Aberrant Activity of Histone–Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis


Abstract
:1. Introduction
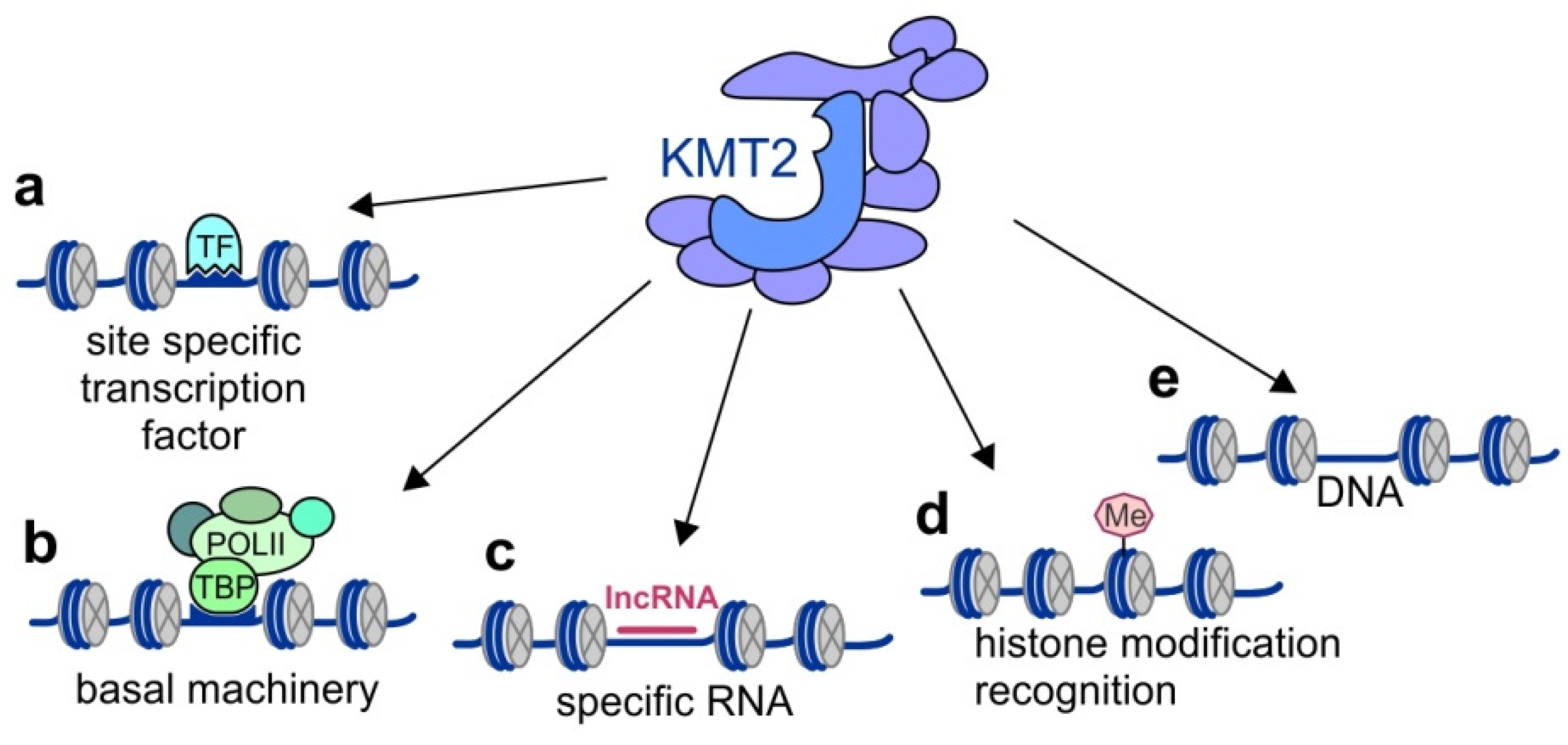
2. Structure of the KMT2 Complexes
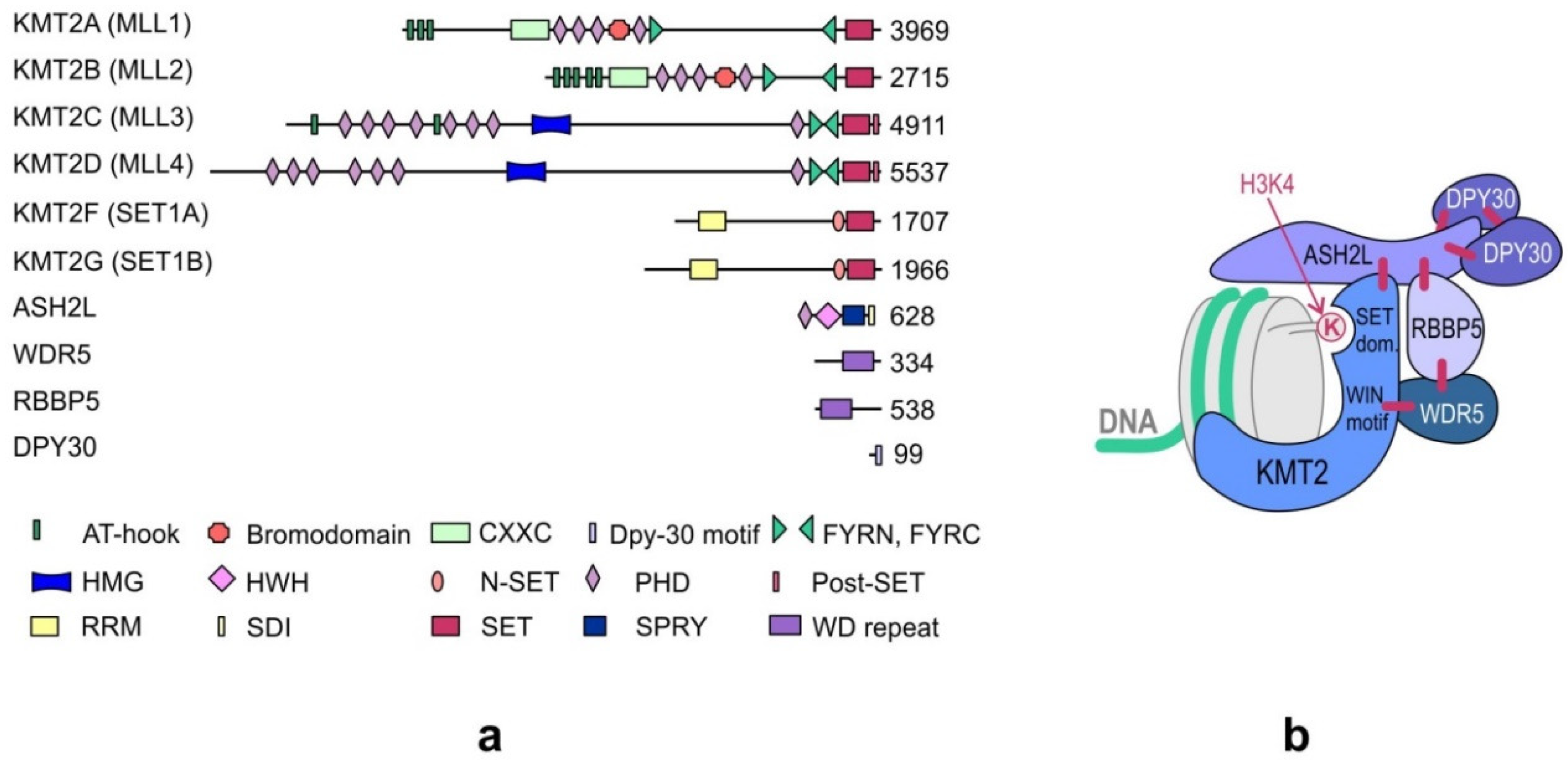
3. Domain Structure of KMT2s
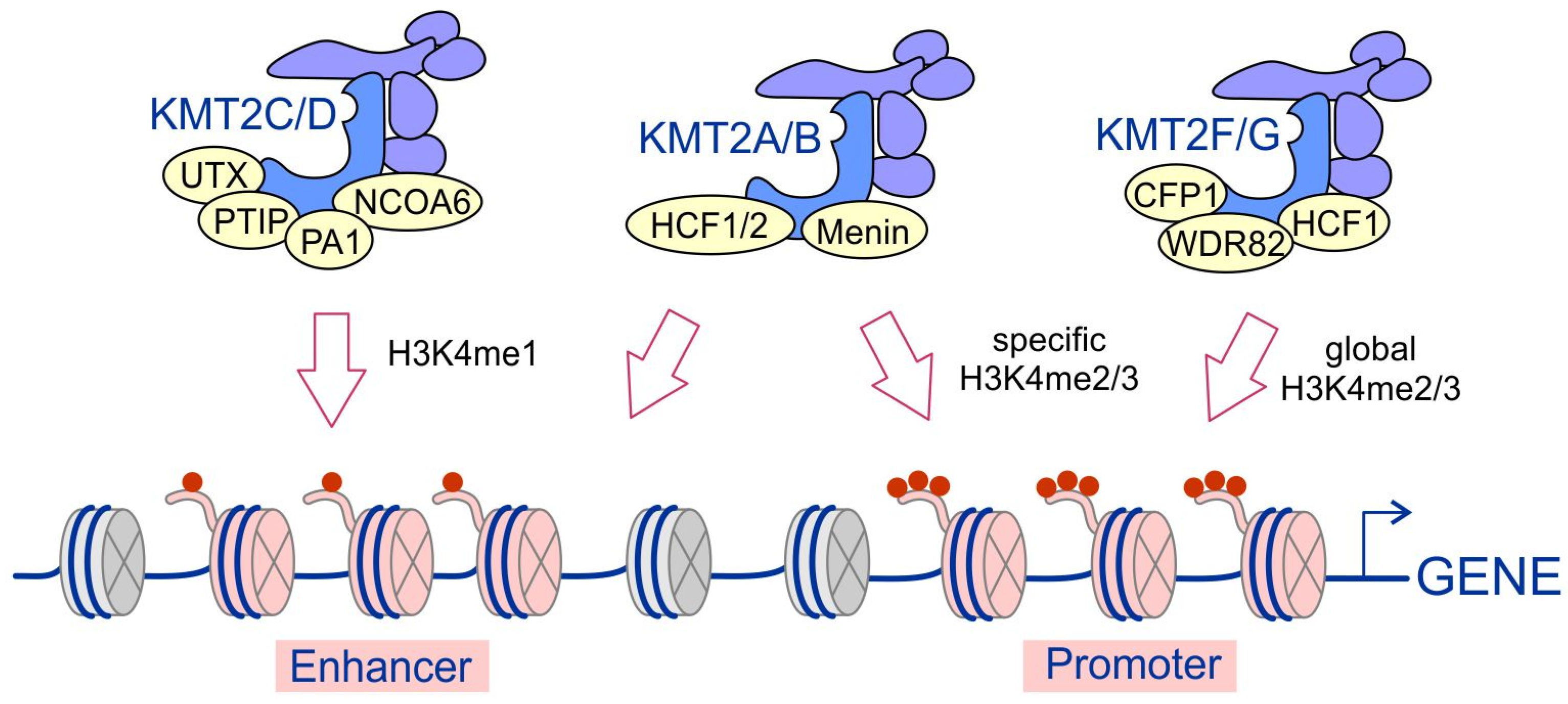
4. Substrate Specificity of KMT2 Complexes and Their Recruitment to Chromatin
5. Mutations in the KMT2 Family in Cancer
5.1. KMT2A (MLL1) and KMT2B (MLL2)
5.2. KMT2C (MLL3) and KMT2D (MLL4)
5.3. KMT2F (SET1A) and KMT2G (SET1B)
6. Roles of the Core Subunits in Oncogenesis
6.1. WDR5
6.2. RBBP5
6.3. ASH2L
6.4. DPY30
7. Therapeutic Strategies Targeting the Aberrant Activity of KMT2 Complexes in Cancers
8. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.A. Coming full circle-from endless complexity to simplicity and back again. Cell 2014, 157, 267–271. [Google Scholar] [CrossRef] [Green Version]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [Green Version]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; et al. New nomenclature for chromatin-modifying enzymes. Cell 2007, 131, 633–636. [Google Scholar] [CrossRef] [Green Version]
- Bulger, M.; Groudine, M. Functional and mechanistic diversity of distal transcription enhancers. Cell 2011, 144, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Ruthenburg, A.J.; Allis, C.D.; Wysocka, J. Methylation of lysine 4 on histone H3: Intricacy of writing and reading a single epigenetic mark. Mol. Cell 2007, 25, 15–30. [Google Scholar] [CrossRef]
- Zentner, G.E.; Scacheri, P.C. The chromatin fingerprint of gene enhancer elements. J. Biol. Chem. 2012, 287, 30888–30896. [Google Scholar] [CrossRef] [Green Version]
- Eissenberg, J.C.; Shilatifard, A. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev. Biol. 2010, 339, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pijnappel, W.W.; Schaft, D.; Roguev, A.; Shevchenko, A.; Tekotte, H.; Wilm, M.; Rigaut, G.; Séraphin, B.; Aasland, R.; Stewart, A.F. The S. cerevisiae SET3 complex includes two histone deacetylases, Hos2 and Hst1, and is a meiotic-specific repressor of the sporulation gene program. Genes Dev. 2001, 15, 2991–3004. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Novera, W.; Zhang, Y.; Deng, L.-W. MLL5 (KMT2E): Structure, function, and clinical relevance. Cell. Mol. Life Sci. 2017, 74, 2333–2344. [Google Scholar] [CrossRef]
- Dou, Y.; Milne, T.A.; Ruthenburg, A.J.; Lee, S.; Lee, J.W.; Verdine, G.L.; Allis, C.D.; Roeder, R.G. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 2006, 13, 713–719. [Google Scholar] [CrossRef]
- Patel, A.; Dharmarajan, V.; Vought, V.E.; Cosgrove, M.S. On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (MLL1) core complex. J. Biol. Chem. 2009, 284, 24242–24256. [Google Scholar] [CrossRef] [Green Version]
- Avdic, V.; Zhang, P.; Lanouette, S.; Groulx, A.; Tremblay, V.; Brunzelle, J.; Couture, J.-F. Structural and biochemical insights into MLL1 core complex assembly. Structure 2011, 19, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Couture, J.-F.; Skiniotis, G. Assembling a COMPASS. Epigenetics 2013, 8, 349–354. [Google Scholar] [CrossRef] [Green Version]
- Southall, S.M.; Wong, P.-S.; Odho, Z.; Roe, S.M.; Wilson, J.R. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks. Mol. Cell 2009, 33, 181–191. [Google Scholar] [CrossRef]
- Hughes, C.M.; Rozenblatt-Rosen, O.; Milne, T.A.; Copeland, T.D.; Levine, S.S.; Lee, J.C.; Hayes, D.N.; Shanmugam, K.S.; Bhattacharjee, A.; Biondi, C.A.; et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol. Cell 2004, 13, 587–597. [Google Scholar] [CrossRef]
- van Nuland, R.; Smits, A.H.; Pallaki, P.; Jansen, P.W.T.C.; Vermeulen, M.; Timmers, H.T.M. Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol. Cell. Biol. 2013, 33, 2067–2077. [Google Scholar] [CrossRef] [Green Version]
- Murai, M.J.; Pollock, J.; He, S.; Miao, H.; Purohit, T.; Yokom, A.; Hess, J.L.; Muntean, A.G.; Grembecka, J.; Cierpicki, T. The same site on the integrase-binding domain of lens epithelium-derived growth factor is a therapeutic target for MLL leukemia and HIV. Blood 2014, 124, 3730–3737. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, A.; Cleary, M.L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.-W.; Hong, T.; Hong, S.; Guo, H.; Yu, H.; Kim, D.; Guszczynski, T.; Dressler, G.R.; Copeland, T.D.; Kalkum, M.; et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 2007, 282, 20395–20406. [Google Scholar] [CrossRef] [Green Version]
- Goo, Y.-H.; Sohn, Y.C.; Kim, D.-H.; Kim, S.-W.; Kang, M.-J.; Jung, D.-J.; Kwak, E.; Barlev, N.A.; Berger, S.L.; Chow, V.T.; et al. Activating Signal Cointegrator 2 Belongs to a Novel Steady-State Complex That Contains a Subset of Trithorax Group Proteins. Mol. Cell. Biol. 2003, 23, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.R.; Kim, D.; Levitan, I.; Dressler, G.R. The BRCT-domain containing protein PTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev. Cell 2007, 13, 580–592. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Tate, C.M.; You, J.-S.; Skalnik, D.G. Identification and characterization of the human Set1B histone H3-Lys4 methyltransferase complex. J. Biol. Chem. 2007, 282, 13419–13428. [Google Scholar] [CrossRef] [Green Version]
- Demers, C.; Chaturvedi, C.-P.; Ranish, J.A.; Juban, G.; Lai, P.; Morle, F.; Aebersold, R.; Dilworth, F.J.; Groudine, M.; Brand, M. Activator-mediated Recruitment of the MLL2 Methyltransferase Complex to the β-globin Locus. Mol. Cell 2007, 27, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.; Li, Y.; Liang, S.; Cui, K.; Salz, T.; Yang, H.; Tang, Z.; Gallagher, P.G.; Qiu, Y.; Roeder, R.; et al. USF1 and hSET1A mediated epigenetic modifications regulate lineage differentiation and HoxB4 transcription. PLoS Genet. 2013, 9, e1003524. [Google Scholar] [CrossRef] [Green Version]
- Aziz, A.; Liu, Q.-C.; Dilworth, F.J. Regulating a master regulator: Establishing tissue-specific gene expression in skeletal muscle. Epigenetics 2010, 5, 691–695. [Google Scholar] [CrossRef] [Green Version]
- Fossati, A.; Dolfini, D.; Donati, G.; Mantovani, R. NF-Y Recruits Ash2L to Impart H3K4 Trimethylation on CCAAT Promoters. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Song, S.; Brand, M.; Dean, A. Nucleosome and transcription activator antagonism at human beta-globin locus control region DNase I hypersensitive sites. Nucleic Acids Res. 2007, 35, 5831–5838. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.C.; Sindhu, K.V.; Li, S.; Nishio, H.; Stoller, J.Z.; Oishi, K.; Puttreddy, S.; Lee, T.J.; Epstein, J.A.; Walsh, M.J.; et al. Transcription factor Ap2δ associates with Ash2l and ALR, a trithorax family histone methyltransferase, to activate Hoxc8 transcription. Proc. Natl. Acad. Sci. USA 2008, 105, 7472–7477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullius, A.; Lüscher-Firzlaff, J.; Costa, I.G.; Walsemann, G.; Forst, A.H.; Gusmao, E.G.; Kapelle, K.; Kleine, H.; Kremmer, E.; Vervoorts, J.; et al. The interaction of MYC with the trithorax protein ASH2L promotes gene transcription by regulating H3K27 modification. Nucleic Acids Res. 2014, 42, 6901–6920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, Y.-S.; Tsai, S.-Y.; Lee, D.-F.; Monk, J.; Su, J.; Ratnakumar, K.; Ding, J.; Ge, Y.; Darr, H.; Chang, B.; et al. Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell 2011, 145, 183–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertero, A.; Madrigal, P.; Galli, A.; Hubner, N.C.; Moreno, I.; Burks, D.; Brown, S.; Pedersen, R.A.; Gaffney, D.; Mendjan, S.; et al. Activin/nodal signaling and NANOG orchestrate human embryonic stem cell fate decisions by controlling the H3K4me3 chromatin mark. Genes Dev. 2015, 29, 702–717. [Google Scholar] [CrossRef] [Green Version]
- Dou, Y.; Milne, T.A.; Tackett, A.J.; Smith, E.R.; Fukuda, A.; Wysocka, J.; Allis, C.D.; Chait, B.T.; Hess, J.L.; Roeder, R.G. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 2005, 121, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kim, D.-H.; Lee, S.; Yang, Q.-H.; Lee, D.K.; Lee, S.-K.; Roeder, R.G.; Lee, J.W. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc. Natl. Acad. Sci. USA 2009, 106, 8513–8518. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Chen, W.-Y.; Shimada, M.; Nguyen, U.T.T.; Kim, J.; Sun, X.-J.; Sengoku, T.; McGinty, R.K.; Fernandez, J.P.; Muir, T.W.; et al. SET1 and p300 act synergistically, through coupled histone modifications, in transcriptional activation by p53. Cell 2013, 154, 297–310. [Google Scholar] [CrossRef] [Green Version]
- Takeda, S.; Chen, D.Y.; Westergard, T.D.; Fisher, J.K.; Rubens, J.A.; Sasagawa, S.; Kan, J.T.; Korsmeyer, S.J.; Cheng, E.H.-Y.; Hsieh, J.J.-D. Proteolysis of MLL family proteins is essential for Taspase1-orchestrated cell cycle progression. Genes Dev. 2006, 20, 2397–2409. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, S.; Chabes, A.L.; Wysocka, J.; Herr, W. E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol. Cell 2007, 27, 107–119. [Google Scholar] [CrossRef]
- Kawabe, Y.-I.; Wang, Y.X.; McKinnell, I.W.; Bedford, M.T.; Rudnicki, M.A. Carm1 regulates Pax7 transcriptional activity through MLL1/2 recruitment during asymmetric satellite stem cell divisions. Cell Stem Cell 2012, 11, 333–345. [Google Scholar] [CrossRef] [Green Version]
- Tschiersch, B.; Hofmann, A.; Krauss, V.; Dorn, R.; Korge, G.; Reuter, G. The protein encoded by the Drosophila position-effect variegation suppressor gene Su(var)3-9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO J. 1994, 13, 3822–3831. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.-D.; Cheng, E.H.-Y.; Korsmeyer, S.J. Taspase1: A threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell 2003, 115, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, R.; Zhou, M.-M. The PHD finger: A versatile epigenome reader. Trends Biochem. Sci. 2011, 36, 364–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, S.S.; Lee, S.-H.; Kan, P.-Y.; Voigt, P.; Ma, L.; Shi, X.; Reinberg, D.; Lee, M.G. Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 2012, 26, 2749–2762. [Google Scholar] [CrossRef] [Green Version]
- Fair, K.; Anderson, M.; Bulanova, E.; Mi, H.; Tropschug, M.; Diaz, M.O. Protein interactions of the MLL PHD fingers modulate MLL target gene regulation in human cells. Mol. Cell. Biol. 2001, 21, 3589–3597. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Osmers, U.; Raman, G.; Schwantes, R.H.; Diaz, M.O.; Bushweller, J.H. The PHD3 Domain of MLL Acts as a CYP33-Regulated Switch between MLL-Mediated Activation and Repression. Biochemistry 2010, 49, 6576–6586. [Google Scholar] [CrossRef]
- Wang, J.; Muntean, A.G.; Wu, L.; Hess, J.L. A subset of mixed lineage leukemia proteins has plant homeodomain (PHD)-mediated E3 ligase activity. J. Biol. Chem. 2012, 287, 43410–43416. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Santillan, D.A.; Koonce, M.; Wei, W.; Luo, R.; Thirman, M.J.; Zeleznik-Le, N.J.; Diaz, M.O. Loss of MLL PHD finger 3 is necessary for MLL-ENL-induced hematopoietic stem cell immortalization. Cancer Res. 2008, 68, 6199–6207. [Google Scholar] [CrossRef] [Green Version]
- Muntean, A.G.; Giannola, D.; Udager, A.M.; Hess, J.L. The PHD fingers of MLL block MLL fusion protein-mediated transformation. Blood 2008, 112, 4690–4693. [Google Scholar] [CrossRef] [Green Version]
- Ansari, K.I.; Mandal, S.S. Mixed lineage leukemia: Roles in gene expression, hormone signaling and mRNA processing. FEBS J. 2010, 277, 1790–1804. [Google Scholar] [CrossRef]
- Fang, L.; Zhang, J.; Zhang, H.; Yang, X.; Jin, X.; Zhang, L.; Skalnik, D.G.; Jin, Y.; Zhang, Y.; Huang, X.; et al. H3K4 Methyltransferase Set1a Is A Key Oct4 Coactivator Essential for Generation of Oct4 Positive Inner Cell Mass. Stem Cells 2016, 34, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Sze, C.C.; Ozark, P.A.; Cao, K.; Ugarenko, M.; Das, S.; Wang, L.; Marshall, S.A.; Rendleman, E.J.; Ryan, C.A.; Zha, D.; et al. Coordinated regulation of cellular identity–associated H3K4me3 breadth by the COMPASS family. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lin, C.; Smith, E.R.; Guo, H.; Sanderson, B.W.; Wu, M.; Gogol, M.; Alexander, T.; Seidel, C.; Wiedemann, L.M.; et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol. Cell. Biol. 2009, 29, 6074–6085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaikkonen, M.U.; Spann, N.J.; Heinz, S.; Romanoski, C.E.; Allison, K.A.; Stender, J.D.; Chun, H.B.; Tough, D.F.; Prinjha, R.K.; Benner, C.; et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol. Cell 2013, 51, 310–325. [Google Scholar] [CrossRef] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Lee, J.-E.; Wang, C.; Xu, S.; Cho, Y.-W.; Wang, L.; Feng, X.; Baldridge, A.; Sartorelli, V.; Zhuang, L.; Peng, W.; et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife 2013, 2, e01503. [Google Scholar] [CrossRef]
- Dreijerink, K.M.A.; Mulder, K.W.; Winkler, G.S.; Höppener, J.W.M.; Lips, C.J.M.; Timmers, H.T.M. Menin links estrogen receptor activation to histone H3K4 trimethylation. Cancer Res. 2006, 66, 4929–4935. [Google Scholar] [CrossRef] [Green Version]
- Ananthanarayanan, M.; Li, Y.; Surapureddi, S.; Balasubramaniyan, N.; Ahn, J.; Goldstein, J.A.; Suchy, F.J. Histone H3K4 trimethylation by MLL3 as part of ASCOM complex is critical for NR activation of bile acid transporter genes and is downregulated in cholestasis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G771–G781. [Google Scholar] [CrossRef] [Green Version]
- Scoville, D.W.; Cyphert, H.A.; Liao, L.; Xu, J.; Reynolds, A.; Guo, S.; Stein, R. MLL3 and MLL4 Methyltransferases Bind to the MAFA and MAFB Transcription Factors to Regulate Islet β-Cell Function. Diabetes 2015, 64, 3772–3783. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Augustin, J.; Hu, J.; Jiang, H. Physical Interactions and Functional Coordination between the Core Subunits of Set1/Mll Complexes and the Reprogramming Factors. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Thomas, L.R.; Wang, Q.; Grieb, B.C.; Phan, J.; Foshage, A.M.; Sun, Q.; Olejniczak, E.T.; Clark, T.; Dey, S.; Lorey, S.; et al. Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol. Cell 2015, 58, 440–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampalli, S.; Li, L.; Mak, E.; Ge, K.; Brand, M.; Tapscott, S.J.; Dilworth, F.J. p38 MAPK signaling regulates recruitment of Ash2L-containing methyltransferase complexes to specific genes during differentiation. Nat. Struct. Mol. Biol. 2007, 14, 1150–1156. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Monckton, E.A.; Godbout, R. Ectopic expression of transcription factor AP-2δ in developing retina: Effect on PSA-NCAM and axon routing. J. Neurochem. 2014, 129, 72–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Skalnik, D.G. Wdr82 is a C-terminal domain-binding protein that recruits the Setd1A Histone H3-Lys4 methyltransferase complex to transcription start sites of transcribed human genes. Mol. Cell. Biol. 2008, 28, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Muntean, A.G.; Tan, J.; Sitwala, K.; Huang, Y.; Bronstein, J.; Connelly, J.A.; Basrur, V.; Elenitoba-Johnson, K.S.J.; Hess, J.L. The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell 2010, 17, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.; Li, Y.; Zhou, L.; Cho, J.; Patel, B.; Terada, N.; Li, Y.; Bungert, J.; Qiu, Y.; Huang, S. HoxBlinc RNA Recruits Set1/MLL Complexes to Activate Hox Gene Expression Patterns and Mesoderm Lineage Development. Cell Rep. 2016, 14, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Ayton, P.M.; Chen, E.H.; Cleary, M.L. Binding to nonmethylated CpG DNA is essential for target recognition, transactivation, and myeloid transformation by an MLL oncoprotein. Mol. Cell. Biol. 2004, 24, 10470–10478. [Google Scholar] [CrossRef] [Green Version]
- Birke, M.; Schreiner, S.; García-Cuéllar, M.-P.; Mahr, K.; Titgemeyer, F.; Slany, R.K. The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res. 2002, 30, 958–965. [Google Scholar] [CrossRef] [Green Version]
- Cierpicki, T.; Risner, L.E.; Grembecka, J.; Lukasik, S.M.; Popovic, R.; Omonkowska, M.; Shultis, D.D.; Zeleznik-Le, N.J.; Bushweller, J.H. Structure of the MLL CXXC domain-DNA complex and its functional role in MLL-AF9 leukemia. Nat. Struct. Mol. Biol. 2010, 17, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Gao, X.; Cao, K.; Morgan, M.A.; Mas, G.; Smith, E.R.; Volk, A.G.; Bartom, E.T.; Crispino, J.D.; Di Croce, L.; et al. Not All H3K4 Methylations Are Created Equal: Mll2/COMPASS Dependency in Primordial Germ Cell Specification. Mol. Cell 2017, 65, 460–475.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, H.K.; Blackledge, N.P.; Klose, R.J. ZF-CxxC domain-containing proteins, CpG islands and the chromatin connection. Biochem. Soc. Trans. 2013, 41, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Bian, C.; Lam, R.; Dong, A.; Min, J. The structural basis for selective binding of non-methylated CpG islands by the CFP1 CXXC domain. Nat. Commun. 2011, 2, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravind, L.; Landsman, D. AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res. 1998, 26, 4413–4421. [Google Scholar] [CrossRef] [Green Version]
- Slany, R.K.; Lavau, C.; Cleary, M.L. The oncogenic capacity of HRX-ENL requires the transcriptional transactivation activity of ENL and the DNA binding motifs of HRX. Mol. Cell. Biol. 1998, 18, 122–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Daniel, J.; Espejo, A.; Lake, A.; Krishna, M.; Xia, L.; Zhang, Y.; Bedford, M.T. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006, 7, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Musselman, C.A.; Khorasanizadeh, S.; Kutateladze, T.G. Towards understanding methyllysine readout. Biochim. Et Biophys. Acta 2014, 1839, 686–693. [Google Scholar] [CrossRef] [Green Version]
- Musselman, C.A.; Lalonde, M.-E.; Côté, J.; Kutateladze, T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef] [Green Version]
- van Ingen, H.; van Schaik, F.M.A.; Wienk, H.; Ballering, J.; Rehmann, H.; Dechesne, A.C.; Kruijzer, J.A.W.; Liskamp, R.M.J.; Timmers, H.T.M.; Boelens, R. Structural insight into the recognition of the H3K4me3 mark by the TFIID subunit TAF3. Structure 2008, 16, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, M.; Mulder, K.W.; Denissov, S.; Pijnappel, W.W.M.P.; van Schaik, F.M.A.; Varier, R.A.; Baltissen, M.P.A.; Stunnenberg, H.G.; Mann, M.; Timmers, H.T.M. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 2007, 131, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ilin, S.; Wang, W.; Duncan, E.M.; Wysocka, J.; Allis, C.D.; Patel, D.J. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 2006, 442, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Wysocka, J.; Swigut, T.; Xiao, H.; Milne, T.A.; Kwon, S.Y.; Landry, J.; Kauer, M.; Tackett, A.J.; Chait, B.T.; Badenhorst, P.; et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 2006, 442, 86–90. [Google Scholar] [CrossRef]
- Kim, S.; Natesan, S.; Cornilescu, G.; Carlson, S.; Tonelli, M.; McClurg, U.L.; Binda, O.; Robson, C.N.; Markley, J.L.; Balaz, S.; et al. Mechanism of Histone H3K4me3 Recognition by the Plant Homeodomain of Inhibitor of Growth 3. J. Biol. Chem. 2016, 291, 18326–18341. [Google Scholar] [CrossRef] [Green Version]
- Peña, P.V.; Davrazou, F.; Shi, X.; Walter, K.L.; Verkhusha, V.V.; Gozani, O.; Zhao, R.; Kutateladze, T.G. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature 2006, 442, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Hong, T.; Walter, K.L.; Ewalt, M.; Michishita, E.; Hung, T.; Carney, D.; Peña, P.; Lan, F.; Kaadige, M.R.; et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 2006, 442, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.-R.; Xu, C.; Fuchs, A.; Mund, A.; Lange, M.; Staege, H.; Schubert, T.; Bian, C.; Dunkel, I.; Eberharter, A.; et al. PHF13 is a molecular reader and transcriptional co-regulator of H3K4me2/3. Elife 2016, 5. [Google Scholar] [CrossRef]
- Morgan, M.A.J.; Rickels, R.A.; Collings, C.K.; He, X.; Cao, K.; Herz, H.-M.; Cozzolino, K.A.; Abshiru, N.A.; Marshall, S.A.; Rendleman, E.J.; et al. A cryptic Tudor domain links BRWD2/PHIP to COMPASS-mediated histone H3K4 methylation. Genes Dev. 2017, 31, 2003–2014. [Google Scholar] [CrossRef]
- Fang, L.; Teng, H.; Wang, Y.; Liao, G.; Weng, L.; Li, Y.; Wang, X.; Jin, J.; Jiao, C.; Chen, L.; et al. SET1A-Mediated Mono-Methylation at K342 Regulates YAP Activation by Blocking Its Nuclear Export and Promotes Tumorigenesis. Cancer Cell 2018, 34, 103–118.e9. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.-S.; Shimazu, T.; Toyokawa, G.; Daigo, Y.; Maehara, Y.; Hayami, S.; Ito, A.; Masuda, K.; Ikawa, N.; Field, H.I.; et al. Enhanced HSP70 lysine methylation promotes proliferation of cancer cells through activation of Aurora kinase B. Nat. Commun. 2012, 3, 1072. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Tian, X.; Peng, C.; Gong, F.; Chen, Y. Crystal Structure of MLL2 Complex Guides the Identification of a Methylation Site on P53 Catalyzed by KMT2 Family Methyltransferases. Structure 2020, 28, 1141–1148.e4. [Google Scholar] [CrossRef]
- Meyer, C.; Hofmann, J.; Burmeister, T.; Gröger, D.; Park, T.S.; Emerenciano, M.; de Oliveira, M.P.; Renneville, A.; Villarese, P.; Macintyre, E.; et al. The MLL recombinome of acute leukemias in 2013. Leukemia 2013, 27, 2165–2176. [Google Scholar] [CrossRef] [PubMed]
- Ziemin-van der Poel, S.; McCabe, N.R.; Gill, H.J.; Espinosa, R.; Patel, Y.; Harden, A.; Rubinelli, P.; Smith, S.D.; LeBeau, M.M.; Rowley, J.D. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc. Natl. Acad. Sci. USA 1991, 88, 10735–10739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Nakamura, T.; Alder, H.; Prasad, R.; Canaani, O.; Cimino, G.; Croce, C.M.; Canaani, E. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell 1992, 71, 701–708. [Google Scholar] [CrossRef]
- Marschalek, R. Systematic Classification of Mixed-Lineage Leukemia Fusion Partners Predicts Additional Cancer Pathways. Ann. Lab. Med. 2016, 36, 85–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tkachuk, D.C.; Kohler, S.; Cleary, M.L. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 1992, 71, 691–700. [Google Scholar] [CrossRef]
- Richardson, C.; Jasin, M. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature 2000, 405, 697–700. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef]
- Roguev, A. The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J. 2001, 20, 7137–7148. [Google Scholar] [CrossRef]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- Aplan, P.D. Chromosomal translocations involving the MLL gene: Molecular mechanisms. DNA Repair 2006, 5, 1265–1272. [Google Scholar] [CrossRef] [Green Version]
- Ayton, P.M.; Cleary, M.L. Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene 2001, 20, 5695–5707. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Cheng, E.H.Y.; Hsieh, J.J.D. MLL fusions: Pathways to leukemia. Cancer Biol. Ther. 2009, 8, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Schneider, B.; Jakob, S.; Strehl, S.; Attarbaschi, A.; Schnittger, S.; Schoch, C.; Jansen, M.W.J.C.; van Dongen, J.J.M.; den Boer, M.L.; et al. The MLL recombinome of acute leukemias. Leukemia 2006, 20, 777–784. [Google Scholar] [CrossRef]
- The Groupe Français de Cytogénétique Oncologique; Huret, J.; Dessen, P.; Bernheim, A. An Atlas on Chromosomes in Hematological Malignancies. Example: 11q23 and MLL partners. Leukemia 2001, 15, 987–989. [Google Scholar] [CrossRef] [Green Version]
- Collins, C.T.; Hess, J.L. Deregulation of the HOXA9/MEIS1 axis in acute leukemia. Curr. Opin. Hematol. 2016, 23, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L Links Histone Methylation to Leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Slany, R.K. The molecular mechanics of mixed lineage leukemia. Oncogene 2016, 35, 5215–5223. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Yokoyama, A. The molecular functions of common and atypical MLL fusion protein complexes. Biochim. Et Biophys. Acta (BBA) Gene Regul. Mech. 2020, 1863, 194548. [Google Scholar] [CrossRef]
- Vlaming, H.; van Leeuwen, F. The upstreams and downstreams of H3K79 methylation by DOT1L. Chromosoma 2016, 125, 593–605. [Google Scholar] [CrossRef]
- Mueller, D.; Bach, C.; Zeisig, D.; Garcia-Cuellar, M.-P.; Monroe, S.; Sreekumar, A.; Zhou, R.; Nesvizhskii, A.; Chinnaiyan, A.; Hess, J.L.; et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007, 110, 4445–4454. [Google Scholar] [CrossRef]
- Cheung, N.; Chan, L.C.; Thompson, A.; Cleary, M.L.; So, C.W.E. Protein arginine-methyltransferase-dependent oncogenesis. Nat. Cell Biol. 2007, 9, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, H.; Taki, T.; Yoshino, H.; Takita, J.; Ida, K.; Ishii, M.; Nishida, K.; Hayashi, Y.; Taniwaki, M.; Bessho, F.; et al. A complex t(1;22;11)(q44;q13;q23) translocation causing MLL-p300 fusion gene in therapy-related acute myeloid leukemia. Eur. J. Haematol. 2008, 81, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Sobulo, O.M.; Borrow, J.; Tomek, R.; Reshmi, S.; Harden, A.; Schlegelberger, B.; Housman, D.; Doggett, N.A.; Rowley, J.D.; Zeleznik-Le, N.J. MLL is fused to CBP, a histone acetyltransferase, in therapy-related acute myeloid leukemia with a t(11;16)(q23;p13.3). Proc. Natl. Acad. Sci. USA 1997, 94, 8732–8737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, N.; Goodman, R.H. CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem. 2001, 276, 13505–13508. [Google Scholar] [CrossRef] [Green Version]
- Benedikt, A.; Baltruschat, S.; Scholz, B.; Bursen, A.; Arrey, T.N.; Meyer, B.; Varagnolo, L.; Müller, A.M.; Karas, M.; Dingermann, T.; et al. The leukemogenic AF4–MLL fusion protein causes P-TEFb kinase activation and altered epigenetic signatures. Leukemia 2011, 25, 135–144. [Google Scholar] [CrossRef]
- Bursen, A.; Schwabe, K.; Rüster, B.; Henschler, R.; Ruthardt, M.; Dingermann, T.; Marschalek, R. The AF4·MLL fusion protein is capable of inducing ALL in mice without requirement of MLL·AF4. Blood 2010, 115, 3570–3579. [Google Scholar] [CrossRef]
- Price, D.H. P-TEFb, a Cyclin-Dependent Kinase Controlling Elongation by RNA Polymerase II. Mol. Cell. Biol. 2000, 20, 2629–2634. [Google Scholar] [CrossRef] [Green Version]
- Thiel, A.T.; Blessington, P.; Zou, T.; Feather, D.; Wu, X.; Yan, J.; Zhang, H.; Liu, Z.; Ernst, P.; Koretzky, G.A.; et al. MLL-AF9-Induced Leukemogenesis Requires Co-Expression of the Wild Type Mll Allele. Cancer Cell 2010, 17, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Liedtke, M.; Cleary, M.L. Therapeutic targeting of MLL. Blood 2009, 113, 6061–6068. [Google Scholar] [CrossRef]
- Chen, W.; Kumar, A.R.; Hudson, W.A.; Li, Q.; Wu, B.; Staggs, R.A.; Lund, E.A.; Sam, T.N.; Kersey, J.H. Malignant transformation initiated by Mll-AF9: Gene dosage and critical target cells. Cancer Cell 2008, 13, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL–AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Rasio, D.; Schichman, S.A.; Negrini, M.; Canaani, E.; Croce, C.M. Complete exon structure of the ALL1 gene. Cancer Res. 1996, 56, 1766–1769. [Google Scholar] [PubMed]
- Reeves, R.; Nissen, M.S. The A.T-DNA-binding domain of mammalian high mobility group I chromosomal proteins. A novel peptide motif for recognizing DNA structure. J. Biol. Chem. 1990, 265, 8573–8582. [Google Scholar] [PubMed]
- Bestor, T.H.; Verdine, G.L. DNA methyltransferases. Curr. Opin. Cell Biol. 1994, 6, 380–389. [Google Scholar] [CrossRef]
- Shin Voo, K.; Carlone, D.L.; Jacobsen, B.M.; Flodin, A.; Skalnik, D.G. Cloning of a Mammalian Transcriptional Activator That Binds Unmethylated CpG Motifs and Shares a CXXC Domain with DNA Methyltransferase, Human Trithorax, and Methyl-CpG Binding Domain Protein 1. Mol. Cell. Biol. 2000, 20, 2108–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, U.; Beckmann, H.; Cashmore, A.R. HAT3.1, a novel Arabidopsis homeodomain protein containing a conserved cysteine-rich region. Plant. J. 1993, 4, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Tamkun, J.W.; Deuring, R.; Scott, M.P.; Kissinger, M.; Pattatucci, A.M.; Kaufman, T.C.; Kennison, J.A. brahma: A regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell 1992, 68, 561–572. [Google Scholar] [CrossRef]
- Ernst, P.; Wang, J.; Huang, M.; Goodman, R.H.; Korsmeyer, S.J. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol. Cell. Biol. 2001, 21, 2249–2258. [Google Scholar] [CrossRef] [Green Version]
- Goto, N.K.; Zor, T.; Martinez-Yamout, M.; Dyson, H.J.; Wright, P.E. Cooperativity in transcription factor binding to the coactivator CREB-binding protein (CBP). The mixed lineage leukemia protein (MLL) activation domain binds to an allosteric site on the KIX domain. J. Biol. Chem. 2002, 277, 43168–43174. [Google Scholar] [CrossRef] [Green Version]
- Dharmarajan, V.; Lee, J.-H.; Patel, A.; Skalnik, D.G.; Cosgrove, M.S. Structural Basis for WDR5 Interaction (Win) Motif Recognition in Human SET1 Family Histone Methyltransferases. J. Biol. Chem. 2012, 287, 27275–27289. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Dharmarajan, V.; Cosgrove, M.S. Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J. Biol. Chem. 2008, 283, 32158–32161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.; Vought, V.E.; Dharmarajan, V.; Cosgrove, M.S. A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J. Biol. Chem. 2008, 283, 32162–32175. [Google Scholar] [CrossRef] [Green Version]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL Targets SET Domain Methyltransferase Activity to Hox Gene Promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Nakamura, T.; Mori, T.; Tada, S.; Krajewski, W.; Rozovskaia, T.; Wassell, R.; Dubois, G.; Mazo, A.; Croce, C.M.; Canaani, E. ALL-1 Is a Histone Methyltransferase that Assembles a Supercomplex of Proteins Involved in Transcriptional Regulation. Mol. Cell 2002, 10, 1119–1128. [Google Scholar] [CrossRef]
- Hsieh, J.J.-D.; Ernst, P.; Erdjument-Bromage, H.; Tempst, P.; Korsmeyer, S.J. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol. Cell. Biol. 2003, 23, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, A.; Kitabayashi, I.; Ayton, P.M.; Cleary, M.L.; Ohki, M. Leukemia proto-oncoprotein MLL is proteolytically processed into 2 fragments with opposite transcriptional properties. Blood 2002, 100, 3710–3718. [Google Scholar] [CrossRef]
- Yokoyama, A.; Wang, Z.; Wysocka, J.; Sanyal, M.; Aufiero, D.J.; Kitabayashi, I.; Herr, W.; Cleary, M.L. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol. Cell. Biol. 2004, 24, 5639–5649. [Google Scholar] [CrossRef] [Green Version]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Economides, M.P.; McCue, D.; Borthakur, G.; Pemmaraju, N. Topoisomerase II inhibitors in AML: Past, present, and future. Expert Opin. Pharmacother. 2019, 20, 1637–1644. [Google Scholar] [CrossRef]
- Super, H.J.; McCabe, N.R.; Thirman, M.J.; Larson, R.A.; Le Beau, M.M.; Pedersen-Bjergaard, J.; Philip, P.; Diaz, M.O.; Rowley, J.D. Rearrangements of the MLL gene in therapy-related acute myeloid leukemia in patients previously treated with agents targeting DNA-topoisomerase II. Blood 1993, 82, 3705–3711. [Google Scholar] [CrossRef] [Green Version]
- Britten, O.; Ragusa, D.; Tosi, S.; Kamel, Y.M. MLL-Rearranged Acute Leukemia with t(4;11)(q21;q23)-Current Treatment Options. Is There a Role for CAR-T Cell Therapy? Cells 2019, 8, 1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirro, J.; Zipf, T.; Pui, C.; Kitchingman, G.; Williams, D.; Melvin, S.; Murphy, S.; Stass, S. Acute mixed lineage leukemia: Clinicopathologic correlations and prognostic significance. Blood 1985, 66, 1115–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stass, S.; Mirro, J.; Melvin, S.; Pui, C.H.; Murphy, S.B.; Williams, D. Lineage switch in acute leukemia. Blood 1984, 64, 701–706. [Google Scholar] [CrossRef]
- Chowdhury, T.; Brady, H.J.M. Insights from clinical studies into the role of the MLL gene in infant and childhood leukemia. Blood Cells Mol. Dis. 2008, 40, 192–199. [Google Scholar] [CrossRef]
- Massoth, L.R.; Hung, Y.P.; Nardi, V.; Nielsen, G.P.; Hasserjian, R.P.; Louissaint, A.; Fisch, A.S.; Deshpande, V.; Zukerberg, L.R.; Lennerz, J.K.; et al. Pan-sarcoma genomic analysis of KMT2A rearrangements reveals distinct subtypes defined by YAP1–KMT2A–YAP1 and VIM – KMT2A fusions. Mod. Pathol. 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.C.; Dou, Y. Hijacked in cancer: The KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 2015, 15, 334–346. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Gui, Y.; Guo, G.; Huang, Y.; Hu, X.; Tang, A.; Gao, S.; Wu, R.; Chen, C.; Li, X.; Zhou, L.; et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011, 43, 875–878. [Google Scholar] [CrossRef]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjöblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [Green Version]
- Ansari, K.I.; Kasiri, S.; Mandal, S.S. Histone methylase MLL1 has critical roles in tumor growth and angiogenesis and its knockdown suppresses tumor growth in vivo. Oncogene 2013, 32, 3359–3370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wend, P.; Fang, L.; Zhu, Q.; Schipper, J.H.; Loddenkemper, C.; Kosel, F.; Brinkmann, V.; Eckert, K.; Hindersin, S.; Holland, J.D.; et al. Wnt/β-catenin signalling induces MLL to create epigenetic changes in salivary gland tumours. EMBO J. 2013, 32, 1977–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Fang, L.; Heuberger, J.; Kranz, A.; Schipper, J.; Scheckenbach, K.; Vidal, R.O.; Sunaga-Franze, D.Y.; Müller, M.; Wulf-Goldenberg, A.; et al. The Wnt-Driven Mll1 Epigenome Regulates Salivary Gland and Head and Neck Cancer. Cell Rep. 2019, 26, 415–428.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaser, S.; Schaft, J.; Lubitz, S.; Vintersten, K.; van der Hoeven, F.; Tufteland, K.R.; Aasland, R.; Anastassiadis, K.; Ang, S.-L.; Stewart, A.F. Multiple epigenetic maintenance factors implicated by the loss of Mll2 in mouse development. Development 2006, 133, 1423–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubitz, S.; Glaser, S.; Schaft, J.; Stewart, A.F.; Anastassiadis, K. Increased Apoptosis and Skewed Differentiation in Mouse Embryonic Stem Cells Lacking the Histone Methyltransferase Mll2. Mol. Biol. Cell. 2007, 18, 2356–2366. [Google Scholar] [CrossRef] [Green Version]
- Huntsman, D.G.; Chin, S.F.; Muleris, M.; Batley, S.J.; Collins, V.P.; Wiedemann, L.M.; Aparicio, S.; Caldas, C. MLL2, the second human homolog of the Drosophila trithorax gene, maps to 19q13.1 and is amplified in solid tumor cell lines. Oncogene 1999, 18, 7975–7984. [Google Scholar] [CrossRef] [Green Version]
- Andreu-Vieyra, C.V.; Chen, R.; Agno, J.E.; Glaser, S.; Anastassiadis, K.; Stewart, A.F.; Matzuk, M.M. MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLoS Biol. 2010, 8. [Google Scholar] [CrossRef]
- Glaser, S.; Lubitz, S.; Loveland, K.L.; Ohbo, K.; Robb, L.; Schwenk, F.; Seibler, J.; Roellig, D.; Kranz, A.; Anastassiadis, K.; et al. The histone 3 lysine 4 methyltransferase, Mll2, is only required briefly in development and spermatogenesis. Epigenet. Chromatin 2009, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.; Li, B.; Li, Y.; Li, Z.; Liu, Z.; Chen, Z.; Wu, J.; Gao, J.; Shen, L. Genomic alterations in advanced gastric cancer endoscopic biopsy samples using targeted next-generation sequencing. Am. J. Cancer Res. 2017, 7, 1540–1553. [Google Scholar] [PubMed]
- Lu, H.; Yang, S.; Zhu, H.; Tong, X.; Xie, F.; Qin, J.; Han, N.; Wu, X.; Fan, Y.; Shao, Y.W.; et al. Targeted next generation sequencing identified clinically actionable mutations in patients with esophageal sarcomatoid carcinoma. BMC Cancer 2018, 18, 251. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.H.; Junck, L.; Druley, T.E.; Gutmann, D.H. NF1 glioblastoma clonal profiling reveals KMT2B mutations as potential somatic oncogenic events. Neurology 2019, 93, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Anastassiadis, K.; Kranz, A.; Stewart, A.F.; Arndt, K.; Waskow, C.; Yokoyama, A.; Jones, K.; Neff, T.; Lee, Y.; et al. MLL2, not MLL1, Plays a Major Role in Sustaining MLL-rearranged Acute Myeloid Leukemia. Cancer Cell 2017, 31, 755–770.e6. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Liu, Y.; Rappaport, A.R.; Kitzing, T.; Schultz, N.; Zhao, Z.; Shroff, A.S.; Dickins, R.A.; Vakoc, C.R.; Bradner, J.E.; et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 2014, 25, 652–665. [Google Scholar] [CrossRef] [Green Version]
- Froimchuk, E.; Jang, Y.; Ge, K. Histone H3 lysine 4 methyltransferase KMT2D. Gene 2017, 627, 337–342. [Google Scholar] [CrossRef]
- Kanda, H.; Nguyen, A.; Chen, L.; Okano, H.; Hariharan, I.K. The Drosophila Ortholog of MLL3 and MLL4, trithorax related, Functions as a Negative Regulator of Tissue Growth. Mol. Cell. Biol. 2013, 33, 1702–1710. [Google Scholar] [CrossRef] [Green Version]
- Santos, M.A.; Faryabi, R.B.; Ergen, A.V.; Day, A.M.; Malhowski, A.; Canela, A.; Onozawa, M.; Lee, J.-E.; Callen, E.; Gutierrez-Martinez, P.; et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature 2014, 514, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Ruault, M.; Brun, M.E.; Ventura, M.; Roizès, G.; De Sario, A. MLL3, a new human member of the TRX/MLL gene family, maps to 7q36, a chromosome region frequently deleted in myeloid leukaemia. Gene 2002, 284, 73–81. [Google Scholar] [CrossRef]
- Fagan, R.J.; Dingwall, A.K. COMPASS Ascending: Emerging clues regarding the roles of MLL3/KMT2C and MLL2/KMT2D proteins in cancer. Cancer Lett. 2019, 458, 56–65. [Google Scholar] [CrossRef]
- Ford, D.J.; Dingwall, A.K. The cancer COMPASS: Navigating the functions of MLL complexes in cancer. Cancer Genet. 2015, 208, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sze, C.C.; Shilatifard, A. MLL3/MLL4/COMPASS Family on Epigenetic Regulation of Enhancer Function and Cancer. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhao, Z.; Ozark, P.A.; Fantini, D.; Marshall, S.A.; Rendleman, E.J.; Cozzolino, K.A.; Louis, N.; He, X.; Morgan, M.A.; et al. Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy. Nat. Med. 2018, 24, 758–769. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Bleeker, F.E.; Lamba, S.; Rodolfo, M.; Daniotti, M.; Scarpa, A.; van Tilborg, A.A.; Leenstra, S.; Zanon, C.; Bardelli, A. Novel Somatic and Germline Mutations in Cancer Candidate Genes in Glioblastoma, Melanoma, and Pancreatic Carcinoma. Cancer Res. 2007, 67, 3545–3550. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.-J.; Yoon, C.; Lee, J.H.; Chang, K.K.; Lin, J.-X.; Kim, Y.-H.; Kook, M.-C.; Aksoy, B.A.; Park, D.J.; Ashktorab, H.; et al. KMT2C Mutations in Diffuse-Type Gastric Adenocarcinoma Promote Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2018, 24, 6556–6569. [Google Scholar] [CrossRef] [Green Version]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Kimball, S.; Liu, H.; Holowatyj, A.; Yang, Z.-Q. Genetic alterations of histone lysine methyltransferases and their significance in breast cancer. Oncotarget 2015, 6, 2466–2482. [Google Scholar] [CrossRef] [Green Version]
- Buck, M.J.; Raaijmakers, L.M.; Ramakrishnan, S.; Wang, D.; Valiyaparambil, S.; Liu, S.; Nowak, N.J.; Pili, R. Alterations in chromatin accessibility and DNA methylation in clear cell renal cell carcinoma. Oncogene 2014, 33, 4961–4965. [Google Scholar] [CrossRef]
- Ibragimova, I.; Dulaimi, E.; Slifker, M.J.; Chen, D.D.; Uzzo, R.G.; Cairns, P. A global profile of gene promoter methylation in treatment-naïve urothelial cancer. Epigenetics 2014, 9, 760–773. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Jerez, A.; Sugimoto, Y.; Makishima, H.; Verma, A.; Jankowska, A.M.; Przychodzen, B.; Visconte, V.; Tiu, R.V.; O’Keefe, C.L.; Mohamedali, A.M.; et al. Loss of heterozygosity in 7q myeloid disorders: Clinical associations and genomic pathogenesis. Blood 2012, 119, 6109–6117. [Google Scholar] [CrossRef] [PubMed]
- Larsson, C.; Cordeddu, L.; Siggens, L.; Pandzic, T.; Kundu, S.; He, L.; Ali, M.A.; Pristovšek, N.; Hartman, K.; Ekwall, K.; et al. Restoration of KMT2C/MLL3 in human colorectal cancer cells reinforces genome-wide H3K4me1 profiles and influences cell growth and gene expression. Clin. Epigenet. 2020, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Chang, C.-C.; Wortham, M.; Chen, L.H.; Kernagis, D.N.; Qin, X.; Cho, Y.-W.; Chi, J.-T.; Grant, G.A.; McLendon, R.E.; et al. Global identification of MLL2-targeted loci reveals MLL2’s role in diverse signaling pathways. Proc. Natl. Acad. Sci. USA 2012, 109, 17603–17608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, S.S.; Zhao, D.; Lin, T.; Gu, B.; Pal, K.; Wu, S.J.; Alam, H.; Lv, J.; Yun, K.; Gopalakrishnan, V.; et al. MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes. Mol. Cell 2018, 70, 825–841.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, H.; Tang, M.; Maitituoheti, M.; Dhar, S.S.; Kumar, M.; Han, C.Y.; Ambati, C.R.; Amin, S.B.; Gu, B.; Chen, T.-Y.; et al. KMT2D Deficiency Impairs Super-Enhancers to Confer a Glycolytic Vulnerability in Lung Cancer. Cancer Cell 2020, 37, 599–617.e7. [Google Scholar] [CrossRef]
- Rahnamoun, H.; Hong, J.; Sun, Z.; Lee, J.; Lu, H.; Lauberth, S.M. Mutant p53 regulates enhancer-associated H3K4 monomethylation through interactions with the methyltransferase MLL4. J. Biol. Chem. 2018, 293, 13234–13246. [Google Scholar] [CrossRef] [Green Version]
- Toska, E.; Osmanbeyoglu, H.U.; Castel, P.; Chan, C.; Hendrickson, R.C.; Elkabets, M.; Dickler, M.N.; Scaltriti, M.; Leslie, C.S.; Armstrong, S.A.; et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 2017, 355, 1324–1330. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Sharma, A.; Dhar, S.S.; Lee, S.-H.; Gu, B.; Chan, C.-H.; Lin, H.-K.; Lee, M.G. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 2014, 74, 1705–1717. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Deng, Z.; Tang, Y.; Deng, Z.; Li, M. Downregulation of KMT2D suppresses proliferation and induces apoptosis of gastric cancer. Biochem. Biophys. Res. Commun. 2018, 504, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Kantidakis, T.; Saponaro, M.; Mitter, R.; Horswell, S.; Kranz, A.; Boeing, S.; Aygün, O.; Kelly, G.P.; Matthews, N.; Stewart, A.; et al. Mutation of cancer driver MLL2 results in transcription stress and genome instability. Genes Dev. 2016, 30, 408–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampias, T.; Karagiannis, D.; Avgeris, M.; Polyzos, A.; Kokkalis, A.; Kanaki, Z.; Kousidou, E.; Tzetis, M.; Kanavakis, E.; Stravodimos, K.; et al. The lysine-specific methyltransferase KMT2C/MLL3 regulates DNA repair components in cancer. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Piunti, A.; Shilatifard, A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 2016, 352. [Google Scholar] [CrossRef] [Green Version]
- Cosmic COSMIC—Catalogue of Somatic Mutations in Cancer. Available online: https://cancer.sanger.ac.uk/cosmic (accessed on 20 October 2020).
- Salz, T.; Li, G.; Kaye, F.; Zhou, L.; Qiu, Y.; Huang, S. hSETD1A regulates Wnt target genes and controls tumor growth of colorectal cancer cells. Cancer Res. 2014, 74, 775–786. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-S.; Lee, M.-H.; Lee, M.-O. Histone methyltransferases regulate the transcriptional expression of ERα and the proliferation of tamoxifen-resistant breast cancer cells. Breast Cancer Res. Treat. 2020, 180, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Salz, T.; Deng, C.; Pampo, C.; Siemann, D.; Qiu, Y.; Brown, K.; Huang, S. Histone Methyltransferase hSETD1A Is a Novel Regulator of Metastasis in Breast Cancer. Mol. Cancer Res. 2015, 13, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Tajima, K.; Yae, T.; Javaid, S.; Tam, O.; Comaills, V.; Morris, R.; Wittner, B.S.; Liu, M.; Engstrom, A.; Takahashi, F.; et al. SETD1A modulates cell cycle progression through a miRNA network that regulates p53 target genes. Nat. Commun. 2015, 6, 8257. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Zheng, Q.; An, J.; Wu, M.; Li, H.; Gui, X.; Pu, H.; Lu, D. SET1A Cooperates With CUDR to Promote Liver Cancer Growth and Hepatocyte-like Stem Cell Malignant Transformation Epigenetically. Mol. Ther. 2016, 24, 261–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajima, K.; Matsuda, S.; Yae, T.; Drapkin, B.J.; Morris, R.; Boukhali, M.; Niederhoffer, K.; Comaills, V.; Dubash, T.; Nieman, L.; et al. SETD1A protects from senescence through regulation of the mitotic gene expression program. Nat. Commun. 2019, 10, 2854. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kumari, N.; Rai, A.; Singh, S.K.; Kakkar, N.; Prasad, R. Expression and clinical significance of COMPASS family of histone methyltransferases in clear cell renal cell carcinoma. Gene 2018, 674, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Oh, H.R.; Choi, M.R.; Gwak, M.; An, C.H.; Chung, Y.J.; Yoo, N.J.; Lee, S.H. Frameshift mutation of a histone methylation-related gene SETD1B and its regional heterogeneity in gastric and colorectal cancers with high microsatellite instability. Hum. Pathol. 2014, 45, 1674–1681. [Google Scholar] [CrossRef]
- Wang, L.; Collings, C.K.; Zhao, Z.; Cozzolino, K.A.; Ma, Q.; Liang, K.; Marshall, S.A.; Sze, C.C.; Hashizume, R.; Savas, J.N.; et al. A cytoplasmic COMPASS is necessary for cell survival and triple-negative breast cancer pathogenesis by regulating metabolism. Genes Dev. 2017, 31, 2056–2066. [Google Scholar] [CrossRef]
- Dias, J.; Nguyen, N.V.; Georgiev, P.; Gaub, A.; Brettschneider, J.; Cusack, S.; Kadlec, J.; Akhtar, A. Structural analysis of the KANSL1/WDR5/KANSL2 complex reveals that WDR5 is required for efficient assembly and chromatin targeting of the NSL complex. Genes Dev. 2014, 28, 929–942. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-L.; Faiola, F.; Xu, M.; Pan, S.; Martinez, E. Human ATAC Is a GCN5/PCAF-containing acetylase complex with a novel NC2-like histone fold module that interacts with the TATA-binding protein. J. Biol. Chem. 2008, 283, 33808–33815. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, V.; Zhang, P.; Chaturvedi, C.-P.; Thornton, J.; Brunzelle, J.S.; Skiniotis, G.; Shilatifard, A.; Brand, M.; Couture, J.-F. Molecular Basis for DPY-30 Association to COMPASS-like and NURF Complexes. Structure 2014, 22, 1821–1830. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xie, W.; Gu, P.; Cai, Q.; Wang, B.; Xie, Y.; Dong, W.; He, W.; Zhong, G.; Lin, T.; et al. Upregulated WDR5 promotes proliferation, self-renewal and chemoresistance in bladder cancer via mediating H3K4 trimethylation. Sci. Rep. 2015, 5, 8293. [Google Scholar] [CrossRef]
- Grebien, F.; Vedadi, M.; Getlik, M.; Giambruno, R.; Grover, A.; Avellino, R.; Skucha, A.; Vittori, S.; Kuznetsova, E.; Smil, D.; et al. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPα N-terminal leukemia. Nat. Chem. Biol. 2015, 11, 571–578. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Banerjee, T.; Vinckevicius, A.; Luo, Q.; Parker, J.B.; Baker, M.R.; Radhakrishnan, I.; Wei, J.-J.; Barish, G.D.; Chakravarti, D. A role for WDR5 in integrating threonine 11 phosphorylation to lysine 4 methylation on histone H3 during androgen signaling and in prostate cancer. Mol. Cell 2014, 54, 613–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Bell, J.L.; Carter, D.; Gherardi, S.; Poulos, R.C.; Milazzo, G.; Wong, J.W.H.; Al-Awar, R.; Tee, A.E.; Liu, P.Y.; et al. WDR5 Supports an N-Myc Transcriptional Complex That Drives a Protumorigenic Gene Expression Signature in Neuroblastoma. Cancer Res. 2015, 75, 5143–5154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, X.; Chen, S.; Wu, J.; Lin, J.; Pan, C.; Ying, X.; Pan, Z.; Qiu, L.; Liu, R.; Geng, R.; et al. PI3K/AKT-mediated upregulation of WDR5 promotes colorectal cancer metastasis by directly targeting ZNF407. Cell Death Dis. 2017, 8, e2686. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.R.; Foshage, A.M.; Weissmiller, A.M.; Tansey, W.P. The MYC-WDR5 Nexus and Cancer. Cancer Res. 2015, 75, 4012–4015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.-Y.; Sun, Z.; Mullokandov, G.; Bosch, A.; Qadeer, Z.A.; Cihan, E.; Rapp, Z.; Parsons, R.; Aguirre-Ghiso, J.A.; Farias, E.F.; et al. Cbx8 Acts Non-canonically with Wdr5 to Promote Mammary Tumorigenesis. Cell Rep. 2016, 16, 472–486. [Google Scholar] [CrossRef] [Green Version]
- Carugo, A.; Genovese, G.; Seth, S.; Nezi, L.; Rose, J.L.; Bossi, D.; Cicalese, A.; Shah, P.K.; Viale, A.; Pettazzoni, P.F.; et al. In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep. 2016, 16, 133–147. [Google Scholar] [CrossRef] [Green Version]
- Neilsen, B.K.; Chakraborty, B.; McCall, J.L.; Frodyma, D.E.; Sleightholm, R.L.; Fisher, K.W.; Lewis, R.E. WDR5 supports colon cancer cells by promoting methylation of H3K4 and suppressing DNA damage. BMC Cancer 2018, 18, 673. [Google Scholar] [CrossRef] [Green Version]
- Peñalosa-Ruiz, G.; Bousgouni, V.; Gerlach, J.P.; Waarlo, S.; van de Ven, J.V.; Veenstra, T.E.; Silva, J.C.R.; van Heeringen, S.J.; Bakal, C.; Mulder, K.W.; et al. WDR5, BRCA1, and BARD1 Co-regulate the DNA Damage Response and Modulate the Mesenchymal-to-Epithelial Transition during Early Reprogramming. Stem Cell Rep. 2019, 12, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Wu, G.; Zhu, J.; Tan, Z.; Shi, D.; Wu, X.; Tang, M.; Li, Z.; Hu, Y.; Zhang, S.; et al. Genotoxic stress-triggered β-catenin/JDP2/PRMT5 complex facilitates reestablishing glutathione homeostasis. Nat. Commun. 2019, 10, 3761. [Google Scholar] [CrossRef]
- Fu, Z.; Chen, C.; Zhou, Q.; Wang, Y.; Zhao, Y.; Zhao, X.; Li, W.; Zheng, S.; Ye, H.; Wang, L.; et al. LncRNA HOTTIP modulates cancer stem cell properties in human pancreatic cancer by regulating HOXA9. Cancer Lett. 2017, 410, 68–81. [Google Scholar] [CrossRef]
- He, W.; Zhong, G.; Jiang, N.; Wang, B.; Fan, X.; Chen, C.; Chen, X.; Huang, J.; Lin, T. Long noncoding RNA BLACAT2 promotes bladder cancer-associated lymphangiogenesis and lymphatic metastasis. J. Clin. Investig. 2018, 128, 861–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, M.; Zhang, Y.; Weng, M.; Hu, Y.; Xuan, Y.; Hu, Y.; Lv, K. lncRNA GCAWKR Promotes Gastric Cancer Development by Scaffolding the Chromatin Modification Factors WDR5 and KAT2A. Mol. Ther. 2018, 26, 2658–2668. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, K.; Malla, P.; Lawrence, H.R.; Chen, Z.; Sinha, C.K.; Malik, R.; Shukla, S.; Kim, J.; Coppola, D.; Lawrence, N.J.; et al. ACK1 Regulates Histone H4 Tyr88-phosphorylation and AR Gene Expression in Castration Resistant Prostate Cancer. Cancer Cell 2017, 31, 790–803.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Bao, J.; Zhu, X.; Dai, G.; Jiang, X.; Jiao, X.; Sheng, H.; Huang, J.; Yu, H. Retinoblastoma Binding Protein 5 Correlates with the Progression in Hepatocellular Carcinoma. BioMed Res. Int. 2018, 2018, 1073432. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, W.; Dai, S.; Zhang, X.; Ban, N.; Fan, S.; Bao, Z.; Sun, J.; Shen, C.; Xia, X.; et al. Expression and clinical role of RBQ3 in gliomas. J. Neurol. Sci. 2015, 359, 177–184. [Google Scholar] [CrossRef]
- Alvarado, A.G.; Thiagarajan, P.S.; Mulkearns-Hubert, E.E.; Silver, D.J.; Hale, J.S.; Alban, T.J.; Turaga, S.M.; Jarrar, A.; Reizes, O.; Longworth, M.S.; et al. Glioblastoma Cancer Stem Cells Evade Innate Immune Suppression of Self-Renewal through Reduced TLR4 Expression. Cell Stem Cell 2017, 20, 450–461.e4. [Google Scholar] [CrossRef] [Green Version]
- Lüscher-Firzlaff, J.; Gawlista, I.; Vervoorts, J.; Kapelle, K.; Braunschweig, T.; Walsemann, G.; Rodgarkia-Schamberger, C.; Schuchlautz, H.; Dreschers, S.; Kremmer, E.; et al. The human trithorax protein hASH2 functions as an oncoprotein. Cancer Res. 2008, 68, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.S.; Qiu, Y.H.; Zhang, N.; Yoo, S.-Y.; Coombes, K.R.; Dent, S.Y.R.; Kornblau, S.M. Low expression of ASH2L protein correlates with a favorable outcome in acute myeloid leukemia. Leuk. Lymphoma 2017, 58, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.; Wu, Y.; Wang, C.; Wang, S.; Sun, H.; Zou, R.; Sun, G.; Song, H.; Liu, W.; Sun, N.; et al. ASH2L is involved in promotion of endometrial cancer progression via upregulation of PAX2 transcription. Cancer Sci. 2020, 111, 2062–2077. [Google Scholar] [CrossRef] [PubMed]
- He, F.-X.; Zhang, L.-L.; Jin, P.-F.; Liu, D.-D.; Li, A.-H. DPY30 regulates cervical squamous cell carcinoma by mediating epithelial-mesenchymal transition (EMT). Oncotargets Ther. 2019, 12, 7139–7147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Shah, K.; Busby, T.; Giles, K.; Khodadadi-Jamayran, A.; Li, W.; Jiang, H. Hijacking a key chromatin modulator creates epigenetic vulnerability for MYC-driven cancer. J. Clin. Investig. 2018, 128, 3605–3618. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Han, M.-E.; Baek, S.-J.; Kim, S.-Y.; Oh, S.-O. Roles of DPY30 in the Proliferation and Motility of Gastric Cancer Cells. PLoS ONE 2015, 10, e0131863. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Li, A.; Zhang, A.; Zhang, S.; Chen, L. DPY30 is required for the enhanced proliferation, motility and epithelial-mesenchymal transition of epithelial ovarian cancer cells. Int. J. Mol. Med. 2018, 42, 3065–3072. [Google Scholar] [CrossRef]
- Lu, K.; Tao, H.; Si, X.; Chen, Q. The Histone H3 Lysine 4 Presenter WDR5 as an Oncogenic Protein and Novel Epigenetic Target in Cancer. Front. Oncol. 2018, 8, 502. [Google Scholar] [CrossRef]
- Chan, A.K.N.; Chen, C.-W. Rewiring the Epigenetic Networks in MLL-Rearranged Leukemias: Epigenetic Dysregulation and Pharmacological Interventions. Front. Cell Dev. Biol. 2019, 7. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective Killing of Mixed Lineage Leukemia Cells by a Potent Small-Molecule DOT1L Inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef]
- Xu, J.; Li, L.; Xiong, J.; denDekker, A.; Ye, A.; Karatas, H.; Liu, L.; Wang, H.; Qin, Z.S.; Wang, S.; et al. MLL1 and MLL1 fusion proteins have distinct functions in regulating leukemic transcription program. Cell Discov. 2016, 2, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Liang, K.; Volk, A.G.; Haug, J.S.; Marshall, S.A.; Woodfin, A.R.; Bartom, E.T.; Gilmore, J.M.; Florens, L.; Washburn, M.P.; Sullivan, K.D.; et al. Therapeutic targeting of MLL degradation pathways in MLL-rearranged leukemia. Cell 2017, 168, 59–72.e13. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.-I.; Robson, S.C.; Chung, C.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Huang, H.; Li, Z.; Li, Y.; Wang, X.; Gurbuxani, S.; Chen, P.; He, C.; You, D.; Zhang, S.; et al. Blockade of miR-150 maturation by MLL-fusion/MYC/LIN-28 is required for MLL-associated leukemia. Cancer Cell 2012, 22, 524–535. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Cheng, X.; Wang, R.; Tan, Y.; Ge, M.; Li, D.; Xu, Q.; Sun, Y.; Zhao, C.; Chen, S.; et al. Restoration of microRNA function impairs MYC-dependent maintenance of MLL leukemia. Leukemia 2020, 34, 2484–2488. [Google Scholar] [CrossRef]
- Cao, F.; Townsend, E.C.; Karatas, H.; Xu, J.; Li, L.; Lee, S.; Liu, L.; Chen, Y.; Ouillette, P.; Zhu, J.; et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol. Cell 2014, 53, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Aho, E.R.; Wang, J.; Gogliotti, R.D.; Howard, G.C.; Phan, J.; Acharya, P.; Macdonald, J.D.; Cheng, K.; Lorey, S.L.; Lu, B.; et al. Displacement of WDR5 from Chromatin by a WIN Site Inhibitor with Picomolar Affinity. Cell Rep. 2019, 26, 2916–2928.e13. [Google Scholar] [CrossRef] [Green Version]
- Aho, E.R.; Weissmiller, A.M.; Fesik, S.W.; Tansey, W.P. Targeting WDR5: A WINning Anti-Cancer Strategy? Epigenet. Insights 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Huang, X.; Lai, C.; Li, L.; Li, T.; Huang, P.; Ouyang, S.; Yan, J.; Cheng, S.; Lei, G.; et al. SET domain containing 1B gene is mutated in primary hepatic neuroendocrine tumors. Int. J. Cancer 2019, 145, 2986–2995. [Google Scholar] [CrossRef]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Aguilar, A.; Xu, T.; Zheng, K.; Huang, L.; Stuckey, J.; Chinnaswamy, K.; Bernard, D.; Fernández-Salas, E.; Liu, L.; et al. Design of the First-in-Class, Highly Potent Irreversible Inhibitor Targeting the Menin-MLL Protein-Protein Interaction. Angew. Chem. Int. Ed. Engl. 2018, 57, 1601–1605. [Google Scholar] [CrossRef]
- Huang, J.; Gurung, B.; Wan, B.; Wan, K.; Hua, X.; Lei, M. The same pocket in menin binds both MLL and JunD, but oppositely regulates transcription. Nature 2012, 482, 542–546. [Google Scholar] [CrossRef]
- Eidahl, J.O.; Crowe, B.L.; North, J.A.; McKee, C.J.; Shkriabai, N.; Feng, L.; Plumb, M.; Graham, R.L.; Gorelick, R.J.; Hess, S.; et al. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res. 2013, 41, 3924–3936. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Li, Q.; Wong, S.H.K.; Huang, M.; Klein, B.J.; Shen, J.; Ikenouye, L.; Onishi, M.; Schneidawind, D.; Buechele, C.; et al. ASH1L Links Histone H3 Lysine 36 Dimethylation to MLL Leukemia. Cancer Discov. 2016, 6, 770–783. [Google Scholar] [CrossRef] [Green Version]
- El Ashkar, S.; Schwaller, J.; Pieters, T.; Goossens, S.; Demeulemeester, J.; Christ, F.; van Belle, S.; Juge, S.; Boeckx, N.; Engelman, A.; et al. LEDGF/p75 is dispensable for hematopoiesis but essential for MLL-rearranged leukemogenesis. Blood 2018, 131, 95–107. [Google Scholar] [CrossRef] [Green Version]
- Milne, T.A. LEDGF: A leukemia-specific target. Blood 2018, 131, 4–5. [Google Scholar] [CrossRef]
- Cermáková, K.; Tesina, P.; Demeulemeester, J.; El Ashkar, S.; Méreau, H.; Schwaller, J.; Rezáčová, P.; Veverka, V.; de Rijck, J. Validation and structural characterization of the LEDGF/p75-MLL interface as a new target for the treatment of MLL-dependent leukemia. Cancer Res. 2014, 74, 5139–5151. [Google Scholar] [CrossRef] [Green Version]
- Mohan, M.; Herz, H.-M.; Takahashi, Y.-H.; Lin, C.; Lai, K.C.; Zhang, Y.; Washburn, M.P.; Florens, L.; Shilatifard, A. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom). Genes Dev. 2010, 24, 574–589. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Jiang, Q.; Lemieux, M.; Jeannotte, L.; Su, L.; Zhang, Y. Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L. Nat. Cell Biol. 2006, 8, 1017–1024. [Google Scholar] [CrossRef] [Green Version]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, A.J.; Deshpande, A.; Sinha, A.U.; Chen, L.; Chang, J.; Cihan, A.; Fazio, M.; Chen, C.-W.; Zhu, N.; Koche, R.; et al. AF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypes. Cancer Cell 2014, 26, 896–908. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Deng, L.; Chen, F.; Yao, Y.; Wu, B.; Wei, L.; Mo, Q.; Song, Y. Inhibition of histone H3K79 methylation selectively inhibits proliferation, self-renewal and metastatic potential of breast cancer. Oncotarget 2014, 5, 10665–10677. [Google Scholar] [CrossRef] [Green Version]
- Junwei, S.; Vakoc, C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef]
- Li, Y.; Wen, H.; Xi, Y.; Tanaka, K.; Wang, H.; Peng, D.; Ren, Y.; Jin, Q.; Dent, S.Y.R.; Li, W.; et al. AF9 YEATS Domain Links Histone Acetylation to DOT1L-Mediated H3K79 Methylation. Cell 2014, 159, 558–571. [Google Scholar] [CrossRef] [Green Version]
- Erb, M.A.; Scott, T.G.; Li, B.E.; Xie, H.; Paulk, J.; Seo, H.-S.; Souza, A.; Roberts, J.M.; Dastjerdi, S.; Buckley, D.L.; et al. Transcription control by the ENL YEATS domain in acute leukaemia. Nature 2017, 543, 270–274. [Google Scholar] [CrossRef] [Green Version]
- Wan, L.; Wen, H.; Li, Y.; Lyu, J.; Xi, Y.; Hoshii, T.; Joseph, J.K.; Wang, X.; Loh, Y.-H.E.; Erb, M.A.; et al. ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 2017, 543, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Christott, T.; Bennett, J.; Coxon, C.; Monteiro, O.; Giroud, C.; Beke, V.; Felce, S.L.; Gamble, V.; Gileadi, C.; Poda, G.; et al. Discovery of a Selective Inhibitor for the YEATS Domains of ENL/AF9. SLAS Discov. Adv. Life Sci. Drug Discov. 2019, 24, 133–141. [Google Scholar] [CrossRef]
- Li, X.; Li, X.-M.; Jiang, Y.; Liu, Z.; Cui, Y.; Fung, K.Y.; van der Beelen, S.H.E.; Tian, G.; Wan, L.; Shi, X.; et al. Structure-guided development of YEATS domain inhibitors by targeting π-π-π stacking. Nat. Chem. Biol. 2018, 14, 1140–1149. [Google Scholar] [CrossRef]
- Gilan, O.; Lam, E.Y.N.; Becher, I.; Lugo, D.; Cannizzaro, E.; Joberty, G.; Ward, A.; Wiese, M.; Fong, C.Y.; Ftouni, S.; et al. Functional interdependence of BRD4 and DOT1L in MLL leukemia. Nat. Struct. Mol. Biol. 2016, 23, 673–681. [Google Scholar] [CrossRef]
- Grembecka, J.; He, S.; Shi, A.; Purohit, T.; Muntean, A.G.; Sorenson, R.J.; Showalter, H.D.; Murai, M.J.; Belcher, A.M.; Hartley, T.; et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat. Chem. Biol. 2012, 8, 277–284. [Google Scholar] [CrossRef]
- Shi, A.; Murai, M.J.; He, S.; Lund, G.; Hartley, T.; Purohit, T.; Reddy, G.; Chruszcz, M.; Grembecka, J.; Cierpicki, T. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood 2012, 120, 4461–4469. [Google Scholar] [CrossRef] [PubMed]
- Dafflon, C.; Craig, V.J.; Méreau, H.; Gräsel, J.; Schacher Engstler, B.; Hoffman, G.; Nigsch, F.; Gaulis, S.; Barys, L.; Ito, M.; et al. Complementary activities of DOT1L and Menin inhibitors in MLL-rearranged leukemia. Leukemia 2017, 31, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Fedorov, O.; Thanasopoulou, A.; Leonards, K.; Jones, K.; Meier, J.; Olzscha, H.; Monteiro, O.; Martin, S.; Philpott, M.; et al. Generation of a Selective Small Molecule Inhibitor of the CBP/p300 Bromodomain for Leukemia Therapy. Cancer Res. 2015, 75, 5106–5119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guenther, M.G.; Lawton, L.N.; Rozovskaia, T.; Frampton, G.M.; Levine, S.S.; Volkert, T.L.; Croce, C.M.; Nakamura, T.; Canaani, E.; Young, R.A. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev. 2008, 22, 3403–3408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, A.B.; Nixon, A.M.L.; Moffat, J. The Mixed Lineage Leukemia (MLL) Fusion–Associated Gene AF4 Promotes CD133 Transcription. Cancer Res. 2012, 72, 1929–1934. [Google Scholar] [CrossRef] [Green Version]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [Green Version]
- Sanjuan-Pla, A.; Bueno, C.; Prieto, C.; Acha, P.; Stam, R.W.; Marschalek, R.; Menéndez, P. Revisiting the biology of infant t(4;11)/MLL-AF4+ B-cell acute lymphoblastic leukemia. Blood 2015, 126, 2676–2685. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Hu, Y.; Jin, Z.; Zhai, Y.; Tan, Y.; Sun, Y.; Zhu, S.; Zhao, C.; Chen, B.; Zhu, J.; et al. TanCAR T cells targeting CD19 and CD133 efficiently eliminate MLL leukemic cells. Leukemia 2018, 32, 2012–2016. [Google Scholar] [CrossRef]
- Bueno, C.; Velasco-Hernandez, T.; Gutiérrez-Agüera, F.; Zanetti, S.R.; Baroni, M.L.; Sánchez-Martínez, D.; Molina, O.; Closa, A.; Agraz-Doblás, A.; Marín, P.; et al. CD133-directed CAR T-cells for MLL leukemia: On-target, off-tumor myeloablative toxicity. Leukemia 2019, 33, 2090–2125. [Google Scholar] [CrossRef]
- Moritz, L.E.; Trievel, R.C. Structure, mechanism, and regulation of polycomb-repressive complex 2. J. Biol. Chem. 2018, 293, 13805–13814. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef]
- Okada-Iwabu, M.; Yamauchi, T.; Iwabu, M.; Honma, T.; Hamagami, K.; Matsuda, K.; Yamaguchi, M.; Tanabe, H.; Kimura-Someya, T.; Shirouzu, M.; et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature 2013, 503, 493–499. [Google Scholar] [CrossRef]
- Akimoto, M.; Maruyama, R.; Kawabata, Y.; Tajima, Y.; Takenaga, K. Antidiabetic adiponectin receptor agonist AdipoRon suppresses tumour growth of pancreatic cancer by inducing RIPK1/ERK-dependent necroptosis. Cell Death Dis. 2018, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H. The complex activities of the SET1/MLL complex core subunits in development and disease. Biochim. Et Biophys. Acta (BBA) Gene Regul. Mech. 2020, 1863, 194560. [Google Scholar] [CrossRef]
- Li, Y.; Han, J.; Zhang, Y.; Cao, F.; Liu, Z.; Li, S.; Wu, J.; Hu, C.; Wang, Y.; Shuai, J.; et al. Structural basis for activity regulation of MLL family methyltransferases. Nature 2016, 530, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Schapira, M.; Tyers, M.; Torrent, M.; Arrowsmith, C.H. WD40 repeat domain proteins: A novel target class? Nat. Rev. Drug Discov. 2017, 16, 773–786. [Google Scholar] [CrossRef]
- Shah, K.K.; Whitaker, R.H.; Busby, T.; Hu, J.; Shi, B.; Wang, Z.; Zang, C.; Placzek, W.J.; Jiang, H. Specific inhibition of DPY30 activity by ASH2L-derived peptides suppresses blood cancer cell growth. Exp. Cell Res. 2019, 382, 111485. [Google Scholar] [CrossRef]
- Yu, W.; Chory, E.J.; Wernimont, A.K.; Tempel, W.; Scopton, A.; Federation, A.; Marineau, J.J.; Qi, J.; Barsyte-Lovejoy, D.; Yi, J.; et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 2012, 3, 1288. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Singer, J.W.; Fleischman, A.; Al-Fayoumi, S.; Mascarenhas, J.O.; Yu, Q.; Agarwal, A. Inhibition of interleukin-1 receptor-associated kinase 1 (IRAK1) as a therapeutic strategy. Oncotarget 2018, 9, 33416–33439. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KMT2A or KMT2B Complex | KMT2C or KMT2D Complex | KMT2F or KMT2G Complex | |
|---|---|---|---|
| Enzyme | KMT2A or KMT2B | KMT2C or KMT2D | KMT2F or KMT2G |
| Core subunits | ASH2L RBBP5 WDR5 DPY30 | ASH2L RBBP5 WDR5 DPY30 | ASH2L RBBP5 WDR5 DPY30 |
| Unique subunits | Menin HCF1 or HCF2 | PTIP PA1 NCOA6 UTX | CFP1 WDR82 HCF1 |
| Mode of Action | Name of Inhibitor | Cellular Outcome | Targeted Cancer Cells | References |
|---|---|---|---|---|
| Targeting core subunits of COMPASS: | MM-401 (microcyclic peptidomimetic) | myeloid differentiation/phenocopying KMT2A deletion | MLL-r leukemia cells in culture | [245,247] |
| Antagonizing the interaction of WDR5 and KMT2A | OICR-9429 (small-molecule antagonist) | Inhibition of proliferation and induction of differentiation | Patient-derived AML cells expressing p30 | [211] |
| Inhibition of cancer cell growth | Various tumor cells with a TP53 gain-of function (GOF) mutation | [154] | ||
| Targeting DPY30 | Cell penetrating peptides (CPPs) derived from ASH2L | Inhibition of cancer cell growth | MLL-r leukemia cells/other MYC-dependent hematologic cancers | [287] |
| Blocking interaction of Menin with KMT2A | MI-463, MI-503 (small-molecule antagonist) | Inhibition of progression of MLL leukemia in vivo vs. normal hematopoiesis | MLL-r leukemia cells/mouse model of MLL leukemia | [249] |
| M-525 (small-molecule antagonist) | Suppression of MLL-regulated gene expression, leukemia cell growth inhibition | Various cell lines derived from MLL-r leukemia (MV4, MOLM-13, MOLM-14) | [250] | |
| MI-2-2 (small-molecule inhibitor) | Inhibition of cell proliferation, downregulation of differentiation | MLL leukemia cells (KMT2A-F4 translocation) | [270,271] | |
| Blocking interaction of KMT2A with LEDGF | CP65 (cyclic peptide) | Impairment of clonogenic growth of primary murine MLL-AF9-expressing leukemic blasts | MLL-AF9 leukemia cells | [256] |
| Targeting DOT1L in KMT2A-rearranged complexes | EPZ004777 (S-adenosylmethionine-competitive inhibitor) | Downregulation of leukemic genes, inhibition of H3K79, inhibition of proliferation | Leukemia cells bearing MLL-r/extension of survival in a mouse MLL xenograft model or complete tumor regression | [237,288] |
| EPZ-5676 (pinometostat) | [259] | |||
| Dissociation of interacting proteins from MYC regulatory elements | I-BET (via BRD4) | Downregulation of MYC-regulated gene expression, inhibition of proliferation | Hematological cancers (MLL-r leukemia) | [242,262,289] |
| A-485 (via the catalytic core of CBP/300) | Inhibition of proliferation | Lineage specific tumor cells (hematological and prostate) | [263] | |
| Inhibitors of YEATS domain of AF9 and ENL (XL-13m) | Downregulation of leukemic gene drivers | MLL-r leukemia cells | [267,268] | |
| Stabilization of wt KMT2A | IRAK1/4 | Inhibition of cancer cells proliferation in vitro/in vivo | MLL-r leukemia cells | [240,290] |
| Restoration of normal gene expression in KMT2C mutant cells | GSK 126 (via a subunit of the polycomb repressive complex 2) | Impairment of cell proliferation, resetting the epigenetic balance of polycomb and compass function | Cells/tumors bearing mutations (PHD domain) in KMT2C | [174] |
| PARP1/2-depdenent DNA repair | Olaparib (PARP1/2 inhibitor) | Synthetic lethality of cancer cells | Cancer cells with low KMT2C levels (bladder cancer) | [195] |
| Pharmacologically contained glycolysis | Glycolytic inhibitors (2-deoxy-D-glucose (2-DG) | Impediment of tumorigenic growth | Lung cancer cells with KMT2D mutations | [187] |
| Suppression of ADIPOR cytoplasmic signaling | AdipoRon (ADIPOR agonist) | Induction of cancer cell death through necroptosis | MIA PaCa-2 tumor cells/cancer cells isolated from patients with pancreatic cancer/TNBC cells | [206,282,283] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poreba, E.; Lesniewicz, K.; Durzynska, J. Aberrant Activity of Histone–Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis. Int. J. Mol. Sci. 2020, 21, 9340. https://doi.org/10.3390/ijms21249340
Poreba E, Lesniewicz K, Durzynska J. Aberrant Activity of Histone–Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis. International Journal of Molecular Sciences. 2020; 21(24):9340. https://doi.org/10.3390/ijms21249340
Chicago/Turabian StylePoreba, Elzbieta, Krzysztof Lesniewicz, and Julia Durzynska. 2020. "Aberrant Activity of Histone–Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis" International Journal of Molecular Sciences 21, no. 24: 9340. https://doi.org/10.3390/ijms21249340
APA StylePoreba, E., Lesniewicz, K., & Durzynska, J. (2020). Aberrant Activity of Histone–Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis. International Journal of Molecular Sciences, 21(24), 9340. https://doi.org/10.3390/ijms21249340