in phagolysosomes using reactive oxygen and nitrogen intermediates (ROI and RNI) [139]. Therefore, the intracellular survival of any phagocytosed pathogen is dependent on its ability to use redox defense mechanisms. In the particular case of M.tb, this homeostasis is assured by mycothiol, an analog of glutathione [140]. Possessing a cysteine-derived sulfhydryl group, its availability and biosynthesis is directly related to the biosynthesis of L-cysteine and the sulfur metabolic pathways [141]. Bearing its crucial role in intracellular survival, these enzymes were observed to be upregulated in several dormancy models, with potential to be used as future drug targets [49,67,142]. Two of these enzymes already have inhibitors described in literature, those being the cysteine synthase CysM and the adenosine-5′-phosphosulfate reductase (APSR) [143,144].
CysM is one of the three pyridoxal-phosphate (PLP)-dependent cysteine synthase present in M.tb. This enzyme catalyzes the synthesis of cysteine using O-phosphoserine and CysO, a sulfur delivery protein [145]. Although two other enzymes can be alternatives towards the biosynthesis of cysteine, the resistance and stability of the intermediate in the catalytic reaction of CysM favors this route under oxidative stress. Therefore, the CysM catalysis is the dominant pathway for the synthesis of cysteine in dormant bacilli [146]. Targeting CysM, a family of inhibitors was developed [143,147]. Since the genomic expression of this enzyme is more prevalent in the dormant state, [67,142] as expected, when tested against active M.tb, these compounds only showed modest activities. However, its promising Kd values (ranging between 0.32 and 8 µM) matched good screening results when they were tested against starved bacilli, with two compounds (6 and 7, Figure 4) showing a 3-log reduction in bacterial count [143].
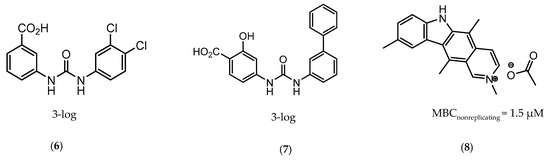
Figure 4.
Selected Cysteine Synthase CysM (6, 7) and Adenosine-5′-phosphosulfate Reductase (APSR) (8) inhibitors, with respective bacterial count and Minimum Bactericidal Concentration (MBC) against non-replicating bacilli values.
The enzymes involved in the sulfate assimilation pathway allow the targeting of a different part of the sulfur mycobacterial metabolism, leading to reduced sulfur levels for the biosynthesis of cysteine and other metabolites [148,149,150]. Furthermore, the expression of these proteins has been identified as upregulated in the dormant state and in situations of oxidative stress [67,69]. Adenosine-5′-phosphosulfate reductase (APSR) is a critical step of this route, reducing the sulfate in adenosine-5′-phosphosulfate (APS) to sulfite and adenosine-5′-monophosphate (AMP) [151,152]. Besides its connections with oxidative stress and mycobacterial survival, this enzyme does not have a homologue in humans, turning it into a potentially attractive and selective therapeutic target [144]. Over the years, inhibitors of APSR have been reported, either discovered by HTS or designed as analogs of its substrate, APS [153,154,155]. However, only in 2016, a series of inhibitors derived from HTS were for the first time screened against nonreplicating M.tb, confirming the potential of this target for dormant bacilli. From this family of inhibitors, two compounds showed minimum bactericidal concentration (MBC) against nonreplicating bacilli of 3 µM and the most promising compound (8, Figure 4) displayed an MBC against nonreplicating bacilli of 1.5 µM. The most potent inhibitors showed no cytotoxicity against VERO monkey kidney cell lines, validating the specificity of this target [144].
4.3. DevR/DevS/DosT System
Oxygen levels in cells are detected through more or less complex biochemical sensors, depending on the organism [156]. In the case of M.tb, its presence or absence is determined by DevS and DosT, two oxygen heme-based sensors, which function like a two-component system along with DevR, a response regulator [157]. By sensing variations in the levels of oxygen in the surrounding media, these promote a series of metabolic changes in the bacillus, initiating the process of latency. DevS and DosT work as receptors that sense the presence of oxygen and regulate their histidine kinase domain accordingly [158]. DosT, the sensor with the lowest oxygen affinity, is the first to be activated in a situation of hypoxia, followed by DevS, in even lower levels of oxygen [159]. These two receptors are then able to phosphorylate DevR, which leads to the upregulation of several genes and several phenotypical changes [160]. The crucial role of this system on initiating the dormancy state supports its importance in the virulence of the pathogen, turning it into a promising potential target for drug discovery [161].
The first DevR inhibitor was discovered in 2009 from a library of 2.5 million compounds, using an in silico pharmacophore-based screening. Compound 9 (Figure 5) was identified as inhibiting the binding of DevR to DNA, displaying activity against hypoxic cultures with a 4-log reduction in bacterial count. However, to achieve these results, a high concentration of this compound was necessary (131 µg/mL) and no inhibition was observed in aerobic or nutrient starved bacilli [162]. More recently, Zheng et al. performed an HTS to identify inhibitors of this system. Five compounds with five different scaffolds fitted in this profile, among them, the known antimalarial drug artemisinin (10, Figure 5) and compounds 11–14 (Figure 5). Although all compounds showed EC50 against a dosR-dependent mutant GFP reporter between 1.2 and 12.7 µM, none was able to efficiently inhibit active M.tb growth. Further studies, revealed that compound 10 downregulated 85 DevR-dependent genes (more than two thirds of all the genes regulated by this regulon) and 157 genes independent from this system, revealing lack of selectivity for the desired target. Compounds 11 and 12 displayed higher specificity, with 48 out of 55 and 76 out of 90, respectively, of the downregulated genes belonging to the DevR-regulated profile. These three compounds were also tested in a combined screening with isoniazid, with an increased sensitization to this drug being observed. The mechanism of action was also explored. While compound 10 led to the alkylation of the heme group of DevS and DosT, compounds 11 and 12 prevented the autophosphorylation of the sensors [163]. Later, compounds 13 and 14 were also further studied, with compound 14 displaying a higher specificity for this system and compound 13 inhibiting a significant portion of these genes but revealing off-target inhibition [164]. The impact of these compounds in survival during NRP state was accessed using a hypoxic model [165]. Compound 13 did not show any impact on survival, suggesting that the portion of genes it inhibits is not essential in the NRP state. On the other hand, compound 14 displayed a 50% survival rate when compared to the positive control. Their mechanism of action was also studied, with compound 13 preventing the binding of DevR to DNA and compound 14 binding to the heme group of DevS, disturbing its electronic spectrum [164].
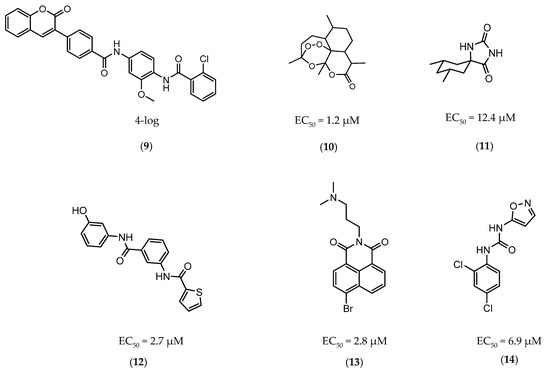
Figure 5.
Selected DevR/DevS/DosT system inhibitors (9, 10, 11, 12, 13, 14), with referred bacterial count and EC50 values.
4.4. Lysine ε-Aminotransferase
Lysine ε-aminotransferase (LAT) is a pyridoxal-5′-phosphate (PLP)-dependent aminotransferase, responsible for the catalysis of the conversion of lysine and α-keto glutaric acid into piperidine-6-carboxylic acid and glutamate, respectively [166,167]. The deletion of the lat gene revealed its important role in maintaining the amino acid pool and, subsequently, the levels of guanosine pentaphosphate, essential in inhibiting the RNA synthesis and promoting survival in a latent state [168]. The Betts model identified LAT as upregulated in dormancy about 41.86 times, turning it into an interesting target against latent M.tb [67].
A few inhibitors have been described by the group of Dharmarajan et al. Their scaffolds were identified through a combination of structure-based and ligand-based methods with the crystallographic structure of LAT. Selected scaffolds were further explored and optimized, generating compounds with potent IC50 for the enzyme and significant inhibition against nutrient starved bacilli. The most promising compounds were compound 15 (Figure 6), with a 2.8-log bacterial reduction and an IC50 of 1.22 µM; compound 16 (Figure 6), with a bacterial reduction of over 2-log and an IC50 of 0.81 µM; and compound 17 (Figure 6), with a 2.9-log reduction in bacterial count and IC50 of 2.62 µM [169,170,171].
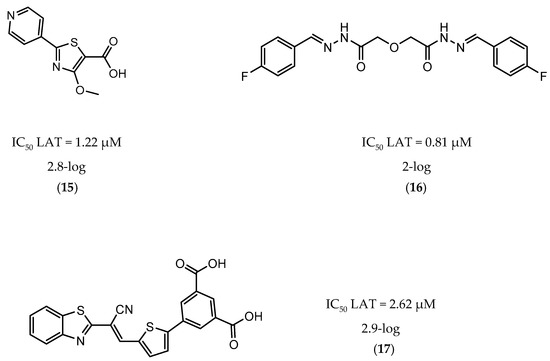
Figure 6.
Selected Lysine ε-aminotransferase (LAT) inhibitors (15, 16, 17), with respective LAT IC50 and bacterial reduction values.
4.5. Other Targets
Besides targets specifically related with the progression and maintenance of dormancy, over the past few years, compounds targeting well-known enzymes essential for M.tb survival have also been tested against nonreplicating bacilli [2]. The enoyl-acyl carrier protein reductase (InhA), decaprenyl-phosphoryl-ribose 2′-epimerase (DprE1), mycocyclosin synthase (Cyp121), and extracellular zinc metalloprotease 1 (Zmp1) are examples of successful screenings.
InhA is part of the fatty acid synthase (FAS)-II system, responsible for the catalysis of the synthesis of fatty acids using Acetyl-CoA [172]. InhA is the target of the first-line drug isoniazid that requires activation from a catalase-peroxidase enzyme, KatG, to act on its target [173,174]. Even though isoniazid is reported as inactive against dormant tuberculosis, [26] Doğan et al. screened their new family of thiourea-based derivatives against nutrient starved mycobacteria. The most promising compound (18, Figure 7) displayed a 2.6-log reduction in bacterial count. Activity against active bacilli and M.tb in infected macrophages was also confirmed [175].
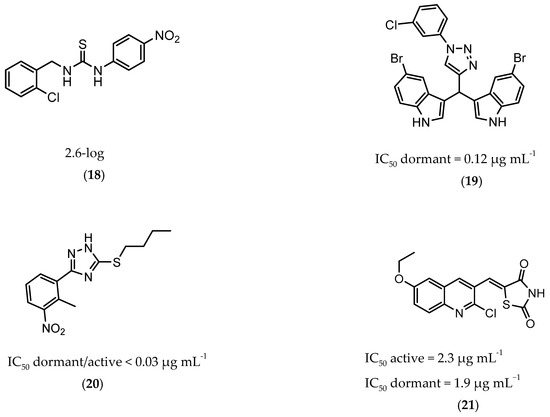
Figure 7.
Selected compounds active against dormant M.tb (18, 19, 20, 21), with referred bacterial count or IC50 for active/dormant bacilli values.
DprE1 is one of the enzymes responsible for the cell wall biosynthesis, catalyzing the formation of decaprenyl-phosphoarabinose from decaprenyl-phosphoribose [176]. This target has been extensively explored as several scaffolds have been discovered and optimized for its inhibition [177,178,179,180]. In 2018, a family of triazole-diindolylmethane derivatives was designed from a molecular hybridization approach, by combining different pharmacophores. All compounds were tested against an attenuated strain (M.tb H37Ra), in both active and dormant state. Compound 19 (Figure 7) proved to be the most potent, with an IC50 of 0.12 µg mL−1 for nonreplicating bacilli [181].
CYP121 is an enzyme from the family cytochrome P450 that catalyzes the formation of mycocyclosin from cyclodityrosine, by coupling two tyrosine residues and generating a carbon-carbon bond [182,183]. This enzyme is essential for the survival of M.tb and cannot be found in any other microorganism or human cell, presenting itself as a specific target for the bacillus [184]. A series of 3-aryl-5-(alkyl-thio)-1H-1,2,4-triazoles derivatives was developed and was found to inhibit CYP121. These compounds were tested against H37Ra, with the most active compound (20, Figure 7) showing IC50 of <0.03 µg mL−1 against both dormant and active bacilli [185].
Zmp1 is an enzyme responsible for preventing the fusion between the phagosome and the lysosome within the macrophage [186,187]. This zinc-peptidase is crucial for the intracellular survival of the bacillus as it averts the phagosome maturation, playing a key role in its pathogenicity [188]. Conjugates of rhodanine and quinoline were synthesized and identified as Zmp1 inhibitors. All compounds were screened against the H37Ra strain in both active and dormant forms. The most promising compound (21) is depicted in Figure 7, showing IC50 of 2.3 µg mL−1 for active M.tb and 1.9 µg mL−1 for dormant M.tb [189].
All compounds referred in the previous section are depicted in Table 1.

Table 1.
Compounds active against dormant M. tuberculosis.
5. Conclusions
The End TB Strategy intends to diminish the deaths from tuberculosis by 95% and sets 2035 as the year for this goal to become a reality [18]. Tuberculosis is nowadays one of the top ten causes of death worldwide and is responsible for one in every three deaths in people infected with HIV [3]. Thus, eradicating this disease in fifteen years is now a major challenge for the scientific community. Resistance is growing on first-line drugs and current drugs require long therapeutic periods. The subsequent low compliance that results from this last factor fails to completely cure the patient and, in most cases, leads to latent tuberculosis that can eventually turn active and restart the problem. Furthermore, the first-line treatment for the latent illness requires once again long treatments, is not effective in dormant bacilli and has been proved to induce its own resistance when used in monotherapy [28].
The goal set up by the WHO of eradicating TB by 2035 can still be accomplished but, this endeavor requires therapies to be highly monitored and more effective drugs to reach the market. More specifically, drugs that efficaciously cure the disease and are active against latent M.tb need to move forward in the pipeline and generate shorter and more effective treatments. More complex and accurate methods to mimic the latent state are needed. Most of the techniques used nowadays only take one latency inducing factor into consideration and it is common that the same drug exhibits contradictory results in different methods. The development of the latent state is complex, not fully understood and involves a series of factors that differently impact genetic modulation. A faithful reproduction will not be easy to accomplish. Most techniques currently used for drug screening against the latent state have not been updated for years and an effort needs to be made by the scientific community in order to generate more accurate mimicking methods that will increase the scientific impact of new compounds.
In order to fully eradicate the disease, this latency needs to be completely understood, specifically targeted, and successfully prevented. Tuberculosis must be addressed as a three-way problem: active disease, resistance to current drugs, and latency. Despite being equally important, research on compounds that are effective in the latent state has not been treated as a priority and, as long as fighting this nonreplicating bacilli is not fully embraced, M.tb will remain a problem to be solved.
Author Contributions
Conceptualization, F.L.; writing—original draft preparation, A.C.; writing—review and editing, A.C., S.G.H., D.F.W., R.M. and F.L.; supervision, F.L.; project administration, F.L.; funding acquisition, D.F.W and F.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by projects UIDB/04138/2020 and UIDP/04138/2020 (Fundação para a Ciência e Tecnologia (FCT), Portugal) and PTDC/MED-FAR/30266/2017 (FCT and FEDER). We also acknowledge FCT for fellowship SFRH/BD/131896/2017 (A.C.). This work was also supported by the Strategic Health Innovation Partnerships (SHIP) initiative of the South African Medical Research Council with funds from the National Treasury under its Economic Competitiveness and Support Package (D.F.W.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Blevins, S.M.; Bronze, M.S. Robert Koch and the ‘golden age’ of bacteriology. Int. J. Infect. Dis. 2010, 14, e744–e751. [Google Scholar] [CrossRef] [PubMed]
- Campanico, A.; Moreira, R.; Lopes, F. Drug discovery in tuberculosis. New drug targets and antimycobacterial agents. Eur. J. Med. Chem. 2018, 150, 525–545. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Mabhula, A.; Singh, V. Drug-resistance in Mycobacterium tuberculosis: Where we stand. MedChemComm 2019, 10, 1342–1360. [Google Scholar] [CrossRef] [PubMed]
- LoBue, P.A.; Mermin, J.H. Latent tuberculosis infection: The final frontier of tuberculosis elimination in the USA. Lancet Infect. Dis. 2017, 17, e327–e333. [Google Scholar] [CrossRef]
- Lazarevic, V.; Nolt, D.; Flynn, J.L. Long-term control of Mycobacterium tuberculosis infection is mediated by dynamic immune responses. J. Immunol. 2005, 175, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Via, L.E.; Lin, P.L.; Ray, S.M.; Carrillo, J.; Allen, S.S.; Eum, S.Y.; Taylor, K.; Klein, E.; Manjunatha, U.; Gonzales, J.; et al. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect. Immun. 2008, 76, 2333–2340. [Google Scholar] [CrossRef]
- Wayne, L.G.; Hayes, L.G. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 1996, 64, 2062–2069. [Google Scholar] [CrossRef]
- Leistikow, R.L.; Morton, R.A.; Bartek, I.L.; Frimpong, I.; Wagner, K.; Voskuil, M.I. The Mycobacterium tuberculosis DosR regulon assists in metabolic homeostasis and enables rapid recovery from nonrespiring dormancy. J. Bacteriol. 2010, 192, 1662–1670. [Google Scholar] [CrossRef]
- Park, H.D.; Guinn, K.M.; Harrell, M.I.; Liao, R.; Voskuil, M.I.; Tompa, M.; Schoolnik, G.K.; Sherman, D.R. Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol. Microbiol. 2003, 48, 833–843. [Google Scholar] [CrossRef]
- Chee, C.B.E.; Reves, R.; Zhang, Y.; Belknap, R. Latent tuberculosis infection: Opportunities and challenges. Respirology 2018, 23, 893–900. [Google Scholar] [CrossRef]
- Comstock, G.W.; Livesay, V.T.; Woolpert, S.F. The prognosis of a positive tuberculin reaction in childhood and adolescence. Am. J. Epidemiol. 1974, 99, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Horsburgh, C.R., Jr.; Rubin, E.J. Clinical practice. Latent tuberculosis infection in the United States. N. Engl. J. Med. 2011, 364, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- CDC. Treatment of Tuberculosis Disease. In Core Curriculum on Tuberculosis: What the Clinician Should Know, 6th ed.; CDC: Atlanta, GA, USA, 2013. [Google Scholar]
- Tiberi, S.; du Plessis, N.; Walzl, G.; Vjecha, M.J.; Rao, M.; Ntoumi, F.; Mfinanga, S.; Kapata, N.; Mwaba, P.; McHugh, T.D.; et al. Tuberculosis: Progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect. Dis. 2018, 18, e183–e198. [Google Scholar] [CrossRef]
- Connolly, L.E.; Edelstein, P.H.; Ramakrishnan, L. Why is long-term therapy required to cure tuberculosis? PLoS Med. 2007, 4, e120. [Google Scholar] [CrossRef]
- Houben, R.M.; Dodd, P.J. The Global Burden of Latent Tuberculosis Infection: A Re-estimation Using Mathematical Modelling. PLoS Med. 2016, 13, e1002152. [Google Scholar] [CrossRef]
- World Health Organization. EndTB Strategy; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Huaman, M.A.; Sterling, T.R. Treatment of Latent Tuberculosis Infection—An Update. Clin. Chest Med. 2019, 40, 839–848. [Google Scholar] [CrossRef]
- Rangaka, M.X.; Wilkinson, R.J.; Boulle, A.; Glynn, J.R.; Fielding, K.; van Cutsem, G.; Wilkinson, K.A.; Goliath, R.; Mathee, S.; Goemaere, E.; et al. Isoniazid plus antiretroviral therapy to prevent tuberculosis: A randomised double-blind, placebo-controlled trial. Lancet 2014, 384, 682–690. [Google Scholar] [CrossRef]
- Zenner, D.; Beer, N.; Harris, R.J.; Lipman, M.C.; Stagg, H.R.; van der Werf, M.J. Treatment of Latent Tuberculosis Infection: An Updated Network Meta-analysis. Ann. Intern. Med. 2017, 167, 248–255. [Google Scholar] [CrossRef]
- Comstock, G.W. How much isoniazid is needed for prevention of tuberculosis among immunocompetent adults? Int. J. Tuberc. Lung Dis. 1999, 3, 847–850. [Google Scholar]
- Kim, H.W.; Kim, J.S. Treatment of Latent Tuberculosis Infection and Its Clinical Efficacy. Tuberc. Respir. Dis. 2018, 81, 6–12. [Google Scholar] [CrossRef]
- Lienhardt, C.; Fielding, K.; Sillah, J.; Tunkara, A.; Donkor, S.; Manneh, K.; Warndorff, D.; McAdam, K.P.; Bennett, S. Risk factors for tuberculosis infection in sub-Saharan Africa: A contact study in The Gambia. Am. J. Respir. Crit. Care Med. 2003, 168, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Rathi, S.K.; Akhtar, S.; Rahbar, M.H.; Azam, S.I. Prevalence and risk factors associated with tuberculin skin test positivity among household contacts of smear-positive pulnionary tuberculosis cases in Umerkot, Pakistan. Int. J. Tuberc. Lung Dis. 2002, 6, 851–857. [Google Scholar] [PubMed]
- Piccaro, G.; Giannoni, F.; Filippini, P.; Mustazzolu, A.; Fattorini, L. Activities of drug combinations against Mycobacterium tuberculosis grown in aerobic and hypoxic acidic conditions. Antimicrob. Agents Chemother. 2013, 57, 1428–1433. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zumla, A.; Nahid, P.; Cole, S.T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 2013, 12, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, S.; Takhar, P.; Baldeviano, C.; Glover, W.; Zhang, Y. Isoniazid induces its own resistance in nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 2100–2104. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Iacobino, A.; Piccaro, G.; Giannoni, F.; Mustazzolu, A.; Fattorini, L. Fighting tuberculosis by drugs targeting nonreplicating Mycobacterium tuberculosis bacilli. Int. J. Mycobacteriol. 2017, 6, 213–221. [Google Scholar]
- Menzies, D.; Adjobimey, M.; Ruslami, R.; Trajman, A.; Sow, O.; Kim, H.; Obeng Baah, J.; Marks, G.B.; Long, R.; Hoeppner, V.; et al. Four Months of Rifampin or Nine Months of Isoniazid for Latent Tuberculosis in Adults. N. Engl. J. Med. 2018, 379, 440–453. [Google Scholar] [CrossRef]
- Menzies, D.; Long, R.; Trajman, A.; Dion, M.J.; Yang, J.; Al Jahdali, H.; Memish, Z.; Khan, K.; Gardam, M.; Hoeppner, V.; et al. Adverse events with 4 months of rifampin therapy or 9 months of isoniazid therapy for latent tuberculosis infection: A randomized trial. Ann. Intern. Med. 2008, 149, 689–697. [Google Scholar] [CrossRef]
- Fatal and severe hepatitis associated with rifampin and pyrazinamide for the treatment of latent tuberculosis infection--New York and Georgia, 2000. MMWR Morb. Mortal. Wkly. Rep. 2001, 50, 289–291.
- Yee, D.; Valiquette, C.; Pelletier, M.; Parisien, I.; Rocher, I.; Menzies, D. Incidence of serious side effects from first-line antituberculosis drugs among patients treated for active tuberculosis. Am. J. Respir. Crit. Care Med. 2003, 167, 1472–1477. [Google Scholar] [CrossRef]
- Petersen, E.; Chakaya, J.; Jawad, F.M.; Ippolito, G.; Zumla, A. Latent tuberculosis infection: Diagnostic tests and when to treat. Lancet Infect. Dis. 2019, 19, 231–233. [Google Scholar] [CrossRef]
- Haas, M.K.; Belknap, R.W. Diagnostic Tests for Latent Tuberculosis Infection. Clin. Chest Med. 2019, 40, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Farhat, M.; Greenaway, C.; Pai, M.; Menzies, D. False-positive tuberculin skin tests: What is the absolute effect of BCG and non-tuberculous mycobacteria? Int. J. Tuberc Lung Dis. 2006, 10, 1192–1204. [Google Scholar] [PubMed]
- Andersen, P.; Munk, M.E.; Pollock, J.M.; Doherty, T.M. Specific immune-based diagnosis of tuberculosis. Lancet 2000, 356, 1099–1104. [Google Scholar] [CrossRef]
- Geluk, A.; van Meijgaarden, K.E.; Franken, K.L.M.C.; Subronto, Y.W.; Wieles, B.; Arend, S.M.; Sampaio, E.P.; de Boer, T.; Faber, W.R.; Naafs, B.; et al. Identification and characterization of the ESAT-6 homologue of Mycobacterium leprae and T-cell cross-reactivity with Mycobacterium tuberculosis. Infect. Immun. 2002, 70, 2544–2548. [Google Scholar] [CrossRef]
- Mack, U.; Migliori, G.B.; Sester, M.; Rieder, H.L.; Ehlers, S.; Goletti, D.; Bossink, A.; Magdorf, K.; Holscher, C.; Kampmann, B.; et al. LTBI: Latent tuberculosis infection or lasting immune responses to M. tuberculosis? A TBNET consensus statement. Eur. Respir. J. 2009, 33, 956–973. [Google Scholar] [CrossRef]
- Sarrazin, H.; Wilkinson, K.A.; Andersson, J.; Rangaka, M.X.; Radler, L.; van Veen, K.; Lange, C.; Wilkinson, R.J. Association between tuberculin skin test reactivity, the memory CD4 cell subset, and circulating FoxP3-expressing cells in HIV-infected persons. J. Infect. Dis. 2009, 199, 702–710. [Google Scholar] [CrossRef]
- Pai, M.; Sotgiu, G. Diagnostics for latent TB infection: Incremental, not transformative progress. Eur. Respir. J. 2016, 47, 704–706. [Google Scholar] [CrossRef]
- Gibson, S.E.R.; Harrison, J.; Cox, J.A.G. Modelling a Silent Epidemic: A Review of the In Vitro Models of Latent Tuberculosis. Pathogens 2018, 7, 88. [Google Scholar] [CrossRef]
- Belton, M.; Brilha, S.; Manavaki, R.; Mauri, F.; Nijran, K.; Hong, Y.T.; Patel, N.H.; Dembek, M.; Tezera, L.; Green, J.; et al. Hypoxia and tissue destruction in pulmonary TB. Thorax 2016, 71, 1145–1153. [Google Scholar] [CrossRef]
- Loebel, R.O.; Shorr, E.; Richardson, H.B. The Influence of Adverse Conditions upon the Respiratory Metabolism and Growth of Human Tubercle Bacilli. J. Bacteriol. 1933, 26, 167–200. [Google Scholar] [CrossRef] [PubMed]
- Nyka, W. Studies on the effect of starvation on mycobacteria. Infect. Immun. 1974, 9, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Guirado, E.; Schlesinger, L.S. Modeling the Mycobacterium tuberculosis Granuloma - the Critical Battlefield in Host Immunity and Disease. Front. Immunol. 2013, 4, 98. [Google Scholar] [CrossRef] [PubMed]
- Wayne, L.G. Dynamics of submerged growth of Mycobacterium tuberculosis under aerobic and microaerophilic conditions. Am. Rev. Respir. Dis. 1976, 114, 807–811. [Google Scholar] [PubMed]
- Wayne, L.G.; Sohaskey, C.D. Nonreplicating persistence of Mycobacterium tuberculosis. Annu. Rev. Microbiol. 2001, 55, 139–163. [Google Scholar] [CrossRef] [PubMed]
- Voskuil, M.I.; Visconti, K.C.; Schoolnik, G.K. Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low-oxygen dormancy. Tuberculosis 2004, 84, 218–227. [Google Scholar] [CrossRef]
- Boon, C.; Li, R.; Qi, R.; Dick, T. Proteins of Mycobacterium bovis BCG induced in the Wayne dormancy model. J. Bacteriol. 2001, 183, 2672–2676. [Google Scholar] [CrossRef]
- Wayne, L.G. In Vitro Model of Hypoxically Induced Nonreplicating Persistence of Mycobacterium tuberculosis. Methods Mol. Med. 2001, 54, 247–269. [Google Scholar]
- Wayne, L.G.; Sramek, H.A. Metronidazole is bactericidal to dormant cells of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1994, 38, 2054–2058. [Google Scholar] [CrossRef]
- Hoff, D.R.; Caraway, M.L.; Brooks, E.J.; Driver, E.R.; Ryan, G.J.; Peloquin, C.A.; Orme, I.M.; Basaraba, R.J.; Lenaerts, A.J. Metronidazole lacks antibacterial activity in guinea pigs infected with Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2008, 52, 4137–4140. [Google Scholar] [CrossRef]
- Klinkenberg, L.G.; Sutherland, L.A.; Bishai, W.R.; Karakousis, P.C. Metronidazole lacks activity against Mycobacterium tuberculosis in an in vivo hypoxic granuloma model of latency. J. Infect. Dis. 2008, 198, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.L.; Dartois, V.; Johnston, P.J.; Janssen, C.; Via, L.; Goodwin, M.B.; Klein, E.; Barry, C.E., 3rd; Flynn, J.L. Metronidazole prevents reactivation of latent Mycobacterium tuberculosis infection in macaques. Proc. Natl. Acad. Sci. USA 2012, 109, 14188–14193. [Google Scholar] [CrossRef] [PubMed]
- Franzblau, S.G.; DeGroote, M.A.; Cho, S.H.; Andries, K.; Nuermberger, E.; Orme, I.M.; Mdluli, K.; Angulo-Barturen, I.; Dick, T.; Dartois, V.; et al. Comprehensive analysis of methods used for the evaluation of compounds against Mycobacterium tuberculosis. Tuberculosis 2012, 92, 453–488. [Google Scholar] [CrossRef] [PubMed]
- Taneja, N.K.; Tyagi, J.S. Resazurin reduction assays for screening of anti-tubercular compounds against dormant and actively growing Mycobacterium tuberculosis, Mycobacterium bovis BCG and Mycobacterium smegmatis. J. Antimicrob. Chemother. 2007, 60, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Aher, R.B.; Sarkar, D. Pharmacophore modeling of pretomanid (PA-824) derivatives for antitubercular potency against replicating and non-replicating Mycobacterium tuberculosis. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bonnett, S.A.; Dennison, D.; Files, M.; Bajpai, A.; Parish, T. A class of hydrazones are active against non-replicating Mycobacterium tuberculosis. PLoS ONE 2018, 13, e0198059. [Google Scholar] [CrossRef]
- Cho, S.; Lee, H.S.; Franzblau, S. Microplate Alamar Blue Assay (MABA) and Low Oxygen Recovery Assay (LORA) for Mycobacterium tuberculosis. Methods Mol. Biol. 2015, 1285, 281–292. [Google Scholar]
- Snewin, V.A.; Gares, M.P.; Gaora, P.O.; Hasan, Z.; Brown, I.N.; Young, D.B. Assessment of immunity to mycobacterial infection with luciferase reporter constructs. Infect. Immun. 1999, 67, 4586–4593. [Google Scholar] [CrossRef]
- Cho, S.H.; Warit, S.; Wan, B.; Hwang, C.H.; Pauli, G.F.; Franzblau, S.G. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 1380–1385. [Google Scholar] [CrossRef]
- Yeware, A.; Sarkar, D. Novel red fluorescence protein based microplate assay for drug screening against dormant Mycobacterium tuberculosis by using paraffin. Tuberculosis 2018, 110, 15–19. [Google Scholar] [CrossRef]
- Carroll, P.; Schreuder, L.J.; Muwanguzi-Karugaba, J.; Wiles, S.; Robertson, B.D.; Ripoll, J.; Ward, T.H.; Bancroft, G.J.; Schaible, U.E.; Parish, T. Sensitive detection of gene expression in mycobacteria under replicating and non-replicating conditions using optimized far-red reporters. PLoS ONE 2010, 5, e9823. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Loebel, R.O.; Shorr, E.; Richardson, H.B. The Influence of Foodstuffs upon the Respiratory Metabolism and Growth of Human Tubercle Bacilli. J. Bacteriol. 1933, 26, 139–166. [Google Scholar] [CrossRef] [PubMed]
- Betts, J.C.; Lukey, P.T.; Robb, L.C.; McAdam, R.A.; Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 2002, 43, 717–731. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.J.; Brown, J.R. Identification of gene targets against dormant phase Mycobacterium tuberculosis infections. BMC Infect. Dis. 2007, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Hampshire, T.; Soneji, S.; Bacon, J.; James, B.W.; Hinds, J.; Laing, K.; Stabler, R.A.; Marsh, P.D.; Butcher, P.D. Stationary phase gene expression of Mycobacterium tuberculosis following a progressive nutrient depletion: A model for persistent organisms? Tuberculosis 2004, 84, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Chinta, K.C.; Saini, V.; Glasgow, J.N.; Mazorodze, J.H.; Rahman, M.A.; Reddy, D.; Lancaster, J.R., Jr.; Steyn, A.J. The emerging role of gasotransmitters in the pathogenesis of tuberculosis. Nitric Oxide 2016, 59, 28–41. [Google Scholar] [CrossRef]
- Voskuil, M.I.; Schnappinger, D.; Visconti, K.C.; Harrell, M.I.; Dolganov, G.M.; Sherman, D.R.; Schoolnik, G.K. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 2003, 198, 705–713. [Google Scholar] [CrossRef]
- Wayne, L.G.; Hayes, L.G. Nitrate reduction as a marker for hypoxic shiftdown of Mycobacterium tuberculosis. Tuber. Lung Dis. 1998, 79, 127–132. [Google Scholar] [CrossRef]
- Campos-Neto, A. Mycobacterium tuberculosis strain 18b, a useful non-virulent streptomycin dependent mutant to study latent tuberculosis as well as for in vivo and in vitro testing of anti-tuberculosis drugs. Tuberculosis 2016, 99, 54–55. [Google Scholar] [CrossRef]
- Hashimoto, T. Experimental studies on the mechanism of infection and immunity in tuberculosis from the analytical standpoint of streptomycin-dependent tubercle bacilli. Isolation and biological characteristics of a streptomycin-dependent mutant, and effect of streptomycin administration on its pathogenicity in guinea-pigs. Kekkaku 1955, 30, 4–8, English summary, 45–46. [Google Scholar] [PubMed]
- Sala, C.; Dhar, N.; Hartkoorn, R.C.; Zhang, M.; Ha, Y.H.; Schneider, P.; Cole, S.T. Simple model for testing drugs against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2010, 54, 4150–4158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sala, C.; Hartkoorn, R.C.; Dhar, N.; Mendoza-Losana, A.; Cole, S.T. Streptomycin-starved Mycobacterium tuberculosis 18b, a drug discovery tool for latent tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 5782–5789. [Google Scholar] [CrossRef] [PubMed]
- Karabanovich, G.; Zemanova, J.; Smutny, T.; Szekely, R.; Sarkan, M.; Centarova, I.; Vocat, A.; Pavkova, I.; Conka, P.; Nemecek, J.; et al. Development of 3,5-Dinitrobenzylsulfanyl-1,3,4-oxadiazoles and Thiadiazoles as Selective Antitubercular Agents Active Against Replicating and Nonreplicating Mycobacterium tuberculosis. J. Med. Chem. 2016, 59, 2362–2380. [Google Scholar] [CrossRef]
- Zhang, M.; Sala, C.; Dhar, N.; Vocat, A.; Sambandamurthy, V.K.; Sharma, S.; Marriner, G.; Balasubramanian, V.; Cole, S.T. In vitro and in vivo activities of three oxazolidinones against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 3217–3223. [Google Scholar] [CrossRef]
- Deb, C.; Lee, C.M.; Dubey, V.S.; Daniel, J.; Abomoelak, B.; Sirakova, T.D.; Pawar, S.; Rogers, L.; Kolattukudy, P.E. A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS ONE 2009, 4, e6077. [Google Scholar] [CrossRef]
- Gold, B.; Warrier, T.; Nathan, C. A multi-stress model for high throughput screening against non-replicating Mycobacterium tuberculosis. Methods Mol. Biol. 2015, 1285, 293–315. [Google Scholar]
- Gold, B.; Deng, H.; Bryk, R.; Vargas, D.; Eliezer, D.; Roberts, J.; Jiang, X.; Nathan, C. Identification of a copper-binding metallothionein in pathogenic mycobacteria. Nature Chem. Biol. 2008, 4, 609–616. [Google Scholar] [CrossRef]
- Gold, B.; Rodriguez, G.M.; Marras, S.A.; Pentecost, M.; Smith, I. The Mycobacterium tuberculosis IdeR is a dual functional regulator that controls transcription of genes involved in iron acquisition, iron storage and survival in macrophages. Mol. Microbiol. 2001, 42, 851–865. [Google Scholar] [CrossRef]
- Hondalus, M.K.; Bardarov, S.; Russell, R.; Chan, J.; Jacobs, W.R., Jr.; Bloom, B.R. Attenuation of and protection induced by a leucine auxotroph of Mycobacterium tuberculosis. Infect. Immun. 2000, 68, 2888–2898. [Google Scholar]
- Ng, V.H.; Cox, J.S.; Sousa, A.O.; MacMicking, J.D.; McKinney, J.D. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: Countering the phagocyte oxidative burst. Mol. Microbiol. 2004, 52, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.M.; Voskuil, M.I.; Gold, B.; Schoolnik, G.K.; Smith, I. ideR, An essential gene in mycobacterium tuberculosis: Role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect. Immun. 2002, 70, 3371–3381. [Google Scholar] [CrossRef] [PubMed]
- Sambandamurthy, V.K.; Wang, X.; Chen, B.; Russell, R.G.; Derrick, S.; Collins, F.M.; Morris, S.L.; Jacobs, W.R. A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat. Med. 2002, 8, 1171–1174. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Festa, R.A.; Ioerger, T.R.; Butler-Wu, S.; Sacchettini, J.C.; Darwin, K.H.; Samanovic, M.I. The Copper-Responsive RicR Regulon Contributes to Mycobacterium tuberculosis Virulence. mBio 2014, 5, e00876-13. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, M.U.; Manzanillo, P.; Cox, J.S. Mycobacterium tuberculosis Senses Host-Derived Carbon Monoxide during Macrophage Infection. Cell Host Microbe 2008, 3, 323–330. [Google Scholar] [CrossRef]
- Timm, J.; Post, F.A.; Bekker, L.-G.; Walther, G.B.; Wainwright, H.C.; Manganelli, R.; Chan, W.-T.; Tsenova, L.; Gold, B.; Smith, I.; et al. Differential expression of iron-, carbon-, and oxygen-responsive mycobacterial genes in the lungs of chronically infected mice and tuberculosis patients. Proc. Natl. Acad. Sci. USA 2003, 100, 14321. [Google Scholar] [CrossRef]
- Walters, S.B.; Dubnau, E.; Kolesnikova, I.; Laval, F.; Daffe, M.; Smith, I. The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol. Microb. 2006, 60, 312–330. [Google Scholar] [CrossRef]
- Dutta, N.K.; Karakousis, P.C. Latent tuberculosis infection: Myths, models, and molecular mechanisms. Microb. Mol. Biol. Rev. MMBR 2014, 78, 343–371. [Google Scholar] [CrossRef]
- Cosma, C.L.; Sherman, D.R.; Ramakrishnan, L. The secret lives of the pathogenic mycobacteria. Annu. Rev. Microbiol. 2003, 57, 641–676. [Google Scholar] [CrossRef]
- Sugawara, I.; Yamada, H.; Mizuno, S. Pathological and immunological profiles of rat tuberculosis. Int. J. Exp. Pathol. 2004, 85, 125–134. [Google Scholar] [CrossRef]
- Gupta, U.D.; Katoch, V.M. Animal models of tuberculosis. Tuberculosis 2005, 85, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Broussard, G.W.; Ennis, D.G. Mycobacterium marinum produces long-term chronic infections in medaka: A new animal model for studying human tuberculosis. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2007, 145, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Luukinen, H.; Hammarén, M.M.; Vanha-Aho, L.M.; Parikka, M. Modeling Tuberculosis in Mycobacterium marinum Infected Adult Zebrafish. J. Vis. Exp. 2018, 140, e58299. [Google Scholar] [CrossRef] [PubMed]
- Swaim, L.E.; Connolly, L.E.; Volkman, H.E.; Humbert, O.; Born, D.E.; Ramakrishnan, L. Mycobacterium marinum infection of adult zebrafish causes caseating granulomatous tuberculosis and is moderated by adaptive immunity. Infect. Immun. 2006, 74, 6108–6117. [Google Scholar] [CrossRef]
- McCune, R.M.; Feldmann, F.M.; McDermott, W. Microbial persistence. II. Characteristics of the sterile state of tubercle bacilli. J. Exp. Med. 1966, 123, 469–486. [Google Scholar] [CrossRef]
- McCune, R.M., Jr.; McDermott, W.; Tompsett, R. The fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. II. The conversion of tuberculous infection to the latent state by the administration of pyrazinamide and a companion drug. J. Exp. Med. 1956, 104, 763–802. [Google Scholar] [CrossRef]
- Ahmad, Z.; Sharma, S.; Khuller, G.K. The potential of azole antifungals against latent/persistent tuberculosis. FEMS Microbiol. Lett. 2006, 258, 200–203. [Google Scholar] [CrossRef]
- Woolhiser, L.; Tamayo, M.H.; Wang, B.; Gruppo, V.; Belisle, J.T.; Lenaerts, A.J.; Basaraba, R.J.; Orme, I.M. In Vivo Adaptation of the Wayne Model of Latent Tuberculosis. Infect Immun. 2007, 75, 2621. [Google Scholar] [CrossRef]
- Horwitz, M.A.; Harth, G. A New Vaccine against Tuberculosis Affords Greater Survival after Challenge than the Current Vaccine in the Guinea Pig Model of Pulmonary Tuberculosis. Infect. Immun. 2003, 71, 1672. [Google Scholar] [CrossRef]
- Nuermberger, E.L.; Yoshimatsu, T.; Tyagi, S.; Bishai, W.R.; Grosset, J.H. Paucibacillary Tuberculosis in Mice after Prior Aerosol Immunization with Mycobacterium bovis BCG. Infect. Immun. 2004, 72, 1065. [Google Scholar] [CrossRef]
- Dutta, N.K.; Illei, P.B.; Jain, S.K.; Karakousis, P.C. Characterization of a Novel Necrotic Granuloma Model of Latent Tuberculosis Infection and Reactivation in Mice. Am. J. Pathol. 2014, 184, 2045–2055. [Google Scholar] [CrossRef] [PubMed]
- Karakousis, P.C.; Yoshimatsu, T.; Lamichhane, G.; Woolwine, S.C.; Nuermberger, E.L.; Grosset, J.; Bishai, W.R. Dormancy Phenotype Displayed by Extracellular Mycobacterium tuberculosis within Artificial Granulomas in Mice. J. Exp. Med. 2004, 200, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.B.; Chen, B.W.; Wang, G.Z.; Fu, L.L.; Shen, X.B.; Su, C.; Du, W.X.; Yang, L.; Xu, M. Recombinant tuberculosis vaccine AEC/BC02 induces antigen-specific cellular responses in mice and protects guinea pigs in a model of latent infection. J. Microbiol. Immunol. Infect. 2015, 48, 597–603. [Google Scholar] [CrossRef]
- Guo-zhi, W.; Balasubramanian, V.; Smith, D.W. The protective and allergenic potency of four BCG substrains in use in China determined in two animal models. Tubercle 1988, 69, 283–291. [Google Scholar] [CrossRef]
- Kashino, S.S.; Napolitano, D.R.; Skobe, Z.; Campos-Neto, A. Guinea pig model of Mycobacterium tuberculosis latent/dormant infection. Microbes Infect. 2008, 10, 1469–1476. [Google Scholar] [CrossRef][Green Version]
- Manabe, Y.C.; Kesavan, A.K.; Lopez-Molina, J.; Hatem, C.L.; Brooks, M.; Fujiwara, R.; Hochstein, K.; Pitt, M.L.; Tufariello, J.; Chan, J.; et al. The aerosol rabbit model of TB latency, reactivation and immune reconstitution inflammatory syndrome. Tuberculosis 2008, 88, 187–196. [Google Scholar] [CrossRef]
- Arrazuria, R.; Juste, R.A.; Elguezabal, N. Mycobacterial Infections in Rabbits: From the Wild to the Laboratory. Transbound. Emerg. Dis. 2017, 64, 1045–1058. [Google Scholar] [CrossRef]
- Subbian, S.; Tsenova, L.; O’Brien, P.; Yang, G.; Kushner, N.L.; Parsons, S.; Peixoto, B.; Fallows, D.; Kaplan, G. Spontaneous latency in a rabbit model of pulmonary tuberculosis. Am. J. Pathol. 2012, 181, 1711–1724. [Google Scholar] [CrossRef]
- Maiello, P.; DiFazio, R.M.; Cadena, A.M.; Rodgers, M.A.; Lin, P.L.; Scanga, C.A.; Flynn, J.L. Rhesus Macaques Are More Susceptible to Progressive Tuberculosis than Cynomolgus Macaques: A Quantitative Comparison. Infect. Immun. 2018, 86, e00505–e00517. [Google Scholar] [CrossRef]
- Capuano, S.V., 3rd; Croix, D.A.; Pawar, S.; Zinovik, A.; Myers, A.; Lin, P.L.; Bissel, S.; Fuhrman, C.; Klein, E.; Flynn, J.L. Experimental Mycobacterium tuberculosis infection of cynomolgus macaques closely resembles the various manifestations of human M. tuberculosis infection. Infect. Immun. 2003, 71, 5831–5844. [Google Scholar] [CrossRef]
- Darrah, P.A.; Zeppa, J.J.; Maiello, P.; Hackney, J.A.; Wadsworth, M.H., 2nd; Hughes, T.K.; Pokkali, S.; Swanson, P.A., 2nd; Grant, N.L.; Rodgers, M.A.; et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature 2020, 577, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Diedrich, C.R.; Mattila, J.T.; Klein, E.; Janssen, C.; Phuah, J.; Sturgeon, T.J.; Montelaro, R.C.; Lin, P.L.; Flynn, J.L. Reactivation of latent tuberculosis in cynomolgus macaques infected with SIV is associated with early peripheral T cell depletion and not virus load. PLoS ONE 2010, 5, e9611. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.L.; Dietrich, J.; Tan, E.; Abalos, R.M.; Burgos, J.; Bigbee, C.; Bigbee, M.; Milk, L.; Gideon, H.P.; Rodgers, M.; et al. The multistage vaccine H56 boosts the effects of BCG to protect cynomolgus macaques against active tuberculosis and reactivation of latent Mycobacterium tuberculosis infection. J. Clin. Investig. 2012, 122, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.L.; Ford, C.B.; Coleman, M.T.; Myers, A.J.; Gawande, R.; Ioerger, T.; Sacchettini, J.; Fortune, S.M.; Flynn, J.L. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat. Med. 2014, 20, 75–79. [Google Scholar] [CrossRef]
- Lin, P.L.; Myers, A.; Smith, L.; Bigbee, C.; Bigbee, M.; Fuhrman, C.; Grieser, H.; Chiosea, I.; Voitenek, N.N.; Capuano, S.V.; et al. Tumor necrosis factor neutralization results in disseminated disease in acute and latent Mycobacterium tuberculosis infection with normal granuloma structure in a cynomolgus macaque model. Arthritis Rheum. 2010, 62, 340–350. [Google Scholar]
- Lin, P.L.; Rodgers, M.; Smith, L.; Bigbee, M.; Myers, A.; Bigbee, C.; Chiosea, I.; Capuano, S.V.; Fuhrman, C.; Klein, E.; et al. Quantitative comparison of active and latent tuberculosis in the cynomolgus macaque model. Infect. Immun. 2009, 77, 4631–4642. [Google Scholar] [CrossRef]
- Peña, J.C.; Ho, W.Z. Monkey models of tuberculosis: Lessons learned. Infect. Immun. 2015, 83, 852–862. [Google Scholar] [CrossRef]
- Pan, L.; Wei, N.; Jia, H.; Gao, M.; Chen, X.; Wei, R.; Sun, Q.; Gu, S.; Du, B.; Xing, A.; et al. Genome-wide transcriptional profiling identifies potential signatures in discriminating active tuberculosis from latent infection. Oncotarget 2017, 8, 112907–112916. [Google Scholar] [CrossRef]
- Seidi, K.; Jahanban-Esfahlan, R. A novel approach to eradicate latent TB: Based on resuscitation promoting factors. J. Med Hypotheses Ideas 2013, 7, 69–74. [Google Scholar] [CrossRef]
- Shi, L.; Sohaskey, C.D.; Kana, B.D.; Dawes, S.; North, R.J.; Mizrahi, V.; Gennaro, M.L. Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc. Natl. Acad. Sci. USA 2005, 102, 15629–15634. [Google Scholar] [CrossRef]
- Sohaskey, C.D.; Wayne, L.G. Role of narK2X and narGHJI in hypoxic upregulation of nitrate reduction by Mycobacterium tuberculosis. J. Bacteriol. 2003, 185, 7247–7256. [Google Scholar] [CrossRef] [PubMed]
- Sohaskey, C.D. Nitrate enhances the survival of Mycobacterium tuberculosis during inhibition of respiration. J. Bacteriol. 2008, 190, 2981–2986. [Google Scholar] [CrossRef]
- Giffin, M.M.; Modesti, L.; Raab, R.W.; Wayne, L.G.; Sohaskey, C.D. ald of Mycobacterium tuberculosis encodes both the alanine dehydrogenase and the putative glycine dehydrogenase. J. Bacteriol. 2012, 194, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Höner Zu Bentrup, K.; Miczak, A.; Swenson, D.L.; Russell, D.G. Characterization of activity and expression of isocitrate lyase in Mycobacterium avium and Mycobacterium tuberculosis. J. Bacteriol. 1999, 181, 7161–7167. [Google Scholar] [CrossRef] [PubMed]
- Giffin, M.M.; Shi, L.; Gennaro, M.L.; Sohaskey, C.D. Role of Alanine Dehydrogenase of Mycobacterium tuberculosis during Recovery from Hypoxic Nonreplicating Persistence. PLoS ONE 2016, 11, e0155522. [Google Scholar] [CrossRef] [PubMed]
- Bhusal, R.P.; Bashiri, G.; Kwai, B.X.C.; Sperry, J.; Leung, I.K.H. Targeting isocitrate lyase for the treatment of latent tuberculosis. Drug Discov. Today 2017, 22, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- May, E.E.; Leitão, A.; Tropsha, A.; Oprea, T.I. A systems chemical biology study of malate synthase and isocitrate lyase inhibition in Mycobacterium tuberculosis during active and NRP growth. Comput. Biol. Chem. 2013, 47, 167–180. [Google Scholar] [CrossRef]
- Saxena, S.; Devi, P.B.; Soni, V.; Yogeeswari, P.; Sriram, D. Identification of novel inhibitors against Mycobacterium tuberculosis L-alanine dehydrogenase (MTB-AlaDH) through structure-based virtual screening. J. Mol. Graph. Model. 2014, 47, 37–43. [Google Scholar] [CrossRef]
- Saxena, S.; Samala, G.; Sridevi, J.P.; Devi, P.B.; Yogeeswari, P.; Sriram, D. Design and development of novel Mycobacterium tuberculosis L-alanine dehydrogenase inhibitors. Eur. J. Med. Chem. 2015, 92, 401–414. [Google Scholar] [CrossRef]
- Reshma, R.S.; Saxena, S.; Bobesh, K.A.; Jeankumar, V.U.; Gunda, S.; Yogeeswari, P.; Sriram, D. Design and development of new class of Mycobacterium tuberculosis L-alanine dehydrogenase inhibitors. Bioorg. Med. Chem. 2016, 24, 4499–4508. [Google Scholar] [CrossRef]
- Samala, G.; Brindha Devi, P.; Saxena, S.; Gunda, S.; Yogeeswari, P.; Sriram, D. Anti-tubercular activities of 5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4-amine analogues endowed with high activity toward non-replicative Mycobacterium tuberculosis. Bioorg. Med. Chem. 2016, 24, 5556–5564. [Google Scholar] [CrossRef] [PubMed]
- Sriram, D.; Yogeeswari, P.; Methuku, S.; Vyas, D.R.; Senthilkumar, P.; Alvala, M.; Jeankumar, V.U. Synthesis of various 3-nitropropionamides as Mycobacterium tuberculosis isocitrate lyase inhibitor. Bioorg. Med. Chem. Lett. 2011, 21, 5149–5154. [Google Scholar] [CrossRef] [PubMed]
- McKinney, J.D.; Höner zu Bentrup, K.; Muñoz-Elías, E.J.; Miczak, A.; Chen, B.; Chan, W.T.; Swenson, D.; Sacchettini, J.C.; Jacobs, W.R., Jr.; Russell, D.G. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 2000, 406, 735–738. [Google Scholar] [CrossRef]
- Kozic, J.; Novotná, E.; Volková, M.; Stolaříková, J.; Trejtnar, F.; Vinšová, J. Synthesis and in vitro antimycobacterial activity of 2-methoxybenzanilides and their thioxo analogues. Eur. J. Med. Chem. 2012, 56, 387–395. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, S.; Deng, Q.; Li, X.; Meng, J.; Guan, Y.; Li, C.; Xiao, C. Identification of a novel inhibitor of isocitrate lyase as a potent antitubercular agent against both active and non-replicating Mycobacterium tuberculosis. Tuberculosis 2016, 97, 38–46. [Google Scholar] [CrossRef]
- Dorhoi, A.; Reece, S.T.; Kaufmann, S.H. For better or for worse: The immune response against Mycobacterium tuberculosis balances pathology and protection. Immunol. Rev. 2011, 240, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Mavi, P.S.; Singh, S.; Kumar, A. Reductive Stress: New Insights in Physiology and Drug Tolerance of Mycobacterium. Antioxid. Redox Signal. 2020, 32, 1348–1366. [Google Scholar] [CrossRef]
- Jothivasan, V.K.; Hamilton, C.J. Mycothiol: Synthesis, biosynthesis and biological functions of the major low molecular weight thiol in actinomycetes. Nat. Prod. Rep. 2008, 25, 1091–1117. [Google Scholar] [CrossRef]
- Schnappinger, D.; Ehrt, S.; Voskuil, M.I.; Liu, Y.; Mangan, J.A.; Monahan, I.M.; Dolganov, G.; Efron, B.; Butcher, P.D.; Nathan, C.; et al. Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J. Exp. Med. 2003, 198, 693–704. [Google Scholar] [CrossRef]
- Brunner, K.; Maric, S.; Reshma, R.S.; Almqvist, H.; Seashore-Ludlow, B.; Gustavsson, A.L.; Poyraz, Ö.; Yogeeswari, P.; Lundbäck, T.; Vallin, M.; et al. Inhibitors of the Cysteine Synthase CysM with Antibacterial Potency against Dormant Mycobacterium tuberculosis. J. Med. Chem. 2016, 59, 6848–6859. [Google Scholar] [CrossRef]
- Palde, P.B.; Bhaskar, A.; Pedró Rosa, L.E.; Madoux, F.; Chase, P.; Gupta, V.; Spicer, T.; Scampavia, L.; Singh, A.; Carroll, K.S. First-in-Class Inhibitors of Sulfur Metabolism with Bactericidal Activity against Non-Replicating, M. tuberculosis. ACS Chem. Biol. 2016, 11, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Schnell, R.; Sriram, D.; Schneider, G. Pyridoxal-phosphate dependent mycobacterial cysteine synthases: Structure, mechanism and potential as drug targets. Biochim. Biophys. Acta 2015, 1854, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, C.M.; Rubin, E.J. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. USA 2003, 100, 12989. [Google Scholar] [CrossRef] [PubMed]
- Brunner, K.; Steiner, E.M.; Reshma, R.S.; Sriram, D.; Schnell, R.; Schneider, G. Profiling of in vitro activities of urea-based inhibitors against cysteine synthases from Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2017, 27, 4582–4587. [Google Scholar] [CrossRef]
- Bhave, D.P.; Muse, W.B., 3rd; Carroll, K.S. Drug targets in mycobacterial sulfur metabolism. Infect. Disord. Drug Targets 2007, 7, 140–158. [Google Scholar] [CrossRef]
- Newton, G.L.; Fahey, R.C. Mycothiol biochemistry. Arch. Microbiol. 2002, 178, 388–394. [Google Scholar] [CrossRef]
- Hatzios, S.K.; Bertozzi, C.R. The regulation of sulfur metabolism in Mycobacterium tuberculosis. PLoS Pathog. 2011, 7, e1002036. [Google Scholar] [CrossRef]
- Williams, S.J.; Senaratne, R.H.; Mougous, J.D.; Riley, L.W.; Bertozzi, C.R. 5’-adenosinephosphosulfate lies at a metabolic branch point in mycobacteria. J. Biol. Chem. 2002, 277, 32606–32615. [Google Scholar] [CrossRef]
- Carroll, K.S.; Gao, H.; Chen, H.; Stout, C.D.; Leary, J.A.; Bertozzi, C.R. A conserved mechanism for sulfonucleotide reduction. PLoS Biol. 2005, 3, e250. [Google Scholar] [CrossRef]
- Paritala, H.; Suzuki, Y.; Carroll, K.S. Design, synthesis and evaluation of Fe-S targeted adenosine 5’-phosphosulfate reductase inhibitors. Nucleosides Nucleotides Nucleic Acids 2015, 34, 199–220. [Google Scholar] [CrossRef]
- Hong, J.A.; Bhave, D.P.; Carroll, K.S. Identification of critical ligand binding determinants in Mycobacterium tuberculosis adenosine-5’-phosphosulfate reductase. J. Med. Chem. 2009, 52, 5485–5495. [Google Scholar] [CrossRef] [PubMed]
- Cosconati, S.; Hong, J.A.; Novellino, E.; Carroll, K.S.; Goodsell, D.S.; Olson, A.J. Structure-based virtual screening and biological evaluation of Mycobacterium tuberculosis adenosine 5’-phosphosulfate reductase inhibitors. J. Med. Chem. 2008, 51, 6627–6630. [Google Scholar] [CrossRef] [PubMed]
- Sousa, E.H.S.; Gilles-Gonzalez, M.-A. Chapter Five—Haem-Based Sensors of O2: Lessons and Perspectives. Adv. Microb. Physiol. 2017, 71, 235–257. [Google Scholar]
- Zheng, H.; Abramovitch, R.B. Inhibiting DosRST as a new approach to tuberculosis therapy. Future Med. Chem. 2020, 12, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, N.; Kapur, V.; Singh, K.K.; Das, T.K.; Sachdeva, S.; Jyothisri, K.; Tyagi, J.S. Characterization of a two-component system, devR-devS, of Mycobacterium tuberculosis. Tuberc. Lung Dis. 2000, 80, 141–159. [Google Scholar] [CrossRef] [PubMed]
- Sousa, E.H.S.; Tuckerman, J.R.; Gonzalez, G.; Gilles-Gonzalez, M.-A. DosT and DevS are oxygen-switched kinases in Mycobacterium tuberculosis. Protein Sci. 2007, 16, 1708–1719. [Google Scholar] [CrossRef]
- Roberts, D.M.; Liao, R.P.; Wisedchaisri, G.; Hol, W.G.; Sherman, D.R. Two sensor kinases contribute to the hypoxic response of Mycobacterium tuberculosis. J. Biol. Chem. 2004, 279, 23082–23087. [Google Scholar] [CrossRef]
- Sousa, E.H.S.; Diógenes, I.C.N.; Lopes, L.G.F.; Moura, J.J.G. Potential therapeutic approaches for a sleeping pathogen: Tuberculosis a case for bioinorganic chemistry. J. Biol. Inorg. Chem. 2020, 25, 685–704. [Google Scholar] [CrossRef]
- Gupta, R.K.; Thakur, T.S.; Desiraju, G.R.; Tyagi, J.S. Structure-based design of DevR inhibitor active against nonreplicating Mycobacterium tuberculosis. J. Med. Chem. 2009, 52, 6324–6334. [Google Scholar] [CrossRef]
- Zheng, H.; Colvin, C.J.; Johnson, B.K.; Kirchhoff, P.D.; Wilson, M.; Jorgensen-Muga, K.; Larsen, S.D.; Abramovitch, R.B. Inhibitors of Mycobacterium tuberculosis DosRST signaling and persistence. Nat. Chem. Biol. 2017, 13, 218–225. [Google Scholar] [CrossRef]
- Zheng, H.; Williams, J.T.; Aleiwi, B.; Ellsworth, E.; Abramovitch, R.B. Inhibiting Mycobacterium tuberculosis DosRST Signaling by Targeting Response Regulator DNA Binding and Sensor Kinase Heme. ACS Chem. Biol. 2020, 15, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Mak, P.A.; Rao, S.P.; Ping Tan, M.; Lin, X.; Chyba, J.; Tay, J.; Ng, S.H.; Tan, B.H.; Cherian, J.; Duraiswamy, J.; et al. A high-throughput screen to identify inhibitors of ATP homeostasis in non-replicating Mycobacterium tuberculosis. ACS Chem. Biol. 2012, 7, 1190–1197. [Google Scholar] [CrossRef]
- Soda, K.; Misono, H.; Yamamoto, T. L-lysine-.alpha.-ketoglutarate aminotransferase. I. Identification of a product, DELTA.1-piperideine-6-carboxylic acid. Biochemistry 1968, 7, 4102–4109. [Google Scholar] [CrossRef] [PubMed]
- Soda, K.; Misono, H. L-lysine-.alpha.-ketoglutarate aminotransferase. II. Purification, crystallization, and properties. Biochemistry 1968, 7, 4110–4119. [Google Scholar] [CrossRef]
- Duan, X.; Li, Y.; Du, Q.; Huang, Q.; Guo, S.; Xu, M.; Lin, Y.; Liu, Z.; Xie, J. Mycobacterium Lysine ε-aminotransferase is a novel alarmone metabolism related persister gene via dysregulating the intracellular amino acid level. Sci. Rep. 2016, 6, 19695. [Google Scholar] [CrossRef] [PubMed]
- Devi, P.B.; Sridevi, J.P.; Kakan, S.S.; Saxena, S.; Jeankumar, V.U.; Soni, V.; Anantaraju, H.S.; Yogeeswari, P.; Sriram, D. Discovery of novel lysine ɛ-aminotransferase inhibitors: An intriguing potential target for latent tuberculosis. Tuberculosis 2015, 95, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Parthiban, B.D.; Saxena, S.; Chandran, M.; Jonnalagadda, P.S.; Yadav, R.; Srilakshmi, R.R.; Perumal, Y.; Dharmarajan, S. Design and Development of Mycobacterium tuberculosis Lysine ɛ-Aminotransferase Inhibitors for Latent Tuberculosis Infection. Chem. Biol. Drug Des. 2016, 87, 265–274. [Google Scholar] [CrossRef]
- Reshma, R.S.; Jeankumar, V.U.; Kapoor, N.; Saxena, S.; Bobesh, K.A.; Vachaspathy, A.R.; Kolattukudy, P.E.; Sriram, D. Mycobacterium tuberculosis lysine-ɛ-aminotransferase a potential target in dormancy: Benzothiazole based inhibitors. Bioorg. Med. Chem. 2017, 25, 2761–2771. [Google Scholar] [CrossRef]
- Rana, P.; Ghouse, S.M.; Akunuri, R.; Madhavi, Y.V.; Chopra, S.; Nanduri, S. FabI (enoyl acyl carrier protein reductase) - A potential broad spectrum therapeutic target and its inhibitors. Eur. J. Med. Chem. 2020, 208, 112757. [Google Scholar] [CrossRef]
- Click, E.S.; Kurbatova, E.V.; Alexander, H.; Dalton, T.L.; Chen, M.P.; Posey, J.E.; Ershova, J.; Cegielski, J.P. Isoniazid and Rifampin-Resistance Mutations Associated with Resistance to Second-Line Drugs and With Sputum Culture Conversion. J. Infect. Dis. 2020, 221, 2072–2082. [Google Scholar] [CrossRef]
- Rivière, E.; Whitfield, M.G.; Nelen, J.; Heupink, T.H.; Van Rie, A. Identifying isoniazid resistance markers to guide inclusion of high-dose isoniazid in tuberculosis treatment regimens. Clin. Microbiol. Infect. 2020, 26, 1332–1337. [Google Scholar] [CrossRef] [PubMed]
- Doğan, Ş.D.; Gündüz, M.G.; Doğan, H.; Krishna, V.S.; Lherbet, C.; Sriram, D. Design and synthesis of thiourea-based derivatives as Mycobacterium tuberculosis growth and enoyl acyl carrier protein reductase (InhA) inhibitors. Eur. J. Med. Chem. 2020, 199, 112402. [Google Scholar]
- Saxena, A.K.; Singh, A. Mycobacterial tuberculosis Enzyme Targets and their Inhibitors. Curr. Top. Med. Chem. 2019, 19, 337–355. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, B.C.; Young, R.J.; Burley, G.A.; Cacho, M.; Torres, P.; Vela-Gonzalez Del Peral, L. Identification of 2-((2,3-dihydrobenzo[b][1,4]dioxin-6-yl)amino)-N-phenylpropanamides as a novel class of potent DprE1 inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127192. [Google Scholar] [CrossRef] [PubMed]
- Hariguchi, N.; Chen, X.; Hayashi, Y.; Kawano, Y.; Fujiwara, M.; Matsuba, M.; Shimizu, H.; Ohba, Y.; Nakamura, I.; Kitamoto, R.; et al. OPC-167832, a Novel Carbostyril Derivative with Potent Antituberculosis Activity as a DprE1 Inhibitor. Antimicrob. Agents Chemother. 2020, 64, e02020-19. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Kong, C.; Fumagalli, M.; Savková, K.; Xu, Y.; Huszár, S.; Sammartino, J.C.; Fan, D.; Chiarelli, L.R.; Mikušová, K.; et al. Design, synthesis and evaluation of covalent inhibitors of DprE1 as antitubercular agents. Eur. J. Med. Chem. 2020, 208, 112773. [Google Scholar] [CrossRef]
- Lechartier, B.; Rybniker, J.; Zumla, A.; Cole, S.T. Tuberculosis drug discovery in the post-post-genomic era. EMBO Mol. Med. 2014, 6, 158–168. [Google Scholar] [CrossRef]
- Danne, A.B.; Choudhari, A.S.; Chakraborty, S.; Sarkar, D.; Khedkar, V.M.; Shingate, B.B. Triazole-diindolylmethane conjugates as new antitubercular agents: Synthesis, bioevaluation, and molecular docking. MedChemComm 2018, 9, 1114–1130. [Google Scholar] [CrossRef]
- Belin, P.; Le Du, M.H.; Fielding, A.; Lequin, O.; Jacquet, M.; Charbonnier, J.B.; Lecoq, A.; Thai, R.; Courçon, M.; Masson, C.; et al. Identification and structural basis of the reaction catalyzed by CYP121, an essential cytochrome P450 in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2009, 106, 7426–7431. [Google Scholar] [CrossRef]
- Ortiz de Montellano, P.R. Potential drug targets in the Mycobacterium tuberculosis cytochrome P450 system. J. Inorg. Biochem. 2018, 180, 235–245. [Google Scholar] [CrossRef]
- Hudson, S.A.; McLean, K.J.; Surade, S.; Yang, Y.Q.; Leys, D.; Ciulli, A.; Munro, A.W.; Abell, C. Application of fragment screening and merging to the discovery of inhibitors of the Mycobacterium tuberculosis cytochrome P450 CYPA121. Angew. Chem. Int. Ed. 2012, 51, 9311–9316. [Google Scholar] [CrossRef] [PubMed]
- Rode, N.D.; Sonawane, A.D.; Nawale, L.; Khedkar, V.M.; Joshi, R.A.; Likhite, A.P.; Sarkar, D.; Joshi, R.R. Synthesis, biological evaluation, and molecular docking studies of novel 3-aryl-5-(alkyl-thio)-1H-1,2,4-triazoles derivatives targeting Mycobacterium tuberculosis. Chem. Biol. Drug Des. 2017, 90, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Petrera, A.; Amstutz, B.; Gioia, M.; Hähnlein, J.; Baici, A.; Selchow, P.; Ferraris, D.M.; Rizzi, M.; Sbardella, D.; Marini, S.; et al. Functional characterization of the Mycobacterium tuberculosis zinc metallopeptidase Zmp1 and identification of potential substrates. Biol. Chem. 2012, 393, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, D.M.; Sbardella, D.; Petrera, A.; Marini, S.; Amstutz, B.; Coletta, M.; Sander, P.; Rizzi, M. Crystal structure of Mycobacterium tuberculosis zinc-dependent metalloprotease-1 (Zmp1), a metalloprotease involved in pathogenicity. J. Biol. Chem. 2011, 286, 32475–32482. [Google Scholar] [CrossRef] [PubMed]
- Vemula, M.H.; Medisetti, R.; Ganji, R.; Jakkala, K.; Sankati, S.; Chatti, K.; Banerjee, S. Mycobacterium tuberculosis Zinc Metalloprotease-1 Assists Mycobacterial Dissemination in Zebrafish. Front. Microbiol. 2016, 7, 1347. [Google Scholar] [CrossRef] [PubMed]
- Subhedar, D.D.; Shaikh, M.H.; Shingate, B.B.; Nawale, L.; Sarkar, D.; Khedkar, V.M.; Kalam Khan, F.A.; Sangshetti, J.N. Quinolidene-rhodanine conjugates: Facile synthesis and biological evaluation. Eur. J. Med. Chem. 2017, 125, 385–399. [Google Scholar] [CrossRef] [PubMed]




























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).