Mutations Causing Mild or No Structural Damage in Interfaces of Multimerization of the Fibrinogen γ-Module More Likely Confer Negative Dominant Behaviors


Abstract
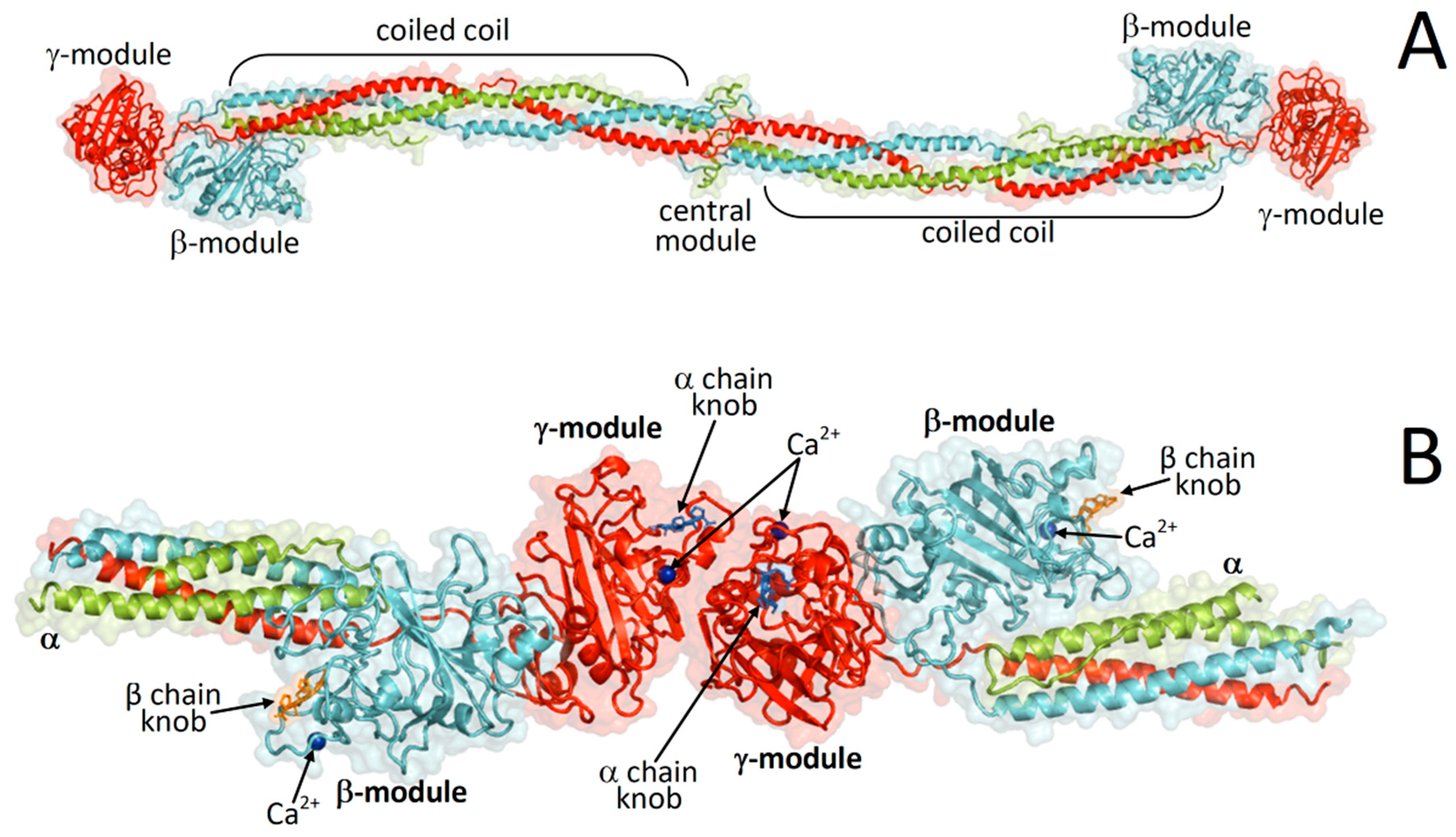
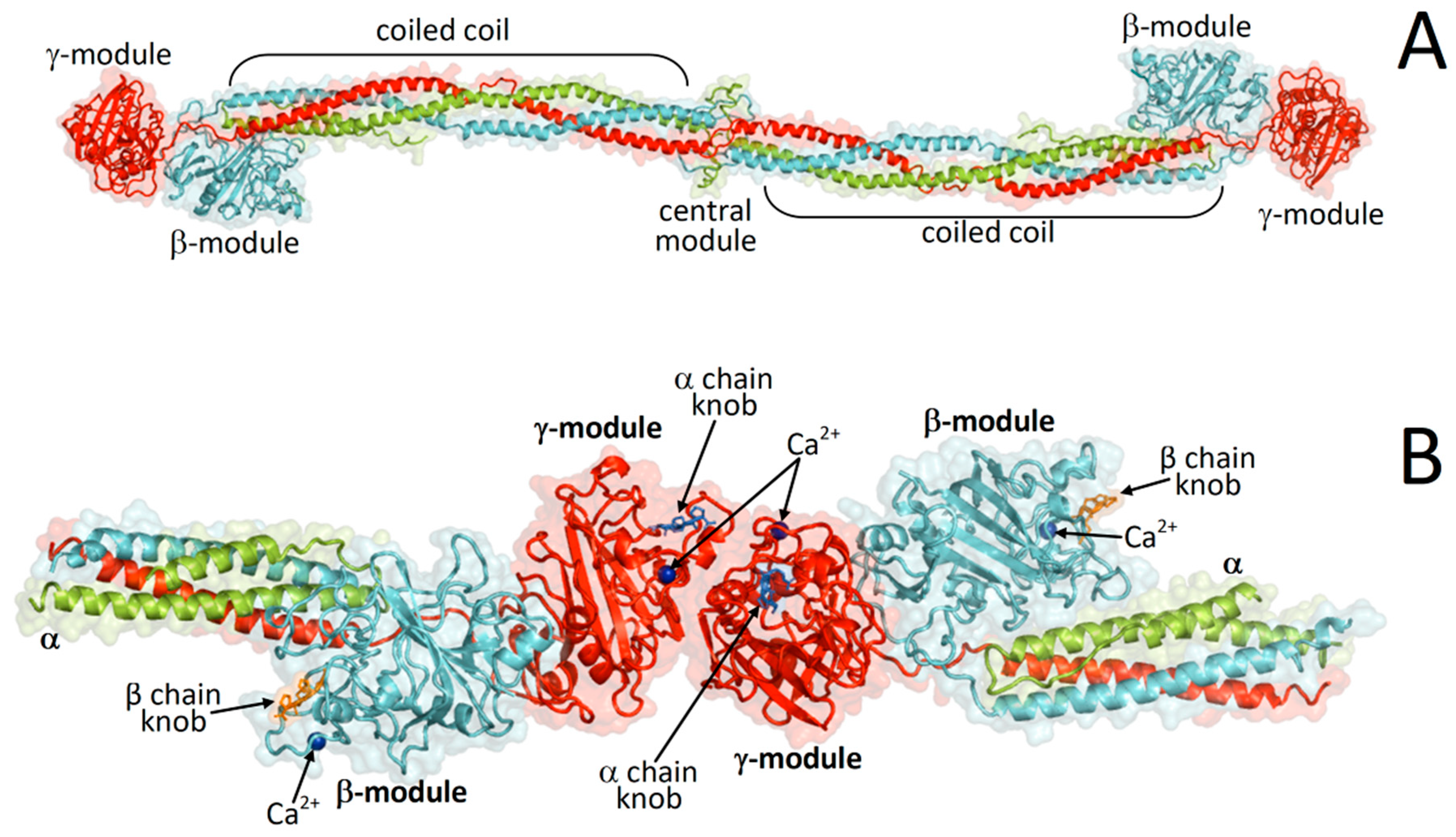
:1. Introduction
2. Results
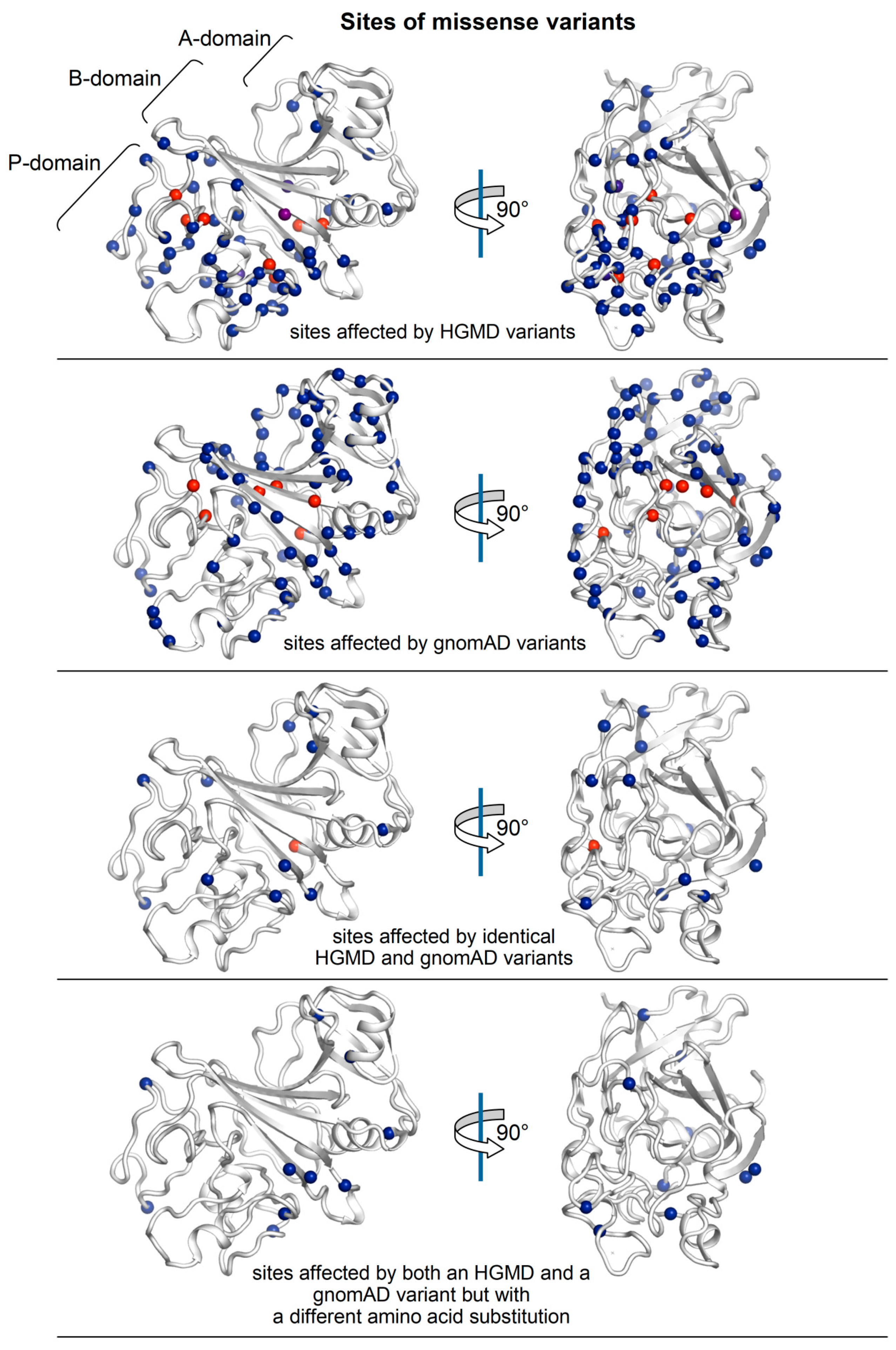
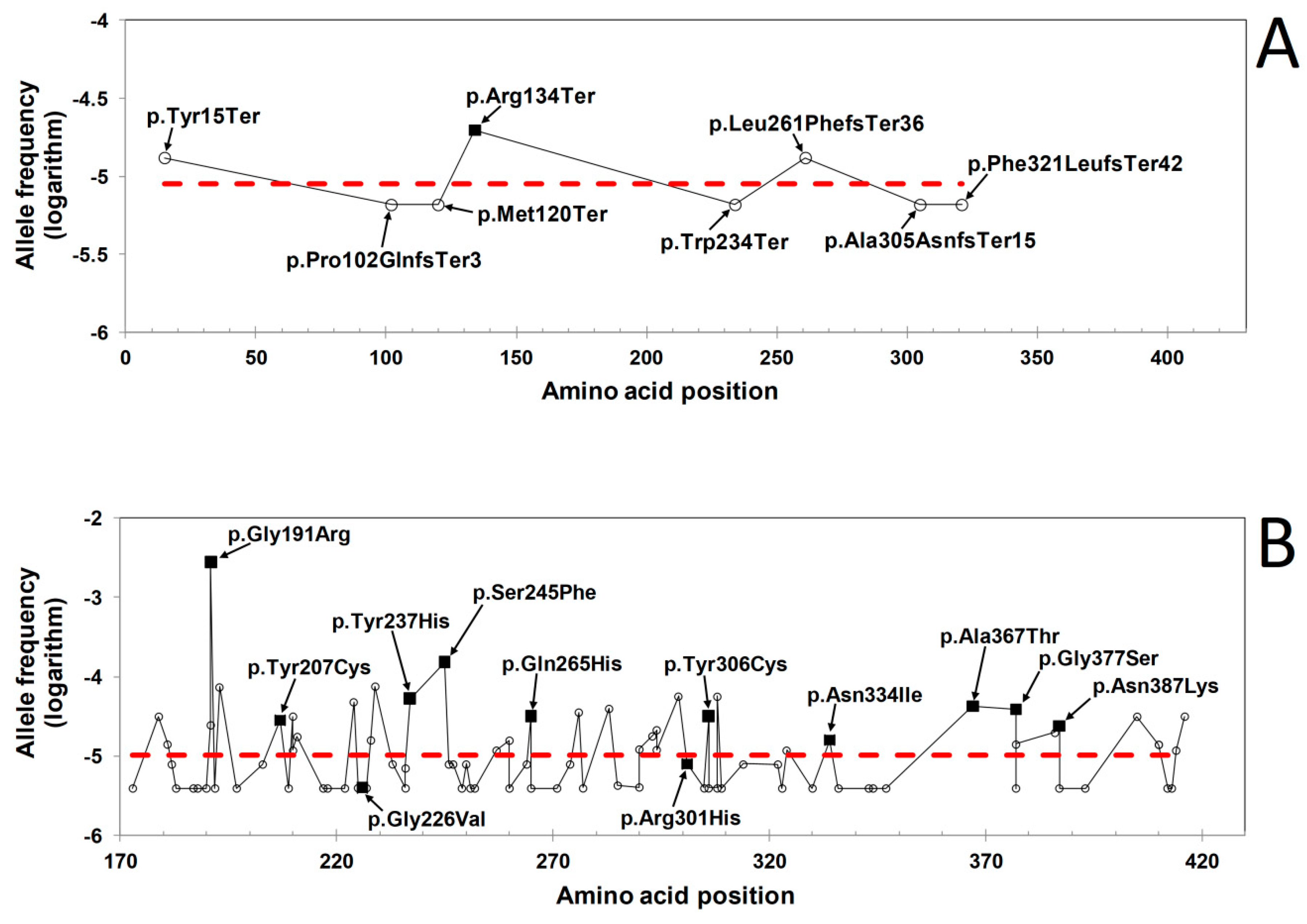
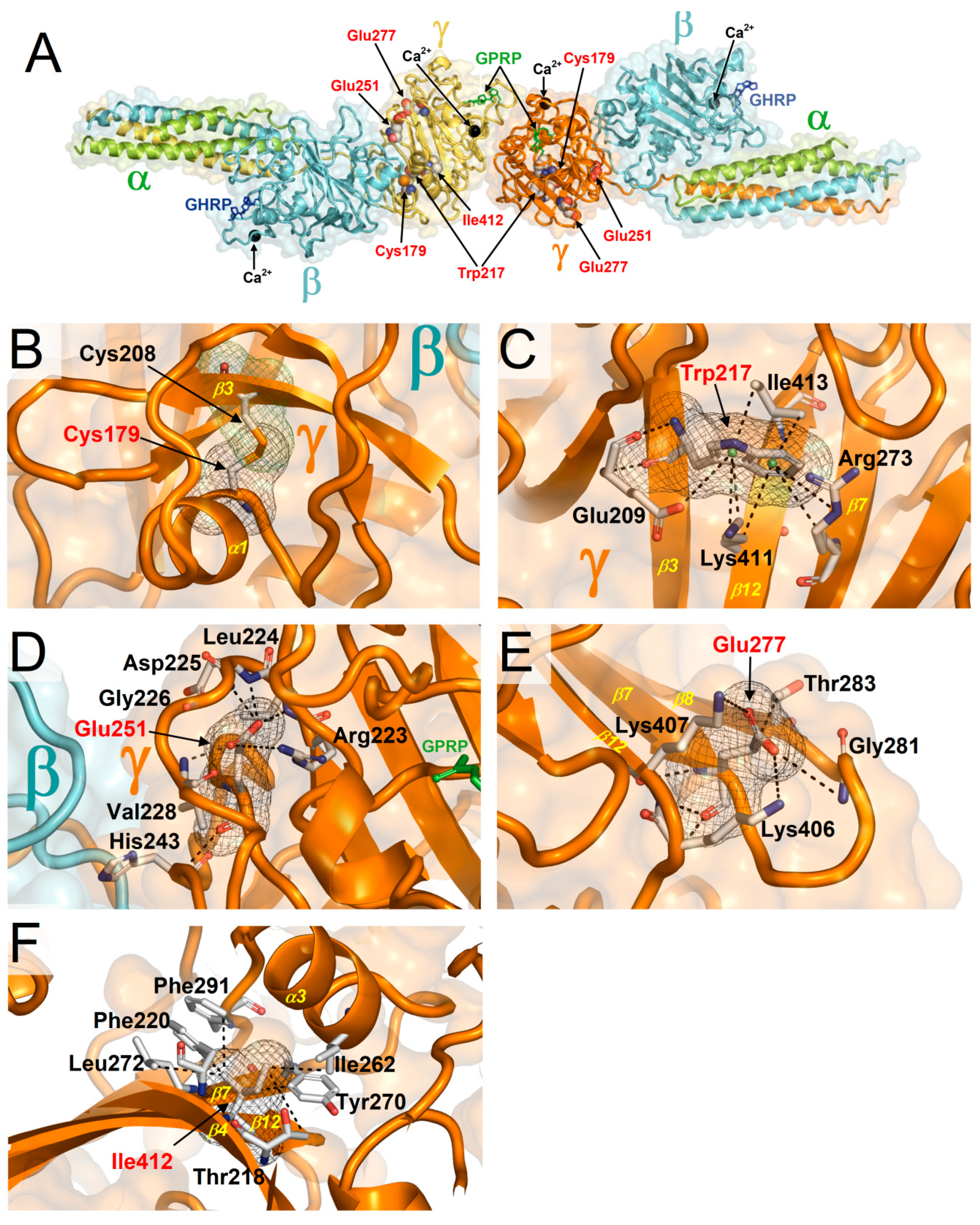
2.1. Distribution of HGMD and GnomAD Missense Variants in the γ-Module
2.2. ∆∆G Values of HGMD and GnomAD Missense Variants Localized in the γ-Module
3. Discussion
3.1. HGMD Variants Predicted to Cause Non-Significant Structural Changes
3.2. GnomAD Variants Predicted to Be Structurally Damaging
3.3. Rationalization of the Missense Variants Shared by the HGMD and GnomAD Databases
4. Materials and Methods
Protein Structural Analysis
5. Conclusions
- (a)
- A number of variants in the healthy population might indeed be pathogenic with an autosomal recessive modality of disease transmission. Variants with such characteristics can especially be those inducing severe protein misfolding leading to the degradation of the protein or at least its inability to recruit the designed interacting fibrinogen protein partners or to undergo undue aggregations, i.e., variants causing full loss of function, or not insinuating into negative domain effects.
- (b)
- Other potentially disease-causing variants found in the healthy population are those with negative dominant effects producing mild and/or difficult to identify disorders and/or late-onset diseases, or disorders caused by specific environmental factors, trauma, etc.
- (c)
- A recurrent observation in this and previous studies is that a number of pathogenic variants hit the protein structure only “softly” but in spots important for protein-protein interactions. Proteins with defects in these critical regions, if not efficiently neutralized by the cellular mechanism of protein degradation or at least by a loss of the protein capacity to bind and recruit other proteins render these variants candidates to be checked as negative dominant mutations. This can be particularly true for proteins engaging in processes of homo- and homo-hetero multimerization.
- (d)
- This study highlights that a significant fraction of gnomAD variants are not neutral (for their co-presence in the HGMD database), and this might be only the tip of the iceberg (as suggested by ∆∆G calculations and protein structural analysis). Care must be exercised for all variants found in the general population and believed as neutral mutations.
Funding
Acknowledgments
Conflicts of Interest
References
- Kollman, J.M.; Pandi, L.; Sawaya, M.R.; Riley, M.; Doolittle, R.F. Crystal structure of human fibrinogen. Biochemistry 2009, 48, 3877–3886. [Google Scholar] [CrossRef] [PubMed]
- Everse, S.J.; Spraggon, G.; Veerapandian, L.; Riley, M.; Doolittle, R.F. Crystal structure of fragment double-D from human fibrin with two different bound ligands. Biochemistry 1998, 37, 8637–8642. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W.; Siebenlist, K.R.; Meh, D.A. The structure and biological features of fibrinogen and fibrin. Ann. N. Y. Acad. Sci. 2001, 936, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W. Hereditary fibrinogen abnormalities. In Williams Hematology, 7th ed.; Mac Grow-Hill: New York, NY, USA, 2006; pp. 1909–1927. [Google Scholar]
- Brennan, S.O.; Wyatt, J.M.; Medicina, D.; Callea, F.; George, P.M. Fibrinogen brescia: Hepatic endoplasmic reticulum storage and hypofibrinogenemia because of a gamma284 Gly→Arg mutation. Am. J. Pathol. 2000, 157, 189–196. [Google Scholar] [CrossRef]
- Callea, F.; De Vos, R.; Pinackat, J.; Favret, M.; Fiaccavento, S.; Ascari, E.; Tortora, O.; Albertini, A.; Henschen, A.; Desmet, V.J. Hereditary hypofibrinogenemia with hepatic storage of fibrinogen. A new endoplasmic reticulum storage disease. In Fibrinogen 2. Biochemistry, Physiology and Clinical Relevance; Lowe, G.D.O., Ed.; Elsevier: Amsterdam, The Netherlands, 1987; pp. 75–78. [Google Scholar]
- Callea, F.; Brisigotti, M.; Fabbretti, G.; Bonino, F.; Desmet, V.J.; Brisiptti, M. Hepatic endoplasmic reticulum storage diseases. Liver 1992, 12, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W.; Siebenlist, K.R.; Diorio, J.P.; Matsuda, M.; Hainfeld, J.F.; Wall, J.S. The role of fibrinogen D domain intermolecular association sites in the polymerization of fibrin and fibrinogen Tokyo II (gamma 275 Arg→Cys). J. Clin. Investig. 1995, 96, 1053–1058. [Google Scholar] [CrossRef]
- Spraggon, G.; Everse, S.J.; Doolittle, R.F. Crystal structures of fragment D from human fibrinogen and its crosslinked counterpart from fibrin. Nat. Cell Biol. 1997, 389, 455–462. [Google Scholar] [CrossRef]
- Mosesson, M.W.; Siebenlist, K.R.; Hainfeld, J.; Wall, J. The Covalent Structure of Factor XIIIa Crosslinked Fibrinogen Fibrils. J. Struct. Biol. 1995, 115, 88–101. [Google Scholar] [CrossRef]
- Siebenlist, K.R.; Meh, D.A.; Mosesson, M.W. Protransglutaminase (factor XIII) mediated crosslinking of fibrinogen and fibrin. Thromb. Haemost. 2001, 86, 1221–1228. [Google Scholar]
- Siebenlist, K.R.; Hernandez, I.; Wall, J.S.; Hainfeld, J.F.; Mosesson, M.W. Fibrinogen assembly and crosslinking on a fibrin fragment E template. Thromb. Haemost. 2002, 87, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Blombäck, B.; Hessel, B.; Hogg, D.; Therkildsen, L. A two-step fibrinogen–fibrin transition in blood coagulation. Nat. Cell Biol. 1978, 275, 501–505. [Google Scholar] [CrossRef]
- Laudano, A.P.; Doolittle, R.F. Synthetic peptide derivatives that bind to fibrinogen and prevent the polymerization of fibrin monomers. Proc. Natl. Acad. Sci. USA 1978, 75, 3085–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Qual. Life Res. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoeldi, J.; Wang, Q.S.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Genome Aggregation Database Consortium; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, 382–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehouck, Y.; Kwasigroch, J.M.; Gilis, D.; Rooman, M. PoPMuSiC 2.1: A web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinform. 2011, 12, 151. [Google Scholar] [CrossRef] [PubMed]
- Parthiban, V.; Gromiha, M.M.; Schomburg, D. CUPSAT: Prediction of protein stability upon point mutations. Nucleic Acids Res. 2006, 34, 239–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Güven, B.; Bellacchio, E.; Sag, E.; Cebi, A.H.; Saygin, I.; Bahadir, A.; Esendagli, G.; Corbeddu, M.; Cakir, M.; Callea, F. Structural characteristics in the g chain variants associated with fibrinogen storage disease suggest the underlying pathogenic mechanism. Int. J. Mol. Sci. 2020, 21, 5139. [Google Scholar] [CrossRef]
- Callea, F.; Giovannoni, I.; Sari, S.; Esendagli, G.; Dalgic, B.; Akyol, G.; Sogo, T.; Al-Hussaini, A.A.; Maggiore, G.; Bartuli, A.; et al. Fibrinogen Gamma Chain mutations provoke Fibrinogen and Apolipoprotein B plasma deficiency and liver storage. Int. J. Mol. Sci. 2017, 18, 2717. [Google Scholar] [CrossRef] [Green Version]
- Neerman-Arbez, M.; Germanos-Haddad, M.; Tzanidakis, K.; Vu, D.; Deutsch, S.; David, A.; Morris, M.A.; De Moerloose, P. Expression and analysis of a split premature termination codon in FGG responsible for congenital afibrinogenemia: Escape from RNA surveillance mechanisms in transfected cells. Blood 2004, 104, 3618–3623. [Google Scholar] [CrossRef]
- Siebenlist, K.R.; Mosesson, M.W.; A Meh, D.; Diorio, J.P.; Albrecht, R.M.; Olson, J.D. Coexisting dysfibrinogenemia (gammaR275C) and factor V Leiden deficiency associated with thromboembolic disease (fibrinogen Cedar Rapids). Blood Coagul. Fibrinolysis 2000, 11, 293–304. [Google Scholar]
- Travlou, A.; Gialeraki, A.; Merkouri, E.; Politou, M.; Sfyridaki, A.; Neerman-Arbez, M. Coexisting dysfibrinogenemia (γArg275His) and FV Leiden associated with thrombosis (Fibrinogen Crete). Thromb. Res. 2010, 126, e162–e164. [Google Scholar] [CrossRef] [PubMed]
- Bednar, D.; Beerens, K.; Sebestova, E.; Bendl, J.; Khare, S.; Chaloupkova, R.; Prokop, Z.; Brezovsky, J.; Baker, D.; Damborsky, J. FireProt: Energy and evolution-based computational design of thermostable multiple-point mutants. PLoS Comput. Biol. 2015, 11, e1004556. [Google Scholar] [CrossRef] [PubMed]
- Schwersensky, M.; Rooman, M.; Pucci, F. Large-scale in silico mutagenesis experiments reveal optimization of genetic code and codon usage for protein mutational robustness. BMC Biol. 2020, 18, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins 1995, 23, 566–579. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | ∆∆G (Kcal/mol) | Interactions a | Secondary Structure b | %SAS c | ||
|---|---|---|---|---|---|---|
| FoldX d | CUPSAT d | PoPMuSiC d | ||||
| 3.9 ± 5.4 e | −1.44 ± 3.06 e | 1.16 ± 0.80 e | ||||
| p.Cys179Arg▲ | 21.8 | −9.16 | 2.90 | α-helix | 0.0 | |
| p.Gly191Arg■▲ | 4.9 | −2.77 | 2.63 | β-strand | 2.1 | |
| p.Phe204Leu | 2.9 | −0.03 | 1.25 | Bβ | β-strand | 12.2 |
| p.Tyr207Cys■ | 1.1 | −4.23 | 2.24 | β-strand | 19.8 | |
| p.Gly226Val■ | 6.2 | −3.98 | 2.35 | coil | 14.7 | |
| p.Trp234Leu | 2.9 | −2.80 | 1.95 | α-helix | 10.7 | |
| p.Tyr237His■ | 3.1 | −1.81 | 1.65 | α-helix | 0.0 | |
| p.Glu239Ala | 0.6 | −0.28 | −0.20 | Bβ | α-helix | 86.4 |
| p.Ser245Phe■ | 0.5 | −3.67 | 0.78 | Bβ | turn | 15.8 |
| p.Trp253Cys | 4.6 | 3.21 | 2.50 | Bβ | β-strand | 0.0 |
| p.Asn256Lys | 3.5 | −5.30 | 0.83 | α-helix | 1.0 | |
| p.Asn256His | 6.6 | −1.75 | −0.07 | α-helix | 1.0 | |
| p.Asn256Asp | 1.1 | −3.27 | 0.76 | α-helix | 1.0 | |
| p.Lys258Thr | 2.0 | 0.20 | 0.50 | Bβ | α-helix | 39.1 |
| p.Gln265His■▲ | 8.1 | −1.17 | 0.44 | 310 helix | 3.0 | |
| p.Trp279Gly | 2.6 | −1.64 | 2.80 | turn | 19.5 | |
| p.Trp279Cys | 2.3 | −1.11 | 2.15 | turn | 19.5 | |
| p.Tyr288Cys | 5.8 | 5.07 | 2.90 | β-strand | 0.0 | |
| p.Gly294Glu▲ | 0.1 | 0.40 | 0.20 | γ | coil | 54.5 |
| p.Arg301Ser▲ | 0.8 | 0.81 | 1.34 | γ | turn | 35.0 |
| p.Arg301His■▲ | 1.7 | −0.79 | 0.84 | γ, knob | turn | 35.0 |
| p.Arg301Cys▲ | 1.3 | −0.81 | 1.26 | γ, knob | turn | 35.0 |
| p.Thr303Arg | −0.5 | −0.74 | 0.96 | γ | β-strand | 43.9 |
| p.Thr303Pro | 2.4 | −0.34 | 2.24 | γ | β-strand | 43.9 |
| p.Ala305Asp▲ | 0.1 | −1.33 | 0.32 | γ | β-strand | 41.5 |
| p.Tyr306Cys■▲ | 1.2 | −6.61 | 0.81 | γ | β-strand | 61.0 |
| p.Gly310Arg | 13.5 | −2.56 | 1.90 | turn | 37.7 | |
| p.Ala315Val | 4.5 | 1.89 | 0.65 | 310 helix | 0.0 | |
| p.Gly318Val | 15.4 | −12.81 | 2.48 | coil | 0.0 | |
| p.Thr331Ala | 1.5 | −0.39 | 1.28 | knob | α-helix | 7.6 |
| p.His333Tyr | 7.6 | −5.85 | 0.46 | turn | 0.8 | |
| p.Asn334Lys | 0.3 | −1.75 | 0.69 | γ | turn | 46.6 |
| p.Asn334Thr | 1.5 | −1.16 | −0.17 | γ | turn | 46.6 |
| p.Asn334Ile■ | 0.5 | −0.18 | −0.04 | γ | turn | 46.6 |
| p.Gly335Asp | 2.9 | −4.01 | 1.42 | γ | turn | 60.7 |
| p.Gly335Cys | 3.1 | −0.13 | 1.42 | γ | turn | 60.7 |
| p.Met336Thr▲ | 2.3 | 0.19 | 1.69 | γ | bridge | 13.0 |
| p.Ser339Asn | 6.0 | −4.03 | 1.19 | bridge | 0.0 | |
| p.Ser339Arg | 16.1 | −1.66 | 0.81 | bridge | 0.0 | |
| p.Ser339Gly | 1.1 | −3.53 | 1.32 | bridge | 0.0 | |
| p.Thr340Ile | 1.8 | −0.83 | 0.62 | bridge | 0.0 | |
| p.Thr340Pro | 2.9 | −12.46 | 1.79 | bridge | 0.0 | |
| p.Asp342Asn | 0.1 | 0.32 | 0.01 | proximal to Ca2+ | turn | 40.1 |
| p.Asp342His | 1.1 | −0.77 | 0.32 | proximal to Ca2+ | turn | 40.1 |
| p.Asp342Gly | 0.9 | 0.11 | 0.92 | proximal to Ca2+ | turn | 40.1 |
| p.Asp344Gly▲ | −0.5 | −0.45 | 0.84 | Ca2+ | turn | 37.3 |
| p.Asp344Val▲ | 0.9 | 0.00 | 0.34 | Ca2+ | turn | 37.3 |
| p.Asp344Tyr▲ | 0.6 | 0.30 | 0.76 | Ca2+ | turn | 37.3 |
| p.Asn345Lys | 1.6 | −2.21 | 0.81 | proximal to Ca2+ | coil | 17.1 |
| p.Asn345Asp | 3.8 | −0.74 | 0.74 | proximal to Ca2+ | coil | 17.1 |
| p.Asp346Glu | 5.9 | 0.07 | 1.16 | Ca2+ | coil | 0.0 |
| p.Asp346Gly | 1.6 | −2.66 | 1.58 | Ca2+ | coil | 0.0 |
| p.Phe348Ile | 5.4 | −1.51 | 1.23 | knob, Ca2+ | turn | 32.8 |
| p.Phe348Cys | 3.0 | 0.26 | 1.70 | knob, Ca2+ | turn | 32.8 |
| p.Asn351Ile | 1.6 | 0.05 | 1.37 | proximal to Ca2+ | turn | 21.0 |
| p.Cys352Ser | 2.9 | −2.86 | 1.99 | proximal to Ca2+ | α-helix | 0.0 |
| p.Cys352Tyr | 32.3 | −2.43 | 0.81 | proximal to Ca2+ | α-helix | 0.0 |
| p.Cys352Phe | 23.1 | −1.55 | 1.19 | proximal to Ca2+ | α-helix | 0.0 |
| p.Ala353Thr | 4.8 | −1.04 | 1.09 | proximal to Ca2+ | α-helix | 0.0 |
| p.Gln355Arg | 0.5 | 1.43 | 0.51 | knob | α-helix | 32.1 |
| p.Asp356Val | −1.8 | −2.62 | 0.57 | knob | α-helix | 0.0 |
| p.Asp356Tyr | 0.5 | −1.45 | −0.02 | knob | α-helix | 0.0 |
| p.Ser358Cys | 1.8 | −7.02 | −0.34 | coil | 0.8 | |
| p.Trp361Arg | 6.4 | 9.09 | 2.81 | coil | 0.8 | |
| p.Met362Ile | 2.9 | −6.89 | 1.13 | turn | 0.0 | |
| p.Asn363Lys | 1.4 | 0.32 | 1.52 | turn | 6.9 | |
| p.Ala367Thr■ | 3.0 | 0.39 | 1.25 | turn | 0.0 | |
| p.Ala367Asp▲ | 6.4 | −0.25 | 2.14 | turn | 0.0 | |
| p.Ala367Val▲ | 2.2 | 0.77 | 0.43 | turn | 0.0 | |
| p.Asn371Ser | 3.1 | −1.72 | 1.41 | turn | 0.0 | |
| p.Asn371Asp | 1.5 | −0.15 | 1.59 | turn | 0.0 | |
| p.Gly372Val | 18.2 | −3.53 | 0.37 | turn | 4.2 | |
| p.Tyr374Cys | 3.1 | −0.33 | 1.73 | coil | 12.4 | |
| p.Gly377Ser■▲ | 3.1 | −0.55 | 1.49 | turn | 92.1 | |
| p.Tyr380Cys | 4.6 | −0.61 | 2.48 | coil | 1.2 | |
| p.Ala383Thr | 0.2 | 1.43 | −0.16 | proximal to knob | turn | 100 |
| p.Ser384Cys | −0.1 | −2.52 | 0.12 | turn | 32.3 | |
| p.Asn387Lys■▲ | −0.3 | 2.27 | 0.59 | proximal to knob | turn | 83.8 |
| p.Tyr389Asn | 1.5 | −2.78 | 1.33 | knob | coil | 40.1 |
| p.Asp390His | 1.8 | −1.96 | 0.65 | knob | coil | 19.9 |
| p.Asp390Val | 3.1 | −2.02 | 0.37 | knob | coil | 19.9 |
| p.Asn391Lys | −0.7 | 3.20 | 1.34 | proximal to knob | coil | 16.1 |
| p.Gly392Ser | 4.6 | −4.01 | 0.81 | coil | 0.0 | |
| p.Trp395Leu | 3.1 | 0.55 | 1.75 | β-strand | 0.0 | |
| p.Thr397Ile | 1.4 | −1.35 | 0.58 | turn | 21.6 | |
| p.Arg401Gly | 2.3 | −1.00 | 2.19 | knob | turn | 28.4 |
| p.Arg401Trp | 2.9 | 3.21 | 0.42 | knob | turn | 28.4 |
| p.Ser404Pro | 6.5 | −1.34 | 2.20 | coil | 0.0 | |
| p.Lys406Asn | 1.9 | −0.39 | 0.98 | coil | 40.3 | |
| Variant | ∆∆G (Kcal/mol) | Interactions a | Secondary Structure b | %SAS c | Allele Frequency | ||
|---|---|---|---|---|---|---|---|
| FoldX d | CUPSAT d | PoPMuSiC d | |||||
| 1.8 ± 2.8 e | −1.45 ± 2.88 e | 0.95 ± 0.86 e | |||||
| p.Asp173Gly | 0.3 | −1.87 | 0.44 | β-strand | 100.0 | 3.98E−06 | |
| p.Cys179Phe▲ | 21.1 | −1.07 | 1.00 | α-helix | 0.0 | 3.18E−05 | |
| p.Asp181Asn | 0.4 | −0.91 | 0.61 | α-helix | 41.8 | 1.42E−05 | |
| p.Ile182Val | 0.7 | −2.46 | 1.40 | α-helix | 0.0 | 7.96E−06 | |
| p.Ala183Val | 2.3 | −0.08 | 0.58 | α-helix | 1.1 | 3.98E−06 | |
| p.Ala187Thr | 0.9 | −1.31 | 0.88 | coil | 2.2 | 3.98E−06 | |
| p.Lys188Arg | −0.7 | −1.41 | 0.04 | coil | 87.9 | 3.98E−06 | |
| p.Ser190Ile | 2.0 | −0.74 | 0.24 | coil | 41.2 | 3.98E−06 | |
| p.Gly191Glu▲ | 5.6 | −11.52 | 3.17 | β-strand | 2.1 | 2.48E−05 | |
| p.Gly191Arg■ | 5.2 | −2.77 | 2.63 | β-strand | 2.1 | 2.77E−03 | |
| p.Leu192Ile | 2.0 | 1.25 | 0.75 | Bβ | β-strand | 10.1 | 3.98E−06 |
| p.Tyr193His | 3.6 | −5.10 | 2.69 | β-strand | 2.6 | 7.43E−05 | |
| p.Pro197Thr | 4.2 | −1.91 | 1.49 | turn | 0.0 | 3.98E−06 | |
| p.Gln203Lys | 0.1 | 0.59 | 0.11 | Bβ | turn | 61.9 | 7.96E−06 |
| p.Tyr207Cys■ | 1.0 | −4.23 | 2.24 | β-strand | 19.8 | 2.83E−05 | |
| p.Glu209Lys | −0.7 | −1.11 | 0.86 | β-strand | 41.4 | 3.98E−06 | |
| p.Ile210Met | 1.1 | 1.37 | 1.31 | β-strand | 0.9 | 3.19E−05 | |
| p.Ile210Ser | 3.7 | 0.12 | 2.99 | β-strand | 0.9 | 1.19E−05 | |
| p.Asp211Asn | 1.2 | −0.54 | 0.43 | turn | 50.6 | 1.77E−05 | |
| p.Trp217Gly | 4.9 | −5.89 | 4.45 | β-strand | 7.2 | 3.98E−06 | |
| p.Thr218Ile | −0.8 | −0.08 | 0.00 | β-strand | 2.8 | 3.98E−06 | |
| p.Lys222Glu | 1.9 | −1.94 | 0.75 | β-strand | 32.2 | 3.98E−06 | |
| p.Leu224Arg | 1.0 | −0.90 | 0.66 | coil | 20.3 | 4.83E−05 | |
| p.Asp225Tyr | 0.6 | −2.57 | 0.50 | Bβ | coil | 55.0 | 4.02E−06 |
| p.Gly226Val■ | 6.1 | −3.98 | 2.35 | coil | 14.7 | 4.01E−06 | |
| p.Ser227Asn | −0.1 | −1.93 | 0.22 | Bβ | coil | 53.1 | 4.01E−06 |
| p.Val228Ala | 1.7 | −3.48 | 2.11 | coil | 10.9 | 1.60E−05 | |
| p.Asp229Asn | 1.2 | −1.07 | 0.16 | coil | 92.6 | 7.61E−05 | |
| p.Asn233Thr | 3.5 | −1.40 | 0.42 | Bβ | coil | 46.9 | 7.99E−06 |
| p.Gln236His | 0.7 | 0.10 | 0.59 | Bβ | α-helix | 54.9 | 3.99E−06 |
| p.Gln236Arg | 0.0 | 0.82 | 0.31 | Bβ | α-helix | 54.9 | 7.10E−06 |
| p.Tyr237His■ | 3.1 | −1.81 | 1.65 | α-helix | 0.0 | 5.32E−05 | |
| p.Ser245Phe■ | 0.2 | −3.67 | 0.78 | Bβ | turn | 15.8 | 1.52E−04 |
| p.Pro246Arg | 1.8 | −0.26 | 0.93 | turn | 56.9 | 7.97E−06 | |
| p.Thr247Ala | −0.5 | 2.35 | 0.44 | turn | 83.0 | 7.97E−06 | |
| p.Thr249Ala | −0.6 | 0.54 | 0.69 | Bβ | coil | 76.8 | 3.98E−06 |
| p.Thr250Ile | 0.4 | −1.59 | −0.04 | Bβ | coil | 36.7 | 7.97E−06 |
| p.Glu251Gly | 2.7 | −3.34 | 1.88 | coil | 0.0 | 3.98E−06 | |
| p.Phe252Leu | 2.5 | −3.86 | 1.60 | Bβ | β-strand | 7.8 | 3.98E−06 |
| p.Glu257Ala | 0.8 | −0.46 | 1.08 | α-helix | 50.4 | 1.19E−05 | |
| p.His260Arg | 1.0 | −0.48 | 0.52 | α-helix | 23.4 | 1.59E−05 | |
| p.His260Asn | 0.9 | −0.15 | 1.24 | α-helix | 23.4 | 3.98E−06 | |
| p.Thr264Pro | 5.7 | −5.49 | 1.66 | α-helix | 36.4 | 7.97E−06 | |
| p.Gln265His■ | 8.8 | −1.17 | 0.44 | 310 helix | 3.0 | 3.19E−05 | |
| p.Gln265Glu▲ | 3.9 | −1.05 | 1.00 | 310 helix | 3.0 | 3.98E−06 | |
| p.Ala271Ser | 1.8 | −4.60 | 0.63 | β-strand | 2.2 | 3.98E−06 | |
| p.Val274Met | 0.0 | −8.19 | 1.78 | β-strand | 0.0 | 7.97E−06 | |
| p.Leu276Met | 0.9 | −9.83 | 1.45 | β-strand | 0.5 | 3.59E−05 | |
| p.Glu277Gly | 1.4 | −2.27 | 1.51 | β-strand | 34.1 | 3.99E−06 | |
| p.Thr283Ile | −0.4 | −4.75 | 0.27 | β-strand | 60.8 | 3.99E−05 | |
| p.Thr285Ala | 0.4 | −1.32 | 1.40 | β-strand | 39.5 | 4.32E−06 | |
| p.Met290Leu | 0.0 | 0.41 | 0.17 | γ | β-strand | 56.5 | 4.10E−06 |
| p.Met290Val | 2.2 | 0.71 | 0.74 | γ | β-strand | 56.5 | 1.23E−05 |
| p.Val293Met | 0.6 | −5.35 | 1.33 | β-strand | 0.6 | 1.80E−05 | |
| p.Gly294Ala▲ | −0.2 | −1.01 | 0.23 | γ | coil | 54.5 | 2.15E−05 |
| p.Gly294Arg▲ | −1.0 | −1.01 | −0.02 | γ | coil | 54.5 | 1.21E−05 |
| p.Lys299Asn | 2.5 | −1.31 | −0.26 | turn | 36.5 | 5.69E−05 | |
| p.Arg301His■ | 1.3 | −0.79 | 0.84 | γ, knob | turn | 35.0 | 8.00E−06 |
| p.Ala305Gly▲ | 0.6 | −0.93 | 0.71 | γ | β-strand | 41.5 | 3.99E−06 |
| p.Tyr306Cys■ | 1.2 | −6.61 | 0.81 | γ | β-strand | 61.0 | 3.19E−05 |
| p.Tyr306His▲ | 1.3 | 2.54 | 0.30 | γ | β-strand | 61.0 | 3.99E−06 |
| p.Ala308Val | 0.9 | −0.98 | 0.05 | β-strand | 16.4 | 3.99E−06 | |
| p.Ala308Thr | 1.7 | 2.44 | 0.17 | β-strand | 16.4 | 5.67E−05 | |
| p.Gly309Asp | 2.3 | −0.64 | 1.42 | β-strand | 71.2 | 3.98E−06 | |
| p.Asp314Asn | −0.2 | −0.51 | 0.26 | coil | 27.1 | 7.97E−06 | |
| p.Gly322Ser | 3.7 | −0.03 | 0.56 | turn | 79.6 | 7.96E−06 | |
| p.Asp323Asn | −0.8 | 1.89 | 0.13 | Knob | turn | 65.5 | 3.98E−06 |
| p.Asp324Glu | 0.0 | 0.26 | 0.82 | γ, knob | turn | 67.9 | 1.19E−05 |
| p.Phe330Leu | 0.4 | −0.92 | 0.90 | γ, knob | α-helix | 30.7 | 3.98E−06 |
| p.Asn334Ile■ | 0.5 | −0.18 | −0.04 | γ | turn | 46.6 | 1.59E−05 |
| p.Met336Leu▲ | −0.6 | −0.84 | 0.98 | γ | bridge | 13.0 | 3.98E−06 |
| p.Asn343Asp | 0.6 | −0.03 | 0.83 | proximal to Ca2+ | bridge | 32.2 | 3.98E−06 |
| p.Asp344Glu▲ | 0.4 | −0.48 | 1.01 | Ca2+ | turn | 37.3 | 3.98E−06 |
| p.Lys347Thr | 2.2 | 0.89 | −0.18 | γ, proximal to Ca2+ | coil | 69.5 | 3.98E−06 |
| p.Ala367Thr■ | 3.2 | 0.39 | 1.25 | turn | 0.0 | 4.24E−05 | |
| p.Gly377Val▲ | 4.5 | −1.64 | 2.14 | turn | 92.1 | 3.89E−05 | |
| p.Gly377Cys▲ | 3.1 | 0.57 | 1.28 | turn | 92.1 | 3.98E−06 | |
| p.Gly377Ser■ | 3.1 | −0.55 | 1.49 | turn | 92.1 | 1.42E−05 | |
| p.Pro386Ser | 1.2 | −1.97 | 0.39 | turn | 93.4 | 1.99E−05 | |
| p.Asn387Lys■ | −0.3 | 2.27 | 0.59 | proximal to knob | turn | 83.8 | 2.39E−05 |
| p.Asn387Ser▲ | 1.0 | 0.24 | −0.10 | proximal to knob | turn | 83.8 | 3.98E−06 |
| p.Ile393Met | 0.2 | −0.23 | 1.41 | coil | 0.9 | 3.98E−06 | |
| p.Met405Val | 3.9 | −4.26 | 0.95 | coil | 0.8 | 3.18E−05 | |
| p.Met410Ile | 2.4 | 5.62 | 0.06 | β-strand | 0.8 | 1.41E−05 | |
| p.Met410Val | 3.4 | 5.50 | 0.59 | β-strand | 0.8 | 1.41E−05 | |
| p.Ile412Thr | 2.2 | −10.75 | 2.93 | β-strand | 3.5 | 3.98E−06 | |
| p.Ile413Val | 0.7 | −1.83 | 1.39 | β-strand | 0.0 | 3.98E−06 | |
| p.Pro414Thr | 3.1 | −3.98 | 1.45 | β-strand | 11.3 | 1.19E−05 | |
| p.Asn416Asp | −0.3 | 1.33 | −0.18 | turn | 95.9 | 3.19E−05 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellacchio, E. Mutations Causing Mild or No Structural Damage in Interfaces of Multimerization of the Fibrinogen γ-Module More Likely Confer Negative Dominant Behaviors. Int. J. Mol. Sci. 2020, 21, 9016. https://doi.org/10.3390/ijms21239016
Bellacchio E. Mutations Causing Mild or No Structural Damage in Interfaces of Multimerization of the Fibrinogen γ-Module More Likely Confer Negative Dominant Behaviors. International Journal of Molecular Sciences. 2020; 21(23):9016. https://doi.org/10.3390/ijms21239016
Chicago/Turabian StyleBellacchio, Emanuele. 2020. "Mutations Causing Mild or No Structural Damage in Interfaces of Multimerization of the Fibrinogen γ-Module More Likely Confer Negative Dominant Behaviors" International Journal of Molecular Sciences 21, no. 23: 9016. https://doi.org/10.3390/ijms21239016
APA StyleBellacchio, E. (2020). Mutations Causing Mild or No Structural Damage in Interfaces of Multimerization of the Fibrinogen γ-Module More Likely Confer Negative Dominant Behaviors. International Journal of Molecular Sciences, 21(23), 9016. https://doi.org/10.3390/ijms21239016