TNF Is Partially Required for Cell-Death-Triggered Skin Inflammation upon Acute Loss of cFLIP

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
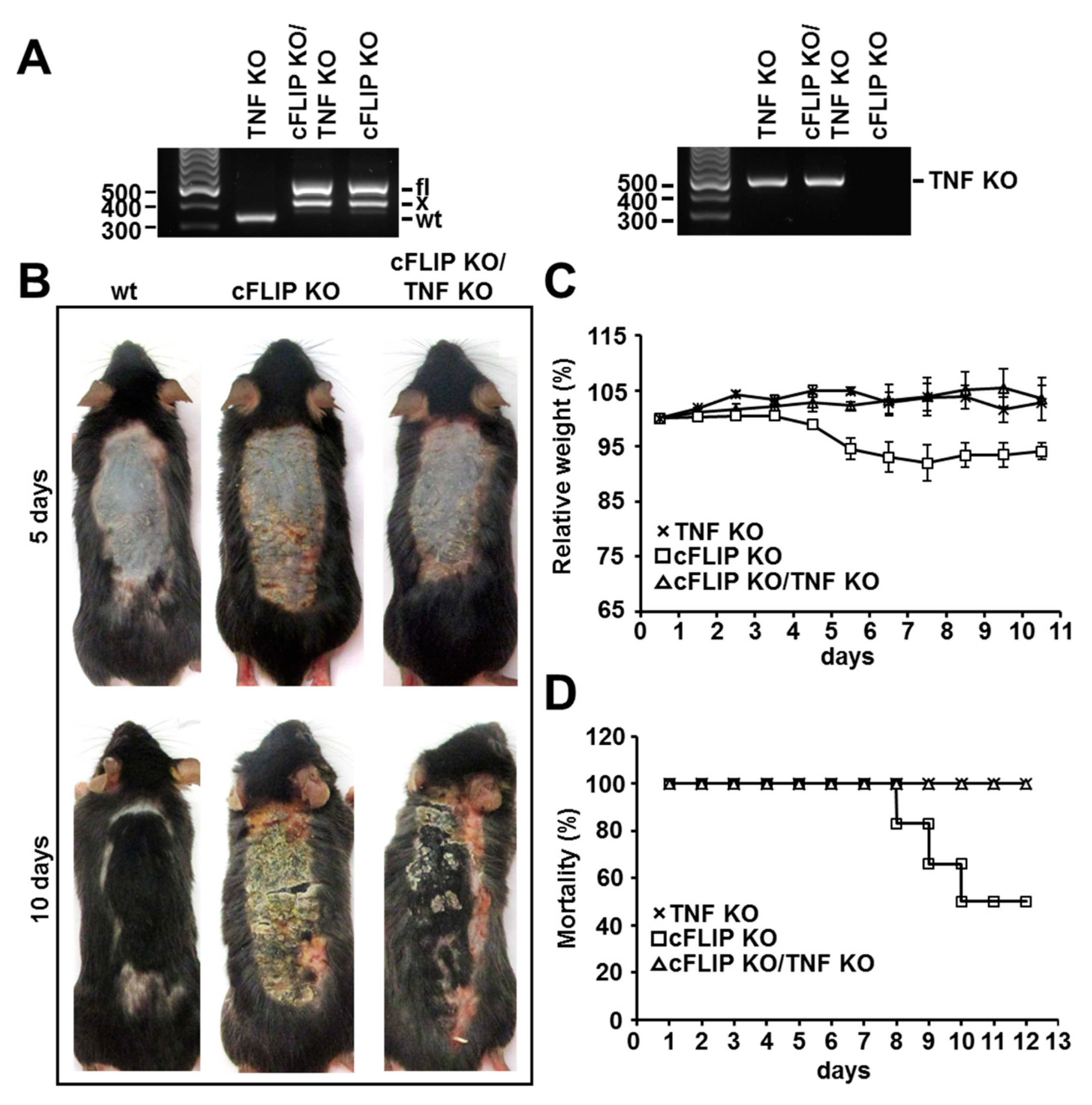
2.1. TNF Deficiency Delays the Development of Inflammatory Skin Disease and Protects from Weight Loss and Mortality upon cFLIP Deletion
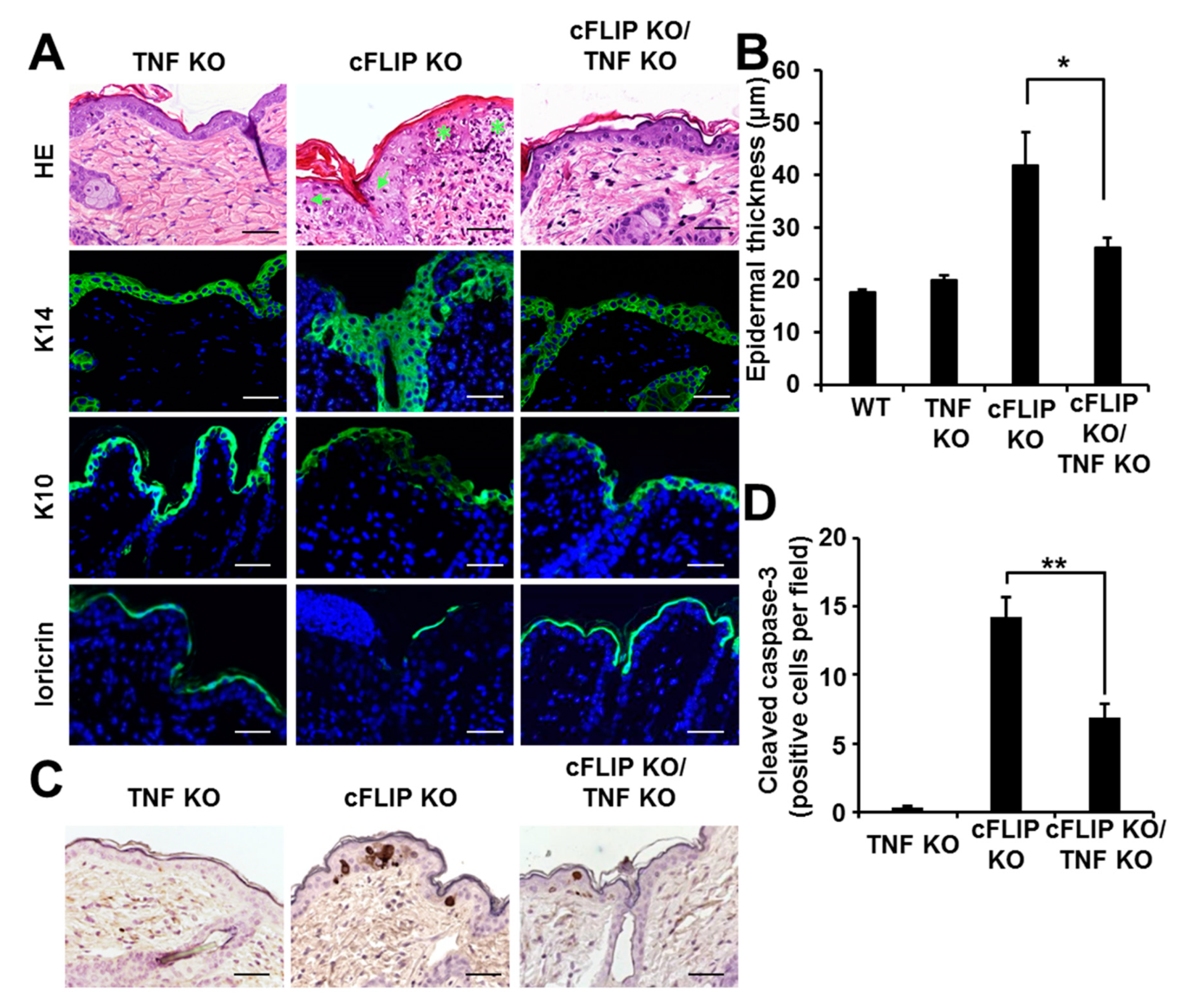
2.2. TNF Deficiency Protects cFLIP-Deficient Epidermis from Hyperproliferation, Dysregulated Differentiation, and Apoptosis
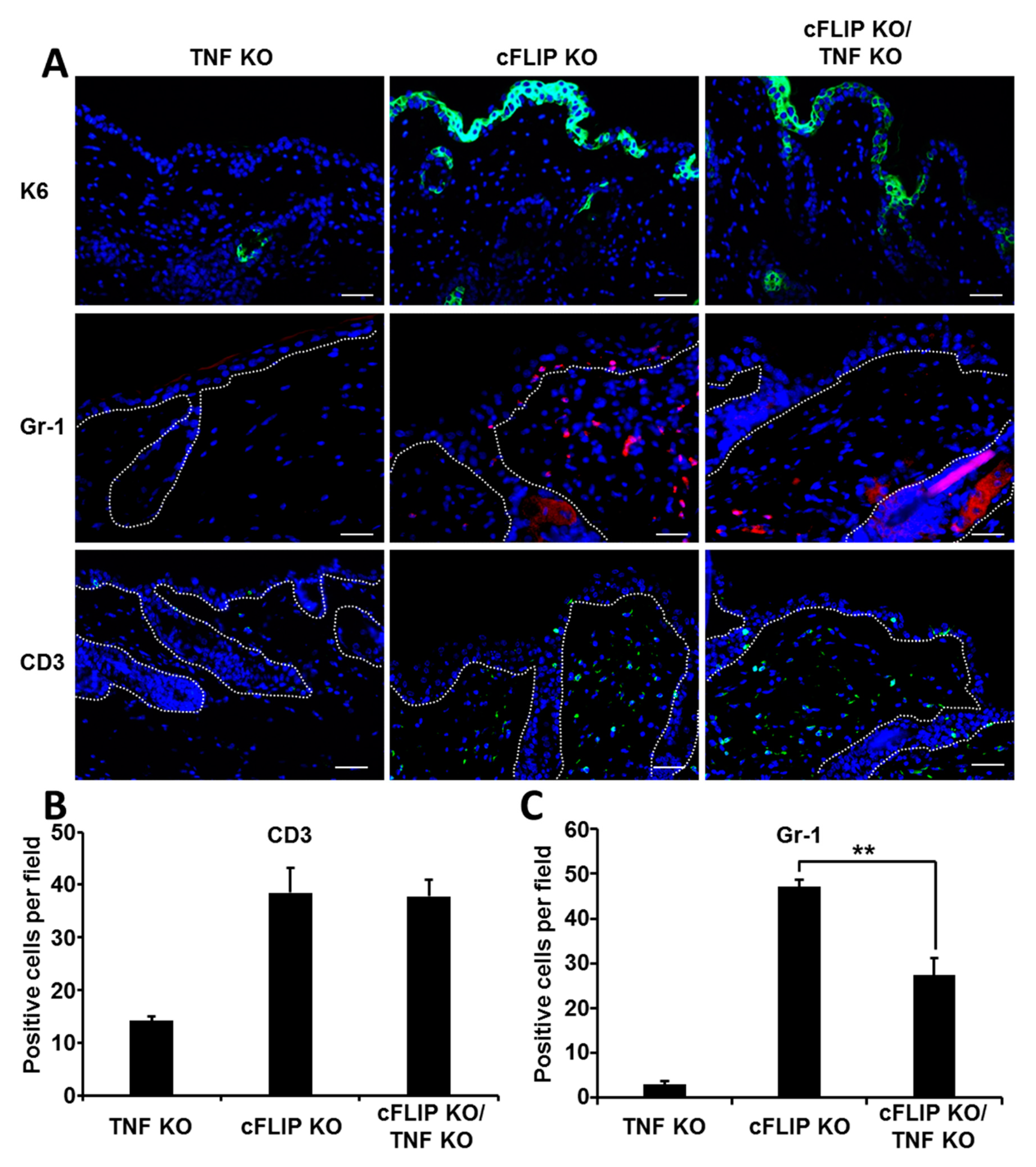
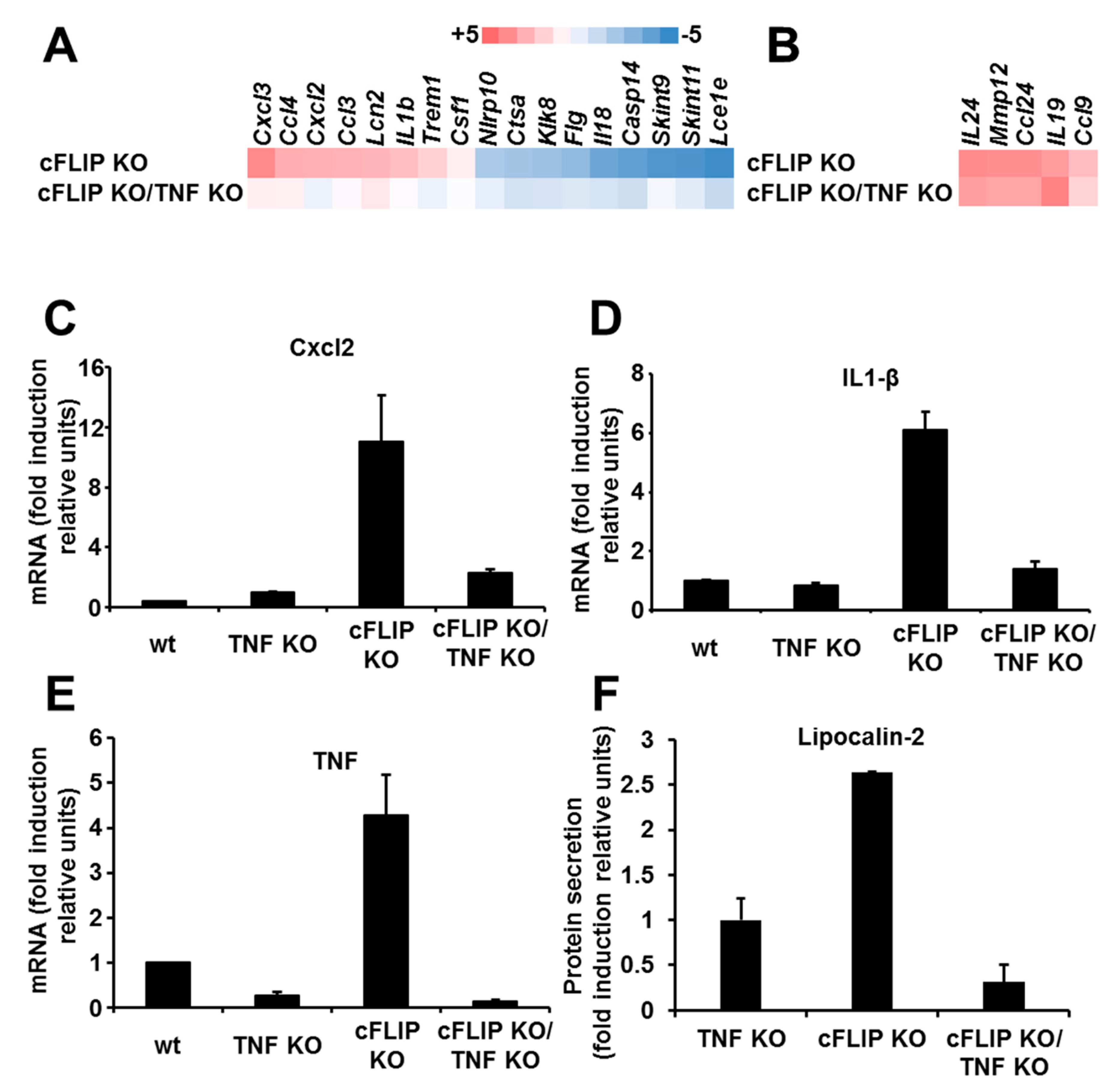
2.3. The Inflammatory Response in the Skin of Epidermal cFLIP KO Is Partially Dependent on TNF
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Genotyping Multiplex PCR
4.3. Histology and Immunohistochemistry
4.4. Primary Keratinocyte Isolation and Immortalization
4.5. Lentiviral Expression of Cre in PKs and Immunoblotting
4.6. Statistical Analysis
4.7. RNA Isolation and Real-Time qPCR
4.8. Affymetrix GeneChip Oligoarray Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Annibaldi, A.; Meier, P. Checkpoints in TNF-Induced Cell Death: Implications in Inflammation and Cancer. Trends Mol. Med. 2018, 24, 49–65. [Google Scholar] [CrossRef]
- Jarosz-Griffiths, H.H.; Holbrook, J.; Lara-Reyna, S.; McDermott, M.F. TNF receptor signalling in autoinflammatory diseases. Int. Immunol. 2019, 31, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Hacker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef]
- Pasparakis, M.; Haase, I.; Nestle, F.O. Mechanisms regulating skin immunity and inflammation. Nat. Rev. Immunol. 2014, 14, 289–301. [Google Scholar] [CrossRef]
- Kovalenko, A.; Kim, J.C.; Kang, T.B.; Rajput, A.; Bogdanov, K.; Dittrich-Breiholz, O.; Kracht, M.; Brenner, O.; Wallach, D. Caspase-8 deficiency in epidermal keratinocytes triggers an inflammatory skin disease. J. Exp. Med. 2009, 206, 2161–2177. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, M.C.; Preukschat, D.; Welz, P.S.; van Loo, G.; Ermolaeva, M.A.; Bloch, W.; Haase, I.; Pasparakis, M. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity 2011, 35, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Panayotova-Dimitrova, D.; Feoktistova, M.; Ploesser, M.; Kellert, B.; Hupe, M.; Horn, S.; Makarov, R.; Jensen, F.; Porubsky, S.; Schmieder, A.; et al. cFLIP regulates skin homeostasis and protects against TNF-induced keratinocyte apoptosis. Cell Rep. 2013, 5, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Weinlich, R.; Oberst, A.; Dillon, C.P.; Janke, L.J.; Milasta, S.; Lukens, J.R.; Rodriguez, D.A.; Gurung, P.; Savage, C.; Kanneganti, T.D.; et al. Protective roles for caspase-8 and cFLIP in adult homeostasis. Cell Rep. 2013, 5, 340–348. [Google Scholar] [CrossRef]
- Dillon, C.P.; Oberst, A.; Weinlich, R.; Janke, L.J.; Kang, T.B.; Ben-Moshe, T.; Mak, T.W.; Wallach, D.; Green, D.R. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 2012, 1, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Panayotova-Dimitrova, D.; Feoktistova, M.; Leverkus, M. RIPping the Skin Apart: Necroptosis Signaling in Toxic Epidermal Necrolysis. J. Investig. Dermatol. 2015, 135, 1940–1943. [Google Scholar] [CrossRef]
- Paradisi, A.; Abeni, D.; Bergamo, F.; Ricci, F.; Didona, D.; Didona, B. Etanercept therapy for toxic epidermal necrolysis. J. Am. Acad. Dermatol. 2014, 71, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Xu, Y. Interleukin-24 Regulates T Cell Activity in Patients with Colorectal Adenocarcinoma. Front. Oncol. 2019, 9, 1401. [Google Scholar] [CrossRef] [PubMed]
- Oral, H.B.; Kotenko, S.V.; Yilmaz, M.; Mani, O.; Zumkehr, J.; Blaser, K.; Akdis, C.A.; Akdis, M. Regulation of T cells and cytokines by the interleukin-10 (IL-10)-family cytokines IL-19, IL-20, IL-22, IL-24 and IL-26. Eur. J. Immunol. 2006, 36, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Eissa, A.; Amodeo, V.; Smith, C.R.; Diamandis, E.P. Kallikrein-related peptidase-8 (KLK8) is an active serine protease in human epidermis and sweat and is involved in a skin barrier proteolytic cascade. J. Biol. Chem. 2011, 286, 687–706. [Google Scholar] [CrossRef] [PubMed]
- Kezic, S.; Jakasa, I. Filaggrin and Skin Barrier Function. Curr. Probl. Dermatol. 2016, 49, 1–7. [Google Scholar] [PubMed]
- Lee, J.H.; Cho, D.H.; Park, H.J. IL-18 and Cutaneous Inflammatory Diseases. Int. J. Mol. Sci. 2015, 16, 29357–29369. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.; Kemperman, P.; Devos, M.; Denecker, G.; Kezic, S.; Yau, N.; Gilbert, B.; Lippens, S.; De Groote, P.; Roelandt, R.; et al. Caspase-14 is required for filaggrin degradation to natural moisturizing factors in the skin. J. Investig. Dermatol. 2011, 131, 2233–2241. [Google Scholar] [CrossRef]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Taraborrelli, L.; Peltzer, N.; Montinaro, A.; Kupka, S.; Rieser, E.; Hartwig, T.; Sarr, A.; Darding, M.; Draber, P.; Haas, T.L.; et al. LUBAC prevents lethal dermatitis by inhibiting cell death induced by TNF, TRAIL and CD95L. Nat. Commun. 2018, 9, 3910. [Google Scholar] [CrossRef]
- Tang, Y.; Tu, H.; Liu, G.; Zheng, G.; Wang, M.; Li, L.; Zhao, X.; Lin, X. RNF31 Regulates Skin Homeostasis by Protecting Epidermal Keratinocytes from Cell Death. J. Immunol. 2018, 200, 4117–4124. [Google Scholar] [CrossRef]
- Kavuri, S.M.; Geserick, P.; Berg, D.; Dimitrova, D.P.; Feoktistova, M.; Siegmund, D.; Gollnick, H.; Neumann, M.; Wajant, H.; Leverkus, M. Cellular FLICE-inhibitory protein (cFLIP) isoforms block CD95- and TRAIL death receptor-induced gene induction irrespective of processing of caspase-8 or cFLIP in the death-inducing signaling complex. J. Biol. Chem. 2011, 286, 16631–16646. [Google Scholar] [CrossRef] [PubMed]
- Lizzul, P.F.; Aphale, A.; Malaviya, R.; Sun, Y.; Masud, S.; Dombrovskiy, V.; Gottlieb, A.B. Differential expression of phosphorylated NF-kappaB/RelA in normal and psoriatic epidermis and downregulation of NF-kappaB in response to treatment with etanercept. J. Investig. Dermatol. 2005, 124, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Chan, K.S.; Carbajal, S.; Clifford, J.; Peavey, M.; Kiguchi, K.; Itami, S.; Nickoloff, B.J.; DiGiovanni, J. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat. Med. 2005, 11, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Ibe, M.; Nakamura, S.; Ishida-Yamamoto, A.; Hashimoto, Y.; Iizuka, H. Extracellular regulated kinase and c-Jun N-terminal kinase are activated in psoriatic involved epidermis. J. Dermatol. Sci. 2002, 30, 94–99. [Google Scholar] [CrossRef]
- Manils, J.; Webb, L.V.; Howes, A.; Janzen, J.; Boeing, S.; Bowcock, A.M.; Ley, S.C. CARD14(E138A) signalling in keratinocytes induces TNF-dependent skin and systemic inflammation. eLife 2020, 9, e56720. [Google Scholar] [CrossRef]
- Ippagunta, S.K.; Gangwar, R.; Finkelstein, D.; Vogel, P.; Pelletier, S.; Gingras, S.; Redecke, V.; Hacker, H. Keratinocytes contribute intrinsically to psoriasis upon loss of Tnip1 function. Proc. Natl. Acad. Sci. USA 2016, 113, E6162–E6171. [Google Scholar] [CrossRef]
- Shao, S.; Cao, T.; Jin, L.; Li, B.; Fang, H.; Zhang, J.; Zhang, Y.; Hu, J.; Wang, G. Increased Lipocalin-2 Contributes to the Pathogenesis of Psoriasis by Modulating Neutrophil Chemotaxis and Cytokine Secretion. J. Investig. Dermatol. 2016, 136, 1418–1428. [Google Scholar] [CrossRef]
- Rickard, J.A.; Anderton, H.; Etemadi, N.; Nachbur, U.; Darding, M.; Peltzer, N.; Lalaoui, N.; Lawlor, K.E.; Vanyai, H.; Hall, C.; et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. eLife 2014, 3, e03464. [Google Scholar] [CrossRef]
- Bamias, G.; Evangelou, K.; Vergou, T.; Tsimaratou, K.; Kaltsa, G.; Antoniou, C.; Kotsinas, A.; Kim, S.; Gorgoulis, V.; Stratigos, A.J.; et al. Upregulation and nuclear localization of TNF-like cytokine 1A (TL1A) and its receptors DR3 and DcR3 in psoriatic skin lesions. Exp. Dermatol. 2011, 20, 725–731. [Google Scholar] [CrossRef]
- Sabour Alaoui, S.; Dessirier, V.; de Araujo, E.; Alexaki, V.I.; Pelekanou, V.; Lkhider, M.; Stathopoulos, E.N.; Castanas, E.; Bagot, M.; Bensussan, A.; et al. TWEAK affects keratinocyte G2/M growth arrest and induces apoptosis through the translocation of the AIF protein to the nucleus. PLoS ONE 2012, 7, e33609. [Google Scholar] [CrossRef]
- Sidler, D.; Wu, P.; Herro, R.; Claus, M.; Wolf, D.; Kawakami, Y.; Kawakami, T.; Burkly, L.; Croft, M. TWEAK mediates inflammation in experimental atopic dermatitis and psoriasis. Nat. Commun. 2017, 8, 15395. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feoktistova, M.; Makarov, R.; Leverkus, M.; Yazdi, A.S.; Panayotova-Dimitrova, D. TNF Is Partially Required for Cell-Death-Triggered Skin Inflammation upon Acute Loss of cFLIP. Int. J. Mol. Sci. 2020, 21, 8859. https://doi.org/10.3390/ijms21228859
Feoktistova M, Makarov R, Leverkus M, Yazdi AS, Panayotova-Dimitrova D. TNF Is Partially Required for Cell-Death-Triggered Skin Inflammation upon Acute Loss of cFLIP. International Journal of Molecular Sciences. 2020; 21(22):8859. https://doi.org/10.3390/ijms21228859
Chicago/Turabian StyleFeoktistova, Maria, Roman Makarov, Martin Leverkus, Amir S. Yazdi, and Diana Panayotova-Dimitrova. 2020. "TNF Is Partially Required for Cell-Death-Triggered Skin Inflammation upon Acute Loss of cFLIP" International Journal of Molecular Sciences 21, no. 22: 8859. https://doi.org/10.3390/ijms21228859
APA StyleFeoktistova, M., Makarov, R., Leverkus, M., Yazdi, A. S., & Panayotova-Dimitrova, D. (2020). TNF Is Partially Required for Cell-Death-Triggered Skin Inflammation upon Acute Loss of cFLIP. International Journal of Molecular Sciences, 21(22), 8859. https://doi.org/10.3390/ijms21228859