Abstract
Pituitary Gonadotropin-Releasing Hormone receptors (GnRH-R) mediate the activity of the hypothalamic decapeptide GnRH, thus playing a key role in the regulation of the reproductive axis. Early-stage prostate cancer (PCa) is dependent on serum androgen levels, and androgen-deprivation therapy (ADT), based on GnRH agonists and antagonists, represents the standard therapeutic approach for PCa patients. Unfortunately, the tumor often progresses towards the more aggressive castration-resistant prostate cancer (CRPC) stage. GnRH receptors are also expressed in CRPC tissues, where their binding to both GnRH agonists and antagonists is associated with significant antiproliferative/proapoptotic, antimetastatic and antiangiogenic effects, mediated by the Gαi/cAMP signaling cascade. GnRH agonists and antagonists are now considered as an effective therapeutic strategy for CRPC patients with many clinical trials demonstrating that the combined use of these drugs with standard therapies (i.e., docetaxel, enzalutamide, abiraterone) significantly improves disease-free survival. In this context, GnRH-based bioconjugates (cytotoxic drugs covalently linked to a GnRH-based decapeptide) have been recently developed. The rationale of this treatment is that the GnRH peptide selectively binds to its receptors, delivering the cytotoxic drug to CRPC cells while sparing nontumor cells. Some of these compounds have already entered clinical trials.
1. Introduction
Gonadotropin-releasing hormone (GnRH) is the hypothalamic decapeptide (pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) known to play a central role in the control of the hypothalamic-pituitary-axis in mammals [1,2,3,4]. It is produced by a small number of hypothalamic neurons and released in a pulsatile way into the hypophyseal circulation to reach the gonadotropes in the anterior pituitary where it binds to its specific receptors (GnRH-R). By binding to these receptors, GnRH triggers the synthesis and release of the two gonadotropoins LH (lutinizing hormone) and FSH (follicle stimulating hormone), thus stimulating gonadal sex steroid hormone production and gamete maturation in both sexes [5,6,7,8].
The pituitary GnRH-R is a protein (328 amino acids) belonging to the GPCR (rhodopsin-like G protein coupled receptor) family. It is encoded by a gene on chromosome 4q13.2 and consists of three exons interrupted by two introns (Table 1). The protein is structurally characterized by a core formed by seven transmembrane domains, an extracellular N-terminal domain (35 amino acids) and a typically short (1–2 amino acids) intracellular C-terminal domain [9,10,11,12]. This feature of GnRH-R has been linked to its slow internalization and desensitization upon hormone stimulation [13,14].

Table 1.
Human Gonadotropin-releasing hormone (GnRH) receptors.
The intracellular signaling pathway triggered by GnRH-R activation at the pituitary level has been widely investigated and is now well characterized. Binding of the decapeptide to its receptors leads to the activation of the Gαq/11 subunit of a heterotrimeric G protein complex, stimulating, in turn, its direct effector phospholipase Cβ (PLCβ). PLCβ catalyzes the formation of diacylglycerol (DAG) and inositol triphosphate (IP3), leading to protein kinase C (PKC) activation and increased cytoplasmic levels of Ca2+ (due to increased ion influx from the extracellular environment and release from intracellular stores), respectively. Interestingly, different PKC isoforms were shown to be involved in these intracellular mechanisms. Activation of these PKC triggers their downstream signaling pathway involving proteins belonging to the MAPK (mitogen-activated protein kinase) cascade. In addition, elevated intracellular Ca2+ levels were also shown to be involved in the MAPK cascade activation [7,11,12,14,15,16,17,18,19,20,21]. In particular, Naor and coworkers recently reported that the PKC isoforms PKCα, PKCβII, PKCδ, PKCε, PKCθ and atypical PKC-ι/λ play differential roles in the ERK1/2, JNK1/2 and p38MAPK phosphorylation in a ligand and cell context-dependent manner. According to these authors, this may be related to “the persistent vs. transient redistribution of the different PKCs into the cell or the redistribution of a specific PKC from the perinuclear zone vs. the plasma membrane” [22,23]. GnRH-R-activated MAPKs then trigger the expression of gonadotropins as well as of GnRH-R genes.
This molecular cascade of events triggered by pituitary GnRH-R activation mediates the key regulatory role of GnRH in the control of the pituitary-gonadal axis functions.
It is now accepted that, in addition to the classical hypothalamic GnRH, other forms of the peptide are present in most vertebrates. In particular, the GnRH-II isoform is a decapeptide conserving the amino acids of GnRH in both the N-terminal (Glp-His-Trp-Ser) and the C-terminal (Pro-Gly-NH2) domains, suggesting that it might bind and activate the classical form of GnRH-R. On the other hand, the GnRH-II amino acid sequence differs from that of GnRH in positions 5, 7 and 8 (His5, Trp7, Tyr8), known to be involved in the biological functions of GnRH [4,12,24,25,26]. These observations stimulated the search for a specific receptor for GnRH-II (GnRH-II-R). However, so far, a full-length GnRH-II-R has been cloned in nonhuman primates [27,28]. In humans, Neill et al. proposed that the GnRH-II-R might be a five transmembrane domain receptor, lacking the transmembrane regions 1 and 2 [29]. Morgan and coworkers suggested the presence, in human tissues, of a nonfunctional form of this receptor, located on chromosome 1q12 and composed of three exons and two introns, as a consequence of a frameshift in exon 1 and a stop codon in exon 2 [30] (Table 1). Another splice variant of GnRH-II-R was reported in human sperm and suggested to have a functional role in male gametogenesis [31]. Thus, the existence of a functional seven transmembrane domain GnRH-II-R in human tissues is still a matter of debate.
A GnRH-III decapeptide was found in sea lamprey (Petromyzon marinus) with an amino acid sequence that differs from that of GnRH in positions 5–8 (His5, Asp6, Trp7, Lys8) [32]. This peptide was reported to be endowed with a very low LH and FSH-stimulating activity in rats [33].
It is now well established that GnRH-R are also expressed in extrapituitary tissues and in several tumor tissues, both related (prostate, breast, ovarian, endometrial tumors) and unrelated (melanoma, glioblastoma, pancreatic, colon, lung, adrenocortical head and neck tumors) to the reproductive system. In cancer cells, this receptor is associated with antiproliferative/proapoptotic effects [4,34,35,36,37,38,39,40,41,42,43,44,45,46].
Interestingly, the GnRH decapeptide was also shown to be expressed in these tumors, demonstrating the existence of an autocrine GnRH/GnRH receptor loop endowed with antitumor activity and supporting the role of this loop as an effective molecular target for anticancer strategies.
2. Prostate Cancer
Prostate cancer (PCa) still represents a major health burden, being the most aggressive tumor and the second most frequent cause of tumor-related deaths among men in western countries [47,48,49]. In the early stages, most PCas are dependent on androgens for their growth and, therefore, patients are treated with androgen-deprivation therapy (ADT, i.e., chemical castration). This therapeutic approach is mainly based on GnRH agonists, either alone or in combination with a drug targeting the androgen receptor signaling (antiandrogens, inhibitors of androgen synthesis).
GnRH agonists (goserelin, leuprorelin, triptorelin) were synthesized based on the observation that native GnRH is endowed with a short half-life, being enzymatically cleaved in the blood at the level of Gly6. Thus, this amino acid is replaced by a D-amino acid in order to obtain analogs resistant to the peptidase degradation. In these analogs, the first amino acids of GnRH are conserved to maintain the biological activity of the native peptide, while the last amino acid (Gly10-amide) is substituted with the residues Pro-NHEt or Pro-Azgly-NH2 to increase the binding affinity to the GnRH-R [50,51,52,53,54,55]. These compounds act by binding to the pituitary GnRH-R and, after the induction of an initial gonadotropin surge, they induce its desensitization, thus suppressing LH and testosterone secretion [56]. To avoid the risk of the initial flare event, together with metabolic dysfunction and increased risk of cardiovascular pathologies, GnRH antagonists were later synthesized (cetrorelix, abarelix, degarelix, ganirelix, ozarelix). They competitively bind to the GnRH-R immediately suppressing gonadotropin secretion. Moreover, they were shown to suppress FSH (follicle stimulating hormone) more rapidly, to lower levels and to a longer period than GnRH agonists [57,58]. These peptides present the Ac-D-Nal-D-Cpa-D-Pal sequence in their N-terminal domain, different D-amino acid derivatives in position 6 and D-Ala at their C-terminal domain [43,59,60,61]. Very recent data from clinical trials and meta-analyses support an improved progression-free survival, overall survival and side effects (cardiovascular diseases) with GnRH antagonists compared with GnRH agonists [55].
Given the key role of the androgen receptor (AR) signaling in PCa growth and progression, several agents targeting this molecular pathway were developed: (i) antiandrogens directly targeting the AR receptor such as bicalutamide, flutamide and nilutamide (first and second generation antiandrogens), and enzalutamide, apalutamide and darolutamide (third generation antiandrogens); (ii) inhibitors of intratumoral androgen synthesis such as finasteride, orteronel and abiraterone [43,62,63,64,65,66,67,68,69].
Unfortunately, the tumor often progresses towards the castration-resistant stage (castration-resistant prostate cancer, CRPC) characterized by uncontrolled progression in the absence of circulating androgens. Chemotherapy (docetaxel), either alone or in combination with antiandrogens (enzalutamide, apalutamide), or with inhibitors of androgen synthesis (abiraterone), represents the standard therapy for CRPC patients [68,70,71,72,73,74,75]. However, undesired side effects and development of drug resistance occur very frequently in these patients.
More recently, immunotherapies (immune checkpoint inhibitors or chimeric antigen receptor T cell therapies - CAR-T) were introduced as a novel therapeutic approach, and they are currently under investigation [76,77]. In particular, in the last two decades, several types of monoclonal antibodies (mAbs) against immune checkpoints were developed and approved by the FDA for the treatment of PCa. These include anti-CTLA-4 (cytotoxic T-lymphocyte-associated protein-4) (ipilimumab, tremelimumab) and anti-PD-1 (programmed death protein-1) (nivolumab, pembrolizumab, lambrolizumab, avelumab, durvalumab) mAbs. Clinical trials have been already performed in PCa patients treated with checkpoint inhibitors, either alone or in combination with antiandrogens [77,78,79,80,81,82,83,84,85,86,87,88]. Unfortunately, despite the initial success in clinical use of these compounds, the efficacy of treatments was reported to be low and the majority of PCa patients showed resistance to these therapies [87,89,90,91].
Studies leading to a better understanding of the molecular mechanisms and signaling pathways involved in PCa development and progression are needed to improve the chemopreventive/treatment strategies for this pathology.
3. GnRH Receptors in Prostate Cancer
3.1. Molecular Structure
The expression of the GnRH-R in human pituitary, as well as its nucleic acid sequence, was first reported by Kakar and coworkers in 1992 [92]. In the same years, it was becoming increasingly clear that the GnRH-R was expressed not only at the pituitary level but also in extrapituitary sites and in cancer tissues, including prostate cancer [34,37,39,44,93,94]. These receptors were first analyzed in terms of binding affinity for GnRH synthetic analogs, leading to contradictory results. In our laboratory, we demonstrated that one single class of low-affinity GnRH binding sites was present in PCa cells, either androgen-dependent or castration-resistant [95,96]. On the other hand, two classes of GnRH binding sites (one low affinity and one high affinity) were demonstrated in human PCa cells as well as in the Dunning R3327 rat model of PCa [97,98], while a single class of high affinity binding sites was observed in Dunning R3327 rats by Pinski and coworkers [99].
Based on these contrasting observations, further studies were performed to characterize this receptor at the molecular level. We could demonstrate the expression of a GnRH receptor, sharing the same mRNA and protein size with the pituitary receptor, in PCa cells [37,100,101,102]. Similar observations were later reported by Bank et al. in PCa cells [103], in the rat R3327 prostate adenocarcinoma and in human prostate biopsies [104,105,106,107,108]. Interestingly, a lower expression of GnRH-R was reported in normal prostate specimens when compared to PCa biopsies [109].
As reported for different types of tumors, the decapeptide GnRH is also expressed in PCa cells, further supporting the existence of a GnRH/GnRH-R autocrine loop involved in the local control of tumor growth and progression [96,100,101,110].
3.2. Antiproliferative/Proapoptotic Activity
The antitumor activity of GnRH-R in PCa cells is now well established. In our, as well as in others laboratories, activation of GnRH-R by means of GnRH agonists was shown to significantly inhibit the proliferation of human androgen-dependent (LNCaP) and CRPC (DU145, PC3) cells, expressing high levels of the GnRH-R, both in vitro [93,95,96,111,112] and in vivo, when subcutaneously inoculated in nude mice [113,114,115]. In line with these observations, GnRH analogs were also reported to inhibit the growth of the rat androgen-independent Dunning R-3327-AT-1 prostate cancer [99] as well as of primary cell cultures from human prostate carcinomas [116]. Moreover, GnRH agonist-based therapy was reported to be associated with longer survival in hormone-refractory PCa patients expressing the GnRH-R [111]. Interestingly, we demonstrated that the classical form of GnRH-R mediates the anticancer activity of CRPC cells, further supporting that a functional GnRH-II-R is not present in humans and that GnRH-II may act through the classical GnRH-R [117].
The antitumor effects of GnRH-R activation have been suggested to be associated not only with a slowdown of the cell cycle progression (mainly at the G2/M checkpoint) but also with induction of apoptosis. Specifically, GnRH agonists were shown to induce apoptosis in CRPC cells by interfering with the activity of both the PI3K pathway, leading to the stimulation of the downstream JNK kinase, and the p38 MAPK signaling cascade [118,119]. The extrinsic apoptotic pathway involving caspase 8 and 3 and p53 phosphorylation, was also reported to be induced by GnRH agonists in primary cultures of human PCas [116,119,120,121,122]. Interestingly, we could demonstrate that GnRH agonists sensitize, and resensitize, to chemotherapy (i.e., docetaxel) in a p53-dependent manner [123]. However, the involvement of the apoptotic pathways in the antitumor activity of GnRH-R in PCa is still a matter of debate [114,124].
Growth factors (EGF, IGF-I) and their receptors play a pivotal role in the growth and progression of tumors, including PCa [101,125,126,127,128,129,130,131]. To elucidate the molecular mechanisms underlying the anticancer effects of GnRH-R, the possible interference of GnRH analogs with the protumoral activity of growth factors has been investigated in different experimental models of PCa.
In our laboratory, it was demonstrated that activation of GnRH-R, in both androgen-dependent and CRPC cells in vitro and in vivo, interferes with the mitogenic activity of the locally expressed EGF stimulatory loop by decreasing the expression of EGF-R and of its downstream signaling molecules (i.e., the transcription factor c-fos) [113,114,128,132]. Similar observations were reported by Iacopino et al., showing that the GnRH agonist leuprolide significantly counteracts EGF-induced ERK1/2 phosphorylation (i.e., activation) in PCa cells [133]. The GnRH-R/EGF receptor interaction in PCa cells was further confirmed by Wells and coworkers [134]. The phosphorylation of the EGF-R at threonine 654 by the endogenous growth factor plays a central role in the receptor activation. To confirm the interference of GnRH-R activation with the EGF mitogenic activity, these authors overexpressed a mutant form of the EGF-R (threonine 654 to alanine) in DU145 CRPC cells. They demonstrated that the GnRH agonist goserelin counteracts the mitogenic activity of EGF in wild type DU145 cells (as expected), but not in cells overexpressing the mutant (i.e., inactive) form of the receptor [134].
The insulin-like growth factor (IGF) system is also known to be deeply involved in PCa growth and progression. This system is composed of two ligands (IGF-I and IGF-II), two receptors (IGFR-IR and IGFR-IIR) and different binding proteins (IGFBP-1 to -6). Mita and coworkers demonstrated that, in human PCa tissues, the expression of IGF-II and IGFBP-2 significantly correlates with pathologic stage lymph node metastasis, histologic differentiation and serum prostate-specific antigen (PSA) levels after hormone therapy [130]. On the other hand, a recent paper reported a significant correlation between IGF-IR expression in human PCa biopsies and tumor stage, suggesting that it may play a role in PCa progression towards an aggressive phenotype [135]. In our laboratory, we demonstrated that, in DU145 cells, activation of the GnRH-R with the agonist goserelin counteracts the mitogenic action of IGF-I in a dose-dependent manner, prevents the growth factor-induced tyrosine phosphorylation of the IGF-IR and decreases the expression of IGF-IR without affecting its binding affinity for the growth factor [37,136].
GnRH antagonists were first developed for the treatment of hormone-dependent PCa, based on their ability to compete with the binding of endogenous GnRH to its pituitary receptors, with the aim to suppress the activity of the pituitary gonadal axis without triggering the initial undesired gonadotropin surge. It was then expected that these compounds might also suppress the activity of the GnRH-R expressed in tumor tissues. Surprisingly, it was found that, in cancer cells expressing the receptor, these compounds exert a significant antiproliferative activity, supporting that they act as agonists on these cells [36,38,39,45,59,137,138]. Specifically, activation of GnRH-R by GnRH antagonists was also reported to suppress the proliferation, and to trigger apoptosis, by interfering with the growth factor receptor intracellular pathways in PCa cells [37,39,41,42,43,93,99,116,139,140,141,142,143]. More recently, Cucchiara and coworkers demonstrated that the GnRH antagonist degarelix significantly reduces the proliferation of C4-2-MDVR (castration- and enzalutamide-resistant) PCa cells expressing the androgen receptor variant AR-V7. Interestingly, degarelix also decreases the expression of the androgen receptor as well as of androgen steroidogenesis pathways in tumor tissues from PCa cell xenografts grown in severe combined immunodeficient (SCID) mice [144].
The observation that, in tumors cells, GnRH-R can be activated not only by GnRH agonists (as expected) but also by GnRH antagonists, pointed out that these receptors might be characterized by specific structural properties according to the cell context in which they are expressed. In particular, Millar et al. proposed the ligand-induced selective signaling concept. This concept foresees that, in different tissues (i.e., pituitary vs. cancer cells), the GnRH-R may adopt different structural conformations associated with selective binding of GnRH analogs and specific intracellular signaling pathways [137,145].
As mentioned above, in addition to the classical GnRH, a structural isoform of the peptide (i.e., GnRH-II) has been discovered in most vertebrates. In particular, GnRH-II is known to be expressed in different human tissues, normal and malignant, including PCa [37,39,146]. This peptide was shown to exert a significant antiproliferative and proapoptotic effect on PCa cells, both androgen-dependent and castration-resistant [117,119,120,146]. It is now generally accepted that the classical form of the GnRH-R mediates the antitumor activity of this peptide, confirming the notion that a functional GnRH-II receptor is lacking in human tissues, although this issue still remains a matter of debate [45,120,147].
More recently, based on docking experiments, a novel ligand and activator of the GnRH-R, GV1001, structurally unrelated to the GnRH decapeptide, has been identified. This peptide is a 16-amino acid fragment of hTERT, the human telomerase reverse transcriptase catalytic subunit. GV1001 was reported to bind and activate GnRH-R, thus triggering an antiproliferative and proapoptotic effect in PCa cells, both in vitro and in vivo, when inoculated in nude mice [148].
Taken together, these observations strongly support that locally expressed GnRH-R mediate the antitumor activity of GnRH decapeptide isoforms, as well as of novel peptides endowed with anticancer effects, in PCa cells.
3.3. Antimetastatic Activity
Locally expressed GnRH-R are also involved in the control of the metastatic behavior of PCa cells. In our laboratory, we demonstrated that GnRH agonists significantly reduce the migratory behavior of CRPC cells towards the extracellular matrix protein vitronectin (by haptotactic assays) as well as their ability to invade a reconstituted basement membrane [149]. We also found that, in these cells, GnRH agonists interfere with the expression and activation (i.e., tyrosine-phosphorylation) of the IGF-IR; counteract the IGF-I-induced phosphorylation of AKT (a kinase known to be involved in the prometastatic activity of the growth factor); abrogate the migratory and invasive behavior triggered by IGF-I; interfere with the effects of the growth factor on actin cytoskeleton organization, expression and cellular localization of integrins (i.e., αvβ3), and cell morphology [149].
In line with these observations, GnRH-R activation was reported to block the invasive behavior of CRPC cells induced by the fibroblast growth factor (FGF) [111]. This antimetastatic activity was suggested to be mediated by the inhibition of the plasminogen activator system by decreasing the enzymatic activity and the secretion of uPA (urokinase-type plasminogen activator) while increasing the expression levels of the protein PAI-1 (plasminogen activator inhibitor type-1) [150]. Enomoto and coworkers showed that both GnRH-I and GnRH-II peptides induce actin cytoskeleton remodeling, and decrease cell migration, through the activation of the classical form of the GnRH receptor [151]. Moreover, the effects of a GnRH agonist (leuprorelin) were also investigated on the expression levels of molecules involved in migration, invasion and cell-cell adhesion in both androgen-dependent (LNCaP) and castration-resistant (PC3) PCa cells. It was found that, in LNCaP cells, the GnRH agonists upregulate the expression of E-cadherin, β- and γ-catenin. On the other hand, the expression of these molecules was not affected in PC3 cells [152].
Interestingly, in addition to their antimetastatic/antiinvasive activity, GnRH-R were shown to be also endowed with antiangiogenic properties. Actually, we could demonstrate that GnRH receptors are also expressed on human umbilical vein endothelial cells (HUVECs), and that GnRH agonists reduce their proliferation and ability to form capillary-like tubes when stimulated by vascular endothelial growth factor (VEGF). These findings suggest that activation of GnRH-R triggers an antiangiogenic effect by counteracting the proangiogenic activity of the growth factor directly at the level of endothelial cells [153].
A potent antimetastatic activity was also reported by treating PCa cells with GnRH antagonists, further supporting the notion that these compounds behave as agonists at the level of the GnRH-R expressed in cancer cells. Treatment of CRPC cells with the GnRH-R antagonist cetrorelix reduces the invasiveness of DU145 cells overexpressing the full-length EGF-R, while increasing the expression levels of cell-cell adhesion molecules such as E-cadherin, α- and β-catenin and p120 [154]. In CRPC cells, Dondi et al. reported that GnRH antagonists are endowed with antimetastatic activities that are similar to those exerted by agonists in CRPC cells [150]. The expression of proteins involved in the cell-cell adhesion and angiogenic processes was also found to be affected by the GnRH antagonist degarelix in BPH (benign prostatic hyperplasia)-1 cells [143].
3.4. Intracellular Signaling Pathways
GnRH analog activities at the pituitary level (activation of the pituitary-gonadal axis) significantly differ from those at the cancer cell level (inhibition of cancer cell growth and metastatic behavior) suggesting that, according to the cell context, GnRH-R can be coupled to different G proteins and, therefore, to specific intracellular signaling pathways and molecular transducers [14,15,16,20,37,42,44,45,137,140,155,156].
As discussed above, GnRH-R activation in pituitary gonadotrope cells stimulates gonadotropin synthesis and release through the Gαq/11/PLC/PKC/MAPK signaling pathway. On the other hand, in cancer cells, and specifically in PCa cells, the GnRH-R has been found to be mainly coupled to a Gαi protein. Activation of this G protein counteracts cAMP accumulation thus triggering antitumor effects [16,20,37,45,102,157,158].
In our laboratory, we demonstrated that in both androgen-dependent (LNCaP) and castration-resistant (DU145) PCa cell lines, pertussis toxin completely abrogates the antiproliferative action of GnRH agonists. These compounds substantially antagonize the pertussis toxin-catalyzed ADP-ribosylation of a Gαi protein. GnRH analogs significantly counteract the forskolin-induced increase of intracellular cAMP levels [102].
Through the activation of the Gαi/cAMP pathway, GnRH analogs were reported to trigger the activity of a phosphotyrosine phosphatase, thus resulting in a decreased phosphorylation of the EGF-R (i.e., inactivation) [159]. In line with this observation, we could show that GnRH agonists significantly counteract the EGF and IGF-I-induced tyrosine phosphorylation of the EGF-R and IGF-I-R respectively, thus interfering with the mitogenic activity of the growth factors in PCa cells [132,149,160].
The GnRH-R-linked Gαi/cAMP pathway was also reported to mediate the effects of GnRH analogs on the MAPK (p38MAPK, ERK, JNK) signaling cascades known to play a pivotal role in cell growth, proliferation and apoptosis [161,162]. Specifically, the p38MAPK pathway was found to mediate the proapoptotic activity of GnRH analogs in BPH-1 cells while the ERK kinase was shown to be activated by GnRH agonists in immortalized human prostate cells engineered to overexpress the GnRH-R. JNK activation was also demonstrated to be involved in the proapoptotic effects of GnRH analogs in CRPC cells [38,42,44,118,119,122,158]. The activation of JNK was suggested to be mediated by inhibition of mixed-lineage kinase 3 (MLK3), the upstream activator of JNK [44,118].
Finally, GnRH analogs were shown to interfere with the activity of the PI3K/AKT intracellular pathway to suppress the proliferative and metastatic features of CRPC [149].
In addition to the Gαi/cAMP pathway, the Gαq/11/PLC/PKC signaling cascade also seems to be involved in the antitumor activity of the GnRH-R in PCa cells [14,118]. Sviridonov and coworkers reported that the PKCα, PKCβII and PKCε kinases are activated by GnRH in PCa cells in a more prolonged way than in gonadotrope cells. This is followed by ERK1/2, p38MAPK and JNK activation (mediated by reduced AKT phosphorylation) and redistribution from the cytosol and Golgi to the plasma membrane. These authors suggest that both a more sustained vs. transient expression, and a different distribution of PKCs in PCa cells vs. gonadotropes, are responsible for the different biological effects elicited by GnRH in these cell types [20,163].
Thus, although the Gαi/cAMP pathway remains the main intracellular signaling cascade coupled to the GnRH-R, additional pathways (i.e., Gαq/11/PLC/PKC) are now accepted to be involved in the antitumor activity of this receptor in PCa cells.
The main signaling pathways activated in GnRH agonist/antagonist-treated PCa cells are summarized in Figure 1.
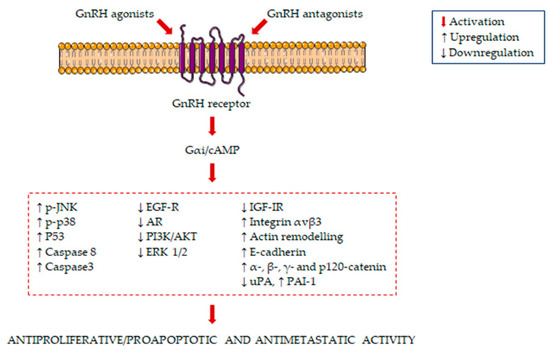
Figure 1.
Molecular mechanisms underlying the antiproliferative/proapoptotic and antimetastatic activity of gonadotropin-releasing hormone receptors (GnRH) agonists and antagonists in prostate cancer (PCa) cells. GnRH agonists and antagonists bind to the GnRH receptor in PCa cells, leading to the activation of Gαi/cAMP. This is followed by the induction of several antiproliferative/proapoptotic and antimetastatic pathways.
4. Emerging Prospective Aspects for New Therapeutic Interventions
4.1. GnRH Agonists and Antagonists
The expression of GnRH-R in PCa cells, specifically CRPC cells, together with their antitumor activity, sustained the hypothesis that they might represent an additional and direct molecular target of GnRH analogs (both agonists and antagonists) in CRPC [43]. Based on this notion, it has been proposed that discontinuation of ADT in CRPC patients could result in a worse outcome of tumor progression. Combination therapies based on ADT treatments together with chemotherapy, enzalutamide (antiandrogen) or abiraterone (inhibitor of androgen synthesis), are highly recommended [55]. Different clinical trials confirm this hypothesis.
It was reported that in CRPC patients receiving a GnRH agonist-based treatment, a high expression of GnRH-R was associated with a better disease-specific survival [111]. A retrospective study by Lawrentschuk et al. reviewed the records of PCa patients who were treated with a GnRH agonist (leuprorelin or goserelin) but underwent disease progression and were then rechallenged with the other GnRH agonist (goserelin or leuprorelin). These authors reported a significant decrease of PSA levels in patients undergoing this GnRH agonist-based switching therapy [164]. Interestingly, Teply and coworkers performed a clinical trial in which PCa patients progressing after an antiandrogen (enzalutamide) treatment were challenged with the bipolar androgen therapy (BAT, low testosterone levels together with a GnRH agonist). The results obtained showed positive clinical responses (as evaluated in terms of PSA levels and radiographic progression-free survival) and a subsequent resensitization to the antiandrogen [165]. In chemotherapy-naive patients with metastatic CRPC, the combined use of a GnRH agonist with docetaxel was shown to improve the median radiographic progression-free survival with respect to the chemotherapeutic drug given alone (nine vs. six months, respectively) [166]. In line with these data, patients who develop CRPC very often continue on a GnRH agonist-based androgen deprivation therapy when starting treatment with chemotherapy [167,168,169].
However, it should be underlined that contrasting results were reported by other clinical trials. For instance, in a large clinical trial (ICELAND), patients with locally advanced or relapsing PCa were treated with a GnRH agonist (leuprorelin), either alone or in combination with the antiandrogen bicalutamide. It was reported that in these patients, continuous androgen deprivation did not improve PSA progression [170].
As discussed in this review, GnRH antagonists bind to locally expressed GnRH-R in CRPC cells triggering the same antitumor effects elicited by GnRH agonists. Moreover, they induce a faster suppression of testosterone levels and are devoid of the undesirable initial testosterone surge. Interestingly, these compounds also induce a more significant and long-lasting reduction of FSH levels when compared with GnRH agonists and were suggested to interfere with the binding of the gonadotropin to its receptors in prostate cancer cells [171]. To this purpose, it must be underlined that both FSH and FSH receptors are expressed in PCa cells and tissues, suggesting their involvement in PCa development [172,173]. Moreover, hifh serum levels of FSU were reported to correlate with metabolic, cardiovascular, skeletal and cognitive effects [174]. Interestingly, in PCa patients, GnRH antagonists were recently reported to induce more significant suppressions of both FSH and PSA levels than those induced by GnRH agonists [175].
In a very recent paper, Abufaraj and coworkers reported that in PCa patients, GnRH antagonist treatments are associated with lower mortality rates and cardiovascular events as compared with GnRH agonists, while there are no differences in musculoskeletal events and fatigue. On the other hand, adverse reactions at the injection site are characteristic features of GnRH antagonists [176]. A lower toxicity of GnRH antagonists vs. agonists has been in additional articles [55,177,178,179,180].
Taken together, these observations paved the way for clinical studies addressing the anticancer potential of GnRH antagonists (i.e., degarelix, at present considered the most efficient antagonist for PCa due to its low histamine-releasing activity) in CRPC patients [181].
In a previous paper, the effects of degarelix were evaluated on PSA levels in patients experiencing a progression towards the CRPC stage after treatment with a GnRH agonist. Unfortunately, only a small number of cases was found to respond to this treatment [182]. On the other hand, a more recent paper reported that in a CRPC case, switching from a GnRH agonist to the antagonist degarelix is associated with a longer control of tumor progression [183]. Similar observations were reported by Atchia and coworkers in a systematic review and meta-analysis. By analyzing the data from thirteen clinical studies, these authors concluded that treatment with degarelix after failure of a GnRH agonist in patients progressing towards the CRPC stage, resulted in decreased or stable PSA levels in patients [184]. Thus, treatment with GnRH antagonists might be considered for patients with disease progression after GnRH agonist therapy. Interestingly, according to Uemura and coworkers, GnRH agonist/antagonist combination treatments represent the mainstay therapeutic approach in CRPC patients in Japan [185].
Clinical trials investigating the efficacy of GnRH analogs, either alone or in combination with standard strategies, in CRPC patients are at present ongoing (see https://www.clinicaltrials.gov/).
4.2. Cytotoxic GnRH-Based Bioconjugates
GnRH receptors expressed in cancer cells, and associated with antitumor activity, are now considered as an interesting molecular target for a new targeted therapeutic approach based on cytotoxic GnRH bioconjugates, compounds in which a GnRH derivative peptide is covalently linked to a cytotoxic drug. The rationale of this targeted therapeutic approach is that the GnRH peptide acts as a targeting moiety to specifically deliver the cytotoxic drug to cancer cells while sparing normal cells that do not express the GnRH receptor. Thus, GnRH binds to its receptor on tumor cell membranes, the bioconjugate is internalized by endocytosis and the cytotoxic drug is then released to enter the nucleus to exert its anticancer activity.
The bioconjugate AEZS-108 (also known as AN-152) consists of doxorubicin (Dox) coupled to the GnRH derivative via an ester bond. It was widely reported to exert a significant antitumor (antiproliferative, antimetastatic and antiangiogenic) activity in different types of cancer [60,186,187,188,189]. Specifically, this bioconjugate was shown to significantly decrease the proliferation and to induce apoptosis in both androgen-sensitive and CRPC cells in vitro and in vivo [187,190,191,192]. After internalization mediated by endocytosis, the ester bond of this compound is cleaved by carboxyilesterases releasing the free drug that, in turn, can accumulate in the nucleus to exert its cytotoxic activity [193]. In line with these observations, in phase I and II clinical trials, Liu and coworkers reported that AEZS-108, associated with acceptable safety properties, decreases PSA levels in CRPC patients facing disease progression after chemotherapy [194,195].
Similar results were obtained with the bioconjugate AN-207 consisting of 2-pyrrolino-doxorubicin coupled to [D-Lys6]-GnRH [187,188].
Novel GnRH-based bioconjugates were developed by coupling [D-Lys6]-GnRH to different anticancer compounds. Karampelas and coworkers developed a GnRH-based bioconjugate in which the decapeptide is linked to a molecule of gemcitabine, a drug with antitumor activity but with a fast metabolic inactivation. They reported that the conjugate exerts a significant antitumor activity in CRPC cells, in vitro and in vivo, associated with a relevant metabolic and pharmacokinetic advantage [196]. Argyros et al. analyzed the effects of a conjugate consisting of [D-Lys6]-GnRH and an analog of the antiangiogenic compound sunitinib (SAN1). These authors found that in mouse xenograft models of CRPC, this compound induces a significant delay in tumor progression when compared with equimolar sunitinib or SAN1 alone. Importantly, no cardiovascular side effects were observed during the treatment [197].
As stated above, the GnRH-III isoform found in sea lamprey is a decapeptide that differs from the classical form of GnRH in amino acids 5–8, with a lysine in position 8 (instead of arginine as in GnRH). GnRH-III binds to mammal GnRH-R at the pituitary level where it is endowed with a very poor gonadotropin releasing activity in mammals. On the other hand, it was shown to bind to GnRH-R expressed in cancer cells to trigger its antitumor effects [33,198].
Based on these observations, different anthracycline-GnRH-III bioconjugates were developed and characterized in terms of chemical and enzymatic stability, as well as in terms of their cytostatic effects. In first-generation bioconjugates, a molecule of daunorubicin (Dau) or Dox was linked to the ε-amino group present in 6Lys of the decapeptide by means of different chemical linkages (ester, oxime, amide bond). For instance, the oxime bond-linked Dau-GnRH-III bioconjugate was shown to possess a high chemical and enzymatic stability (in human serum as well as in the presence of rat liver lysosomal homogenates) as well as significant antitumor effects in several cancer cells, both in vitro and in vivo [199,200,201,202].
To increase the cytotoxic activity of these compounds, multifunctional anticancer drug-GnRH-III bioconjugates were developed with the aim to increase the stability and the antitumor effects of these compounds (i.e., chemical modifications of the targeting decapeptide and attachment of more than one cytotoxic drug). Leurs and coworkers reported the synthesis and development of a bifunctional [4Lys]-GnRH-III containing two lysine amino acids (in position 4 and 8) coupled with two Dau molecules through their ε-amino groups. The same authors also synthesized and characterized a bioconjugate in which the GnRH-III peptide was used as a scaffold and a second molecule of Lys was attached to the amino group of 8Lys to provide the binding sites for two Dau molecules [203,204]. Similarly, GnRH-III based multifunctional delivery systems obtained by linking two different anticancer drugs (i.e., Dau and metotrexate) or composed by Dau-GnRH-III derivative dimers were developed [205,206]. More recently, the development and the anticancer effects of different oxime bond-linked Dau-GnRH-III bioconjugates containing different unnatural amino acids were reported by Schuster and coworkers [207]. A high enzymatic stability, as well as a significant cytotoxic activity in different types of cancer cells, both in vitro and in vivo, was reported for all these compounds [203,204,205,206,207].
In our laboratory we investigated the anticancer activity of two GnRH-III bioconjugates on CRPC cells (Dau-GnRH-III, in which Dau is bound to 8Lys of GnRH-III; Dau-[4Lys(Ac)]-GnRH-III, in which 4Ser of the Dau-GnRH-III conjugate is replaced by an acethylated lysine). We demonstrated that, after a rapid internalization, both Dau-GnRH-III and Dau-[4Lys(Ac)]-GnRH-III exert a significant antiproliferative/proapoptotic activity, that is counteracted by the cotreatment with a GnRH-R antagonist or by silencing of the classical form of the GnRH-R [208].
Although further studies (both in vitro and in vivo) are needed to confirm their lack of toxicity as well as their anticancer effects, these observations support an important role of GnRH bioconjugates as a novel delivery approach of cytotoxic drugs in targeted cancer therapy.
Current GnRH-R-targeted strategies for the management of both androgen responsive PCa and CRPC are illustrated in Figure 2.
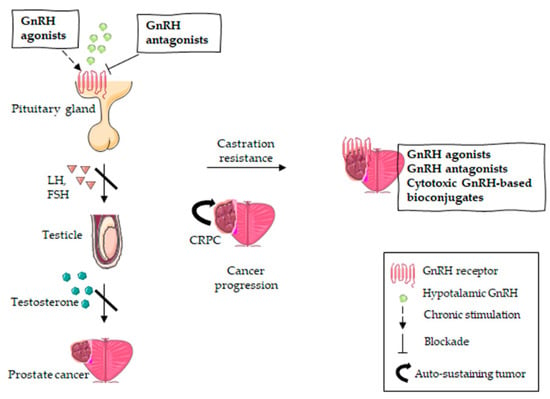
Figure 2.
GnRH-R-targeted therapies in PCa treatment. Androgen-responsive PCa is targeted with GnRH antagonists (direct inhibition of the pituitary GnRH receptor) or GnRH agonists (chronic stimulation inducing receptor desensitization), resulting in the suppression of the pituitary axis. The inhibition of androgen production leads to tumor regression. However, PCa can become castration-resistant, which means tumor progression occurs despite the suppression of the pituitary axis. Given the expression of GnRH receptors on CRPC cells, the latter might be further specifically targeted with GnRH agonists, GnRH antagonists or cytotoxic GnRH-based bioconjugates.
5. Conclusions
Pituitary GnRH-R mediate the pivotal role of the hypothalamic decapeptide GnRH in the control of the pituitary-gonadal axis functions. It has been widely shown that GnRH-R, sharing the same gene sequence, as well as mRNA and protein size, with the pituitary receptor, are also expressed in different tumors, such as PCa. Several data from the literature, from in vitro and in vivo studies, report that in PCa cells, and specifically in CRPC cells, GnRH agonists are associated with significant antiproliferative/proapoptotic, antimetastatic and antiangiogenic activities. Interestingly, GnRH antagonists were demonstrated to trigger the same antitumor effects. Millar and coworkers proposed that GnRH-R may adopt different structural conformations according to the cell context in which they are expressed, thus being associated with selective binding of GnRH analogs [137,145].
The opposite role of GnRH-R at the pituitary (stimulation of gonadotropin synthesis/secretion) and at the cancer level (antiproliferative effects) is related to the different intracellular signaling pathways associated with these receptors. In PCa cells these receptors are mainly associated with the Gαi/cAMP pathway, triggering the activity of a tyrosine phosphatase, and subsequent inactivation of tyrosine kinase receptors (i.e., EGF-R and IGF-I-R), and finally interfering with downstream different molecular pathways, such as the MAPK and PI3K/AKT signaling cascades.
Based on these molecular observations, GnRH agonists and antagonists, in combination with docetaxel, are currently considered as an effective therapeutic approach for chemotherapy-naive CRPC patients.
GnRH-based bioconjugates are now considered as a novel targeted therapeutic approach for CRPC. These are drugs in which a cytotoxic compound is covalently linked to a GnRH derivative peptide. It is believed that by binding to its receptors in cancer cells, the GnRH peptide can specifically deliver the chemotherapeutic drug to cancer cells while sparing normal cells not expressing the GnRH receptors. In this context, the most studied bioconjugate is AEZS-108 (AN-152), consisting of a molecule of doxorubicin linked to [D-Lys6]-GnRH. This bioconjugate was demonstrated to exert a significant antitumor activity in CRPC cells in vitro and in vivo. Results from phase I and II clinical studies support these experimental observations. Bioconjugates based on different isoforms of the GnRH peptide, such as GnRH-III, are also under investigation.
Further studies are needed to obtain novel compounds with a high specific efficacy and low toxicity to improve the treatment options for CRPC patients.
Author Contributions
Conceptualization, P.L.; Visualization, F.F., M.M. and M.M.M.; Writing—original draft preparation, F.F., M.M., M.R. and P.L.; writing—review and editing, F.F., M.M., M.M.M., M.R., R.M.M. and P.L.; funding acquisition, P.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MIUR Progetto di Eccellenza (Department of Pharmacological and Biomolecular Sciences, Università degli Studi di Milano). F.F. was supported by an AIRC fellowship for Italy.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Baba, Y.; Matsuo, H.; Schally, A.V. Structure of the porcine LH- and FSH-releasing hormone. II. Confirmation of the proposed structure by conventional sequential analyses. Biochem. Biophys. Res. Commun. 1971, 44, 459–463. [Google Scholar] [CrossRef]
- Schally, A.; Arimura, A.; Baba, Y.; Nair, R.; Matsuo, H.; Redding, T.; Debeljuk, L.; White, W. Isolation and properties of the FSH and LH-releasing hormone. Biochem. Biophys. Res. Commun. 1971, 43, 393–399. [Google Scholar] [CrossRef]
- Conn, P.P.M.; Crowley, W.F. Gonadotropin-releasing hormone and its analogs. Annu. Rev. Med. 1994, 45, 391–405. [Google Scholar] [CrossRef]
- Tzoupis, H.; Nteli, A.; Androutsou, M.-E.; Tselios, T. Gonadotropin-releasing hormone and GnRH receptor: Structure, function and drug development. Curr. Med. Chem. 2020, 27, 6136–6158. [Google Scholar] [CrossRef] [PubMed]
- Stopa, E.G.; Koh, E.T.; Svendsen, C.N.; Rogers, W.T.; Schwaber, J.S.; King, J.C. Computer-assisted mapping of immunoreactive mammalian gonadotropin-releasing hormone in adult human basal forebrain and amygdala. Endocrinology 1991, 128, 3199–3207. [Google Scholar] [CrossRef] [PubMed]
- Seeburg, P.H.; Mason, A.J.; Stewart, T.A.; Nikolics, K. The mammalian GnRH gene and its pivotal role in reproduction. Recent Prog. Horm. Res. 1987, 43, 69–98. [Google Scholar] [PubMed]
- Maggi, R.; Cariboni, A.M.; Marelli, M.M.; Moretti, R.M.; Andrè, V.; Marzagalli, M.; Limonta, P. GnRH and GnRH receptors in the pathophysiology of the human female reproductive system. Hum. Reprod. Update 2015, 22, 358–381. [Google Scholar] [CrossRef] [PubMed]
- Limonta, P.; Marelli, M.M.; Moretti, R.; Marzagalli, M.; Fontana, F.; Maggi, R. GnRH in the human female reproductive axis. Vitam. Horm. 2018, 107, 27–66. [Google Scholar] [CrossRef]
- Neill, J.D. GnRH and GnRH receptor genes in the human genome. Endocrinology 2002, 143, 737–743. [Google Scholar] [CrossRef]
- Kakar, S.S.; Malik, M.; Winters, S.J.; Mazhawidza, W. Gonadotropin-releasing hormone receptors: Structure, expression, and signaling transduction. Vitam. Horm. 2004, 69, 151–207. [Google Scholar] [CrossRef]
- Millar, R.P.; Lu, Z.-L.; Pawson, A.J.; Flanagan, C.A.; Morgan, K.; Maudsley, S.R. Gonadotropin-releasing hormone receptors. Endocr. Rev. 2004, 25, 235–275. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.K.; Leung, P.C. Molecular biology of gonadotropin-releasing hormone (GnRH)-I, GnRH-II, and their receptors in humans. Endocr. Rev. 2005, 26, 283–306. [Google Scholar] [CrossRef] [PubMed]
- Fana, N.C.; Eui-Bae, J.; Chun, P.; Olofssonb, J.I.; Krisingera, J.; Leunga, P.C. The human gonadotropin-releasing hormone (GnRH) receptor gene: Cloning, genomic organization and chromosomal assignment. Mol. Cell. Endocrinol. 1994, 103, R1–R6. [Google Scholar] [CrossRef]
- McArdle, C.A.; Franklin, J.; Green, L.; Hislop, J.N. Signalling, cycling and desensitisation of gonadotrophin-releasing hormone receptors. J. Endocrinol. 2002, 173, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kraus, S.; Naor, Z.; Seger, R. Intracellular signaling pathways mediated by the gonadotropin-releasing hormone (GnRH) receptor. Arch. Med. Res. 2001, 32, 499–509. [Google Scholar] [CrossRef]
- Aguilar-Rojas, A. Human gonadotropin-releasing hormone receptor-activated cellular functions and signaling pathways in extra-pituitary tissues and cancer cells (Review). Oncol. Rep. 2009, 22, 981–990. [Google Scholar] [CrossRef]
- Naor, Z. Signaling by G-protein-coupled receptor (GPCR): Studies on the GnRH receptor. Front. Neuroendocr. 2009, 30, 10–29. [Google Scholar] [CrossRef]
- McArdle, C.A. Gonadotropin-releasing hormone receptor signaling: Biased and unbiased. Mini-Rev. Med. Chem. 2012, 12, 841–850. [Google Scholar] [CrossRef]
- Naor, Z.; Huhtaniemi, I. Interactions of the GnRH receptor with heterotrimeric G proteins. Front. Neuroendocr. 2013, 34, 88–94. [Google Scholar] [CrossRef]
- Sviridonov, L.; Dobkin-Bekman, M.; Shterntal, B.; Przedecki, F.; Formishell, L.; Kravchook, S.; Navi, L.R.-B.; Bar-Lev, T.H.; Kazanietz, M.G.; Yao, Z.; et al. Differential signaling of the GnRH receptor in pituitary gonadotrope cell lines and prostate cancer cell lines. Mol. Cell. Endocrinol. 2013, 369, 107–118. [Google Scholar] [CrossRef]
- Janjic, M.M.; Stojilkovic, S.S.; Bjelobaba, I. Intrinsic and regulated gonadotropin-releasing hormone receptor gene transcription in mammalian pituitary gonadotrophs. Front. Endocrinol. 2017, 8, 221. [Google Scholar] [CrossRef] [PubMed]
- Bjelobaba, I.; Stojilkovic, S.S.; Naor, Z. Editorial: Gonadotropin-releasing hormone receptor signaling and functions. Front. Endocrinol. 2018, 9, 143. [Google Scholar] [CrossRef] [PubMed]
- Mugami, S.; Dobkin-Bekman, M.; Navi, L.R.; Naor, Z. Differential roles of PKC isoforms (PKCs) in GnRH stimulation of MAPK phosphorylation in gonadotrope derived cells. Mol. Cell. Endocrinol. 2018, 463, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Cheon, K.W.; Lee, H.S.; Parhar, I.S.; Kang, I.S. Expression of the second isoform of gonadotrophin-releasing hormone (GnRH-II) in human endometrium throughout the menstrual cycle. Mol. Hum. Reprod. 2001, 7, 447–452. [Google Scholar] [CrossRef]
- Millar, R.P. GnRHs and GnRH receptors. Anim. Reprod. Sci. 2005, 88, 5–28. [Google Scholar] [CrossRef]
- Schneider, J.S.; Rissman, E.F. Gonadotropin-releasing hormone II: A multi-purpose neuropeptide. Integr. Comp. Biol. 2008, 48, 588–595. [Google Scholar] [CrossRef][Green Version]
- Neill, J.D.; Duck, L.; Sellers, J.C.; Musgrove, L.C. A gonadotropin-releasing hormone (GnRH) receptor specific for GnRH II in primates. Biochem. Biophys. Res. Commun. 2001, 282, 1012–1018. [Google Scholar] [CrossRef]
- Millar, R.; Lowe, S.; Conklin, D.; Pawson, A.; Maudsley, S.; Troskie, B.; Ott, T.; Millar, M.; Lincoln, G.; Sellar, R.; et al. A novel mammalian receptor for the evolutionarily conserved type II GnRH. Proc. Natl. Acad. Sci. USA 2001, 98, 9636–9641. [Google Scholar] [CrossRef]
- Neill, J.D.; Musgrove, L.C.; Duck, L.W. Newly recognized GnRH receptors: Function and relative role. Trends Endocrinol. Metab. 2004, 15, 383–392. [Google Scholar] [CrossRef]
- Morgan, K.; Conklin, D.; Pawson, A.J.; Sellar, R.; Ott, T.R.; Millar, R.P. A transcriptionally active human type II gonadotropin-releasing hormone receptor gene homolog overlaps two genes in the antisense orientation on chromosome 1q.12. Endocrinology 2003, 144, 423–436. [Google Scholar] [CrossRef]
- Van Biljon, W.; Wykes, S.; Scherer, S.; Krawetz, S.; Hapgood, J. Type II gonadotropin-releasing hormone receptor transcripts in human sperm. Biol. Reprod. 2002, 67, 1741–1749. [Google Scholar] [CrossRef] [PubMed]
- Sower, S.A.; Chiang, Y.C.; Lovas, S.; Conlon, J.M. Primary structure and biological activity of a third gonadotropin-releasing hormone from lamprey brain. Endocrinology 1993, 132, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, M.; Vincze, B.; Horvath, J.E.; Seprodi, J. Structure-activity study on the LH- and FSH-releasing and anticancer effects of gonadotropin-releasing hormone (GnRH)-III analogs. Peptides 2007, 28, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Imai, A.; Ohno, T.; Iida, K.; Fuseya, T.; Furui, T.; Tamaya, T. Presence of gonadotropin-releasing hormone receptor and its messenger ribonucleic acid in endometrial carcinoma and endometrium. Gynecol. Oncol. 1994, 55, 144–148. [Google Scholar] [CrossRef]
- Schally, A.V.; Comaru-Schally, A.M.; Nagy, A.; Kovacs, M.; Szepeshazi, K.; Plonowski, A.; Varga, J.L.; Halmos, G. Hypothalamic hormones and cancer. Front. Neuroendocr. 2001, 22, 248–291. [Google Scholar] [CrossRef]
- Gründker, C.; Emons, G. Role of gonadotropin-releasing hormone (GnRH) in ovarian cancer. Reprod. Biol. Endocrinol. 2003, 1, 65. [Google Scholar] [CrossRef]
- Limonta, P.; Moretti, R.M.; Marelli, M.M.; Motta, M. The biology of gonadotropin hormone-releasing hormone: Role in the control of tumor growth and progression in humans. Front. Neuroendocr. 2003, 24, 279–295. [Google Scholar] [CrossRef]
- Harrison, G.S.; Wierman, M.E.; Nett, T.M.; Glode, L.M. Gonadotropin-releasing hormone and its receptor in normal and malignant cells. Endocr. Relat. Cancer 2004, 11, 725–748. [Google Scholar] [CrossRef]
- Marelli, M.M.; Moretti, R.M.; Januszkiewicz-Caulier, J.; Motta, M.; Limonta, P. Gonadotropin-releasing hormone (GnRH) receptors in tumors: A new rationale for the therapeutical application of GnRH analogs in cancer patients? Curr. Cancer Drug Targets 2006, 6, 257–269. [Google Scholar] [CrossRef]
- So, W.-K.; Cheng, J.-C.; Poon, S.-L.; Leung, P.C.K. Gonadotropin-releasing hormone and ovarian cancer: A functional and mechanistic overview. FEBS J. 2008, 275, 5496–5511. [Google Scholar] [CrossRef]
- Limonta, P.; Marelli, M.M.; Mai, S.; Motta, M.; Martini, L.; Moretti, R.M. GnRH receptors in cancer: From cell biology to novel targeted therapeutic strategies. Endocr. Rev. 2012, 33, 784–811. [Google Scholar] [CrossRef] [PubMed]
- Limonta, P.; Manea, M. Gonadotropin-releasing hormone receptors as molecular therapeutic targets in prostate cancer: Current options and emerging strategies. Cancer Treat. Rev. 2013, 39, 647–663. [Google Scholar] [CrossRef] [PubMed]
- Manea, M.; Marelli, M.; Moretti, R.M.; Maggi, R.; Marzagalli, M.; Limonta, P. Targeting hormonal signaling pathways in castration resistant prostate cancer. Recent Pat. Anti-Cancer Drug Discov. 2014, 9, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Rojas, A.; Pérez-Solis, M.A.; Maya-Núñez, G. The gonadotropin-releasing hormone system: Perspectives from reproduction to cancer (Review). Int. J. Oncol. 2016, 48, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Gründker, C.; Emons, G. The Role of gonadotropin-releasing hormone in cancer cell proliferation and metastasis. Front. Endocrinol. 2017, 8, 187. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Rojas, A.; Maya-Núñez, G.; Huerta-Reyes, M.; Pérez-Solis, M.A.; Silva-García, R.; Guillen, N.; Olivo-Marin, J.-C. Activation of human gonadotropin-releasing hormone receptor promotes down regulation of ARHGAP18 and regulates the cell invasion of MDA-MB-231 cells. Mol. Cell. Endocrinol. 2018, 460, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P. Epidemiology of prostate cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef]
- Barsouk, A.; Padala, S.A.; Vakiti, A.; Mohammed, A.; Saginala, K.; Thandra, K.C.; Rawla, P.; Barsouk, A. Epidemiology, staging and management of prostate cancer. Med. Sci. 2020, 8, 28. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Schally, A.V.; Coy, D.H.; Arimura, A. LH-RH agonists and antagonists. Int. J. Gynecol. Obstet. 1980, 18, 318–324. [Google Scholar] [CrossRef]
- Sealfon, S.C.; Weinstein, H.; Millar, R.P. Molecular mechanisms of ligand interaction with the gonadotropin-releasing hormone receptor. Endocr. Rev. 1997, 18, 180–205. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D. Hormonal therapy in prostate cancer: Historical approaches. Rev. Urol. 2004, 6, S3–S11. [Google Scholar] [PubMed]
- Moreau, J.-P.; Delavault, P.; Blumberg, J. Luteinizing hormone-releasing hormone agonists in the treatment of prostate cancer: A review of their discovery, development, and place in therapy. Clin. Ther. 2006, 28, 1485–1508. [Google Scholar] [CrossRef] [PubMed]
- Rove, K.O.; Crawford, E.D. Traditional androgen ablation approaches to advanced prostate cancer: New insights. Can. J. Urol. 2014, 21, 14–21. [Google Scholar]
- Van Poppel, H.; Abrahamsson, P. Considerations for the use of gonadotropin-releasing hormone agonists and antagonists in patients with prostate cancer. Int. J. Urol. 2020, 27, 830–837. [Google Scholar] [CrossRef]
- Wu, Y.; Rosenberg, J.E.; Taplin, M.-E. Novel agents and new therapeutics in castration-resistant prostate cancer. Curr. Opin. Oncol. 2011, 23, 290–296. [Google Scholar] [CrossRef]
- Trachtenberg, J.; Gittleman, M.; Steidle, C.; Barzell, W.; Friedel, W.; Pessis, D.; Fotheringham, N.; Campion, M.; Garnick, M.B. A phase 3, multicenter, open label, randomized study of abarelix versus leuprolide plus daily antiandrogen in men with prostate cancer. J. Urol. 2002, 167, 1670–1674. [Google Scholar] [CrossRef]
- Klotz, L.; Boccon-Gibod, L.; Shore, N.D.; Andreou, C.; Persson, B.-E.; Cantor, P.; Jensen, J.-K.; Olesen, T.K.; Schröder, F.H. The efficacy and safety of degarelix: A 12-month, comparative, randomized, open-label, parallel-group phase III study in patients with prostate cancer. BJU Int. 2008, 102, 1531–1538. [Google Scholar] [CrossRef]
- Mező, G.; Manea, M. Luteinizing hormone-releasing hormone antagonists. Expert Opin. Ther. Pat. 2009, 19, 1771–1785. [Google Scholar] [CrossRef]
- Liu, S.; Liu, S.; Pinski, J. Luteinizing hormone-releasing hormone receptor targeted agents for prostate cancer. Expert Opin. Investig. Drugs 2011, 20, 769–778. [Google Scholar] [CrossRef]
- Tan, O.; Bukulmez, O. Biochemistry, molecular biology and cell biology of gonadotropin-releasing hormone antagonists. Curr. Opin. Obstet. Gynecol. 2011, 23, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Schasfoort, E.; van de Beek, C.; Newling, D. Safety and efficacy of a non-steroidal anti-androgen, based on results of a post marketing surveillance of nilutamide. Prostate Cancer Prostatic Dis. 2001, 4, 112–117. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Odonnell, A.G.; Judson, I.; Dowsett, M.; Raynaud, F.I.; Dearnaley, D.P.; Mason, M.G.; Harland, S.J.; Robbins, A.S.; Halbert, G.; Nutley, B.; et al. Hormonal impact of the 17α-hydroxylase/C17,20-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br. J. Cancer 2004, 90, 2317–2325. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, M.; Hara, T.; Hitaka, T.; Kaku, T.; Takeuchi, T.; Takahashi, J.; Asahi, S.; Miki, H.; Tasaka, A.; Kusaka, M. Orteronel (TAK-700), a novel non-steroidal 17,20-lyase inhibitor: Effects on steroid synthesis in human and monkey adrenal cells and serum steroid levels in cynomolgus monkeys. J. Steroid Biochem. Mol. Biol. 2012, 129, 115–128. [Google Scholar] [CrossRef]
- Fizazi, K.; Albiges, L.; Loriot, Y.; Massard, C. ODM-201: A new-generation androgen receptor inhibitor in castration-resistant prostate cancer. Expert Rev. Anticancer. Ther. 2015, 15, 1007–1017. [Google Scholar] [CrossRef]
- Gibbons, J.A.; de Vries, M.; Krauwinkel, W.; Ohtsu, Y.; Noukens, J.; van der Walt, J.-S.; Mol, R.; Mordenti, J.; Ouatas, T. Pharmacokinetic drug interaction studies with enzalutamide. Clin. Pharmacokinet. 2015, 54, 1057–1069. [Google Scholar] [CrossRef]
- Markham, A. Apalutamide: First global approval. Drugs 2018, 78, 699–705. [Google Scholar] [CrossRef]
- Saranyutanon, S.; Srivastava, S.K.; Pai, S.; Singh, S.; Singh, A.P. Therapies targeted to androgen receptor signaling axis in prostate cancer: Progress, challenges, and hope. Cancers 2019, 12, 51. [Google Scholar] [CrossRef]
- Crawford, E.D.; Stanton, W.; Mandair, D. Darolutamide: An evidenced-based review of its efficacy and safety in the treatment of prostate cancer. Cancer Manag. Res. 2020, 12, 5667–5676. [Google Scholar] [CrossRef]
- Kassem, L.; Shohdy, K.S.; Abdel-Rahman, O. Abiraterone acetate/androgen deprivation therapy combination versus docetaxel/androgen deprivation therapy combination in advanced hormone-sensitive prostate cancer: A network meta-analysis on safety and efficacy. Curr. Med. Res. Opin. 2018, 34, 903–910. [Google Scholar] [CrossRef]
- Gravis, G. Systemic treatment for metastatic prostate cancer. Asian J. Urol. 2019, 6, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Saad, F.; Shore, N.; Zhang, T.; Sharma, S.; Cho, H.K.; Jacobs, I.A. Emerging therapeutic targets for patients with advanced prostate cancer. Cancer Treat. Rev. 2019, 76, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Adashek, J.J.; Reed, J.P.; Tandon, A.; Freedland, S.J.; Posadas, E.; Bhowmick, N.; Chung, L.W.; Freeman, M.; Figlin, R.A.; Gong, J. Combination androgen receptor inhibition and docetaxel in metastatic castration-sensitive prostate cancer: The next step in first-line treatment? Clin. Genitourin. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Farha, N.G.; Kasi, A. Docetaxel; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Sathianathen, N.J.; Koschel, S.; Thangasamy, I.A.; Teh, J.; Alghazo, O.; Butcher, G.; Howard, H.; Kapoor, J.; Lawrentschuk, N.; Siva, S.; et al. Indirect comparisons of efficacy between combination approaches in metastatic hormone-sensitive prostate cancer: A systematic review and network meta-analysis. Eur. Urol. 2020, 77, 365–372. [Google Scholar] [CrossRef]
- Vitkin, N.; Nersesian, S.; Siemens, D.R.; Koti, M. The tumor immune contexture of prostate cancer. Front. Immunol. 2019, 10, 603. [Google Scholar] [CrossRef] [PubMed]
- Jafari, S.; Hatami-Baroogh, L.; Kahroba, H.; Hejazi, M.S.; Maleki-Dizaji, N.; Barghi, S.; Kiaie, S.H.; Jadidi-Niaragh, F. Clinical application of immune checkpoints in targeted immunotherapy of prostate cancer. Cell. Mol. Life Sci. 2020, 77, 3693–3710. [Google Scholar] [CrossRef] [PubMed]
- Fong, L.; Kwek, S.S.; O’Brien, S.; Kavanagh, B.; McNeel, D.G.; Weinberg, V.; Lin, A.M.; Rosenberg, J.; Ryan, C.J.; Rini, B.I.; et al. Potentiating endogenous antitumor immunity to prostate cancer through combination immunotherapy with CTLA4 blockade and GM-CSF. Cancer Res. 2009, 69, 609–615. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; van den Eertwegh, A.J.M.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef]
- Graff, J.N.; Alumkal, J.J.; Drake, C.G.; Thomas, G.V.; Redmond, W.L.; Farhad, M.; Cetnar, J.P.; Ey, F.S.; Bergan, R.C.; Slottke, R.; et al. Early evidence of anti-PD-1 activity in enzalutamide-resistant prostate cancer. Oncotarget 2016, 7, 52810–52817. [Google Scholar] [CrossRef]
- Graff, J.N.; Baciarello, G.; Armstrong, A.J.; Higano, C.S.; Iversen, P.L.; Flaig, T.W.; Forer, D.; Parli, T.; Phung, D.; Tombal, B.; et al. Efficacy and safety of enzalutamide in patients 75 years or older with chemotherapy-naive metastatic castration-resistant prostate cancer: Results from PREVAIL. Ann. Oncol. 2016, 27, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Modena, A.; Ciccarese, C.; Iacovelli, R.; Brunelli, M.; Montironi, R.; Fiorentino, M.; Tortora, G.; Massari, F. Immune checkpoint inhibitors and prostate cancer: A new frontier? Oncol. Rev. 2016, 10, 293. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gao, J.; Ward, J.F.; Pettaway, C.A.; Shi, L.Z.; Subudhi, S.K.; Vence, L.M.; Zhao, H.; Chen, J.; Chen, H.; Efstathiou, E.; et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017, 23, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Velho, P.I.; Antonarakis, E.S. PD-1/PD-L1 pathway inhibitors in advanced prostate cancer. Expert Rev. Clin. Pharmacol. 2018, 11, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Claps, M.; Mennitto, A.; Guadalupi, V.; Sepe, P.; Stellato, M.; Zattarin, E.; Gillessen, S.S.; Sternberg, C.N.; Berruti, A.; De Braud, F.G.M.; et al. Immune-checkpoint inhibitors and metastatic prostate cancer therapy: Learning by making mistakes. Cancer Treat. Rev. 2020, 88, 102057. [Google Scholar] [CrossRef] [PubMed]
- Fay, E.; Graff, J.N. Immunotherapy in prostate cancer. Cancers 2020, 12, 1752. [Google Scholar] [CrossRef]
- Kim, T.J.; Koo, K.C. Current status and future perspectives of checkpoint inhibitor immunotherapy for prostate cancer: A comprehensive review. Int. J. Mol. Sci. 2020, 21, 5484. [Google Scholar] [CrossRef]
- Kittai, A.; Meshikhes, M.; Aragon-Ching, J.B. Ipilimumab: A potential immunologic agent in the treatment of metastatic castration-resistant prostate cancer. Cancer Biol. Ther. 2014, 15, 1299–1300. [Google Scholar] [CrossRef][Green Version]
- Hansen, A.R.; Massard, C.; Ott, P.A.; Haas, N.; Lopez, J.S.; Ejadi, S.; Wallmark, J.M.; Keam, B.; Delord, J.-P.; Aggarwal, R.; et al. Pembrolizumab for advanced prostate adenocarcinoma: Findings of the KEYNOTE-028 study. Ann. Oncol. 2018, 29, 1807–1813. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: Multicohort, open-label phase II KEYNOTE-199 study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef]
- Kakar, S.S.; Musgrove, L.C.; Devor, D.C.; Sellers, J.C.; Neill, J.D. Cloning, sequencing, and expression of human gonadotropin releasing hormone (GnRH) receptor. Biochem. Biophys. Res. Commun. 1992, 189, 289–295. [Google Scholar] [CrossRef]
- Limonta, P.; Marelli, M.M.; Moretti, R.M. LHRH analogues as anticancer agents: Pituitary and extrapituitary sites of action. Expert Opin. Investig. Drugs 2001, 10, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Völker, P.; Gründker, C.; Schmidt, O.; Schulz, K.-D.; Emons, G. Expression of receptors for luteinizing hormone-releasing hormone in human ovarian and endometrial cancers: Frequency, autoregulation, and correlation with direct antiproliferative activity of luteinizing hormone-releasing hormone analogues. Am. J. Obstet. Gynecol. 2002, 186, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Limonta, P.; Dondi, D.; Moretti, R.M.; Maggi, R.; Motta, M. Antiproliferative effects of luteinizing hormone-releasing hormone agonists on the human prostatic cancer cell line LNCaP. J. Clin. Endocrinol. Metab. 1992, 75, 207–212. [Google Scholar] [PubMed]
- Dondi, D.; Limonta, P.; Moretti, R.M.; Marelli, M.M.; Garattini, E.; Motta, M. Antiproliferative effects of luteinizing hormone-releasing hormone (LHRH) agonists on human androgen-independent prostate cancer cell line DU 145: Evidence for an autocrine-inhibitory LHRH loop. Cancer Res. 1994, 54, 4091–4095. [Google Scholar] [PubMed]
- Qayum, A.; Gullick, W.; Clayton, R.; Sikora, K.; Waxman, J. The effects of gonadotrophin releasing hormone analogues in prostate cancer are mediated through specific tumour receptors. Br. J. Cancer 1990, 62, 96–99. [Google Scholar] [CrossRef]
- Srkalovic, G.; Bokser, L.; Radulovic, S.; Korkut, E.; Schally, A.V. Receptors for luteinizing hormone-releasing hormone (LHRH) in dunning R3327 prostate cancers and rat anterior pituitaries after treatment with a sustained delivery system of LHRH antagonist SB-75. Endocrinology 1990, 127, 3052–3060. [Google Scholar] [CrossRef]
- Pinski, J.; Reile, H.; Halmos, G.; Groot, K.; Schally, A.V. Inhibitory effects of analogs of luteinizing hormone-releasing hormone on the growth of the androgen-independent dunning R-3327-AT-1 rat prostate cancer. Int. J. Cancer 1994, 59, 51–55. [Google Scholar] [CrossRef]
- Limonta, P.; Dondi, D.; Moretti, R.M.; Fermo, D.; Garattini, E.; Motta, M. Expression of luteinizing hormone-releasing hormone mRNA in the human prostatic cancer cell line LNCaP. J. Clin. Endocrinol. Metab. 1993, 76, 797–800. [Google Scholar]
- Limonta, P.; Dondi, D.; Marelli, M.M.; Moretti, R.M.; Negri-Cesi, P.; Motta, M. Growth of the androgen-dependent tumor of the prostate: Role of androgens and of locally expressed growth modulatory factors. J. Steroid Biochem. Mol. Biol. 1995, 53, 401–405. [Google Scholar] [CrossRef]
- Limonta, P.; Moretti, R.M.; Montagnani Marelli, M.; Dondi, D.; Parenti, M.; Motta, M. The luteinizing hormone-releasing hormone receptor in human prostate cancer cells: Messenger ribonucleic acid expression, molecular size, and signal transduction pathway. Endocrinology 1999, 140, 5250–5256. [Google Scholar] [CrossRef] [PubMed]
- Bahk, J.Y.; Hyun, J.S.; Lee, H.; Kim, M.O.; Cho, G.J.; Lee, B.H.; Choi, W.S. Expression of gonadotropin-releasing hormone (GnRH) and GnRH receptor mRNA in prostate cancer cells and effect of GnRH on the proliferation of prostate cancer cells. Urol. Res. 1998, 26, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Halmos, G.; Arencibia, J.M.; Schally, A.V.; Davis, R.; Bostwick, D.G. High incidence of receptors for luteinizing hormone-releasing hormone (LHRH) and LHRH receptor gene expression in human prostate cancers. J. Urol. 2000, 163, 623–629. [Google Scholar] [CrossRef]
- Straub, B.; Müller, M.; Krause, H.; Schrader, M.; Goessl, C.; Heicappell, R.; Miller, K. Increased incidence of luteinizing hormone-releasing hormone receptor gene messenger RNA expression in hormone-refractory human prostate cancers. Clin. Cancer Res. 2001, 7, 2340–2343. [Google Scholar] [PubMed]
- Tieva, A.; Stattin, P.; Wikstrom, P.; Bergh, A.; Damber, J.E. Gonadotropin-releasing hormone receptor expression in the human prostate. Prostate 2001, 47, 276–284. [Google Scholar] [CrossRef]
- Szabó, J.; Végh, A.; Rácz, G.; Szende, B. Immunohistochemical demonstration of gonadotropin-releasing hormone receptors in prostate carcinoma. Urol. Oncol. Semin. Orig. Investig. 2005, 23, 399–401. [Google Scholar] [CrossRef]
- Szabó, J.; Bartók, K.; Krenács, T.; Szepesváry, Z.; Szende, B. GnRH receptor and androgen receptor status and outcome of advanced prostate carcinomas. Anticancer Res. 2009, 29, 681–684. [Google Scholar]
- Bono, A.V.; Salvadore, M.; Celato, N. Gonadotropin-releasing hormone receptors in prostate tissue. Anal. Quant. Cytol. Histol. 2002, 24, 221–227. [Google Scholar]
- Limonta, P.; Moretti, R.M.; Dondi, D.; Marelli, M.M.; Motta, M. Androgen-dependent prostatic tumors: Biosynthesis and possible actions of LHRH. J. Steroid Biochem. Mol. Biol. 1994, 49, 347–350. [Google Scholar] [CrossRef]
- Gnanapragasam, V.J.; Darby, S.; Khan, M.; Lock, W.; Robson, C.; Leung, H.Y. Evidence that prostate gonadotropin-releasing hormone receptors mediate an anti-tumourigenic response to analogue therapy in hormone refractory prostate cancer. J. Pathol. 2005, 206, 205–213. [Google Scholar] [CrossRef]
- Sundaram, S.; Durairaj, C.; Kadam, R.; Kompella, U.B. Luteinizing hormone-releasing hormone receptor-targeted deslorelin-docetaxel conjugate enhances efficacy of docetaxel in prostate cancer therapy. Mol. Cancer Ther. 2009, 8, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Marelli, M.M.; Moretti, R.M.; Dondi, D.; Limonta, P.; Motta, M. Effects of LHRH agonists on the growth of human prostatic tumor cells: “In vitro” and “in vivo” studies. Arch. Ital. Urol. Androl. 1997, 69, 257–263. [Google Scholar]
- Dondi, D.; Moretti, R.M.; Marelli, M.M.; Pratesi, G.; Polizzi, D.; Milani, M.; Motta, M.; Limonta, P. Growth-inhibitory effects of luteinizing hormone-releasing hormone (LHRH) agonists on xenografts of the DU 145 human androgen-independent prostate cancer cell line in nude mice. Int. J. Cancer 1998, 76, 506–511. [Google Scholar] [CrossRef]
- Limonta, P.; Pratesi, G.; Moretti, R.M.; Marelli, M.M.; Motta, M.; Dondi, D. Comments on inhibition of growth of androgen-independent DU-145 prostate cancer in vivo by luteinising hormone-releasing hormone antagonist cetrorelix and bombesin antagonists RC-3940-II and RC-3950-II, Jungwirth et al., Eur. J. Cancer 1997, 33(7), 1141–1148. Eur. J. Cancer 1998, 34, 1134–1135. [Google Scholar] [PubMed]
- Castellón, E.; Clementi, M.; Hitschfeld, C.; Sánchez, C.A.; Benitez, D.; Saenz, L.; Contreras, H.; Huidobro, C. Effect of leuprolide and cetrorelix on cell growth, apoptosis, and GnRH receptor expression in primary cell cultures from human prostate carcinoma. Cancer Investig. 2006, 24, 261–268. [Google Scholar] [CrossRef]
- Marelli, M.M.; Moretti, R.M.; Mai, S.; Januszkiewicz-Caulier, J.; Motta, M.; Limonta, P. Type I gonadotropin-releasing hormone receptor mediates the antiproliferative effects of GnRH-II on prostate cancer cells. J. Clin. Endocrinol. Metab. 2009, 94, 1761–1767. [Google Scholar] [CrossRef]
- Kraus, S.; Levy, G.; Hanoch, T.; Naor, Z.; Seger, R. Gonadotropin-releasing hormone induces apoptosis of prostate cancer cells: Role of c-Jun NH2-terminal kinase, protein kinase B, and extracellular signal-regulated kinase pathways. Cancer Res. 2004, 64, 5736–5744. [Google Scholar] [CrossRef]
- Kraus, S.; Naor, Z.; Seger, R. Gonadotropin-releasing hormone in apoptosis of prostate cancer cells. Cancer Lett. 2006, 234, 109–123. [Google Scholar] [CrossRef]
- Maiti, K.; Oh, D.Y.; Moon, J.S.; Acharjee, S.; Li, J.H.; Bai, D.G.; Park, H.S.; Lee, K.; Lee, Y.C.; Jung, N.C.; et al. Differential effects of gonadotropin-releasing hormone (GnRH)-I and GnRH-II on prostate cancer cell signaling and death. J. Clin. Endocrinol. Metab. 2005, 90, 4287–4298. [Google Scholar] [CrossRef][Green Version]
- Clementi, M.; Sánchez, C.A.; Benitez, D.A.; Contreras, H.R.; Huidobro, C.; Cabezas, J.; Acevedo, C.; Castellón, E.A. Gonadotropin releasing hormone analogs induce apoptosis by extrinsic pathway involving p53 phosphorylation in primary cell cultures of human prostatic adenocarcinomas. Prostate 2009, 69, 1025–1033. [Google Scholar] [CrossRef]
- Morgan, K.; Stavrou, E.; Leighton, S.P.; Miller, N.; Sellar, R.; Millar, R.P. Elevated GnRH receptor expression plus GnRH agonist treatment inhibits the growth of a subset of papillomavirus 18-immortalized human prostate cells. Prostate 2010, 71, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.M.; Marelli, M.M.; Taylor, D.M.; Martini, P.G.V.; Marzagalli, M.; Limonta, P. Gonadotropin-releasing hormone agonists sensitize, and resensitize, prostate cancer cells to docetaxel in a p53-dependent manner. PLoS ONE 2014, 9, e93713. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, C.; Iacopino, F.; Lama, G.; Capucci, S.; Zelano, G.; Boca, M.; Pistilli, A.; Sica, G. Apoptosis-related gene expression affected by a GnRH analogue without induction of programmed cell death in LNCaP cells. Anticancer Res. 2004, 24, 2729–2738. [Google Scholar] [PubMed]
- Limonta, P.; Moretti, R.M.; Dondi, D.; Marelli, M.M.; Motta, M. The EGF/TGF-alpha system as an autocrine growth-stimulatory loop in LNCaP cells. Endocr. Relat. Cancer 1994, 1, 5–13. [Google Scholar]
- Dondi, D.; Moretti, R.M.; Marelli, M.M.; Motta, M.; Limonta, P. Growth factors in steroid-responsive prostatic tumor cells. Steroids 1996, 61, 222–225. [Google Scholar] [CrossRef]
- Kimura, G.; Kasuya, J.; Giannini, S.; Honda, Y.; Mohan, S.; Kawachi, M.H.; Akimoto, M.; Fujita-Yamaguchi, Y. Insulin-like growth factor (IGF) system components in human prostatic cancer cell-lines: LNCaP, DU145, and PC-3 cells. Int. J. Urol. 1996, 3, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Motta, M.; Dondi, D.; Moretti, R.M.; Marelli, M.M.; Pimpinelli, F.; Maggi, R.; Limonta, P. Role of growth factors, steroid and peptide hormones in the regulation of human prostatic tumor growth. J. Steroid Biochem. Mol. Biol. 1996, 56, 107–111. [Google Scholar] [CrossRef]
- Kaplan, P.J.; Mohan, S.; Cohen, P.; Foster, B.A.; Greenberg, N.M. The insulin-like growth factor axis and prostate cancer: Lessons from the transgenic adenocarcinoma of mouse prostate (TRAMP) model. Cancer Res. 1999, 59, 2203–2209. [Google Scholar]
- Mita, K.; Nakahara, M.; Usui, T. Expression of the insulin-like growth factor system and cancer progression in hormone-treated prostate cancer patients. Int. J. Urol. 2000, 7, 321–329. [Google Scholar] [CrossRef][Green Version]
- Cardillo, M.R.; Monti, S.; Di Silverio, F.; Gentile, V.; Sciarra, F.; Toscano, V. Insulin-like growth factor (IGF)-I, IGF-II and IGF type I receptor (IGFR-I) expression in prostatic cancer. Anticancer Res. 2003, 23, 3825–3835. [Google Scholar]
- Moretti, R.M.; Marelli, M.M.; Dondi, D.; Poletti, A.; Martini, L.; Motta, M.; Limonta, P. Luteinizing hormone-releasing hormone agonists interfere with the stimulatory actions of epidermal growth factor in human prostatic cancer cell lines, LNCaP and DU 145. J. Clin. Endocrinol. Metab. 1996, 81, 3930–3937. [Google Scholar] [PubMed]
- Iacopino, F.; Lama, G.; Angelucci, C.; Sica, G. Leuprorelin acetate affects ERK1/2 activity in prostate cancer cells. Int. J. Oncol. 2006, 29, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.; Souto, J.C.S.; Solava, J.; Kassis, J.; Bailey, K.J.; Turner, T. Luteinizing hormone-releasing hormone agonist limits DU-145 prostate cancer growth by attenuating epidermal growth factor receptor signaling. Clin. Cancer Res. 2002, 8, 1251–1257. [Google Scholar] [PubMed]
- Ahearn, T.U.; Peisch, S.; Pettersson, A.; Ebot, E.M.; Zhou, C.K.; Graff, R.E.; Sinnott, J.A.; Fazli, L.; Judson, G.L.; Bismar, T.A.; et al. Expression of IGF/insulin receptor in prostate cancer tissue and progression to lethal disease. Carcinogenesis 2018, 39, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Marelli, M.M.; Moretti, R.M.; Dondi, D.; Motta, M.; Limonta, P. Luteinizing hormone-releasing hormone agonists interfere with the mitogenic activity of the insulin-like growth factor system in androgen-independent prostate cancer cells. Endocrinology 1999, 140, 329–334. [Google Scholar] [CrossRef][Green Version]
- Millar, R.P.; Pawson, A.J.; Morgan, K.; Rissman, E.F.; Lu, Z. Diversity of actions of GnRHs mediated by ligand-induced selective signaling. Front. Neuroendocr. 2008, 29, 17–35. [Google Scholar] [CrossRef]
- Doroszko, M.; Chrusciel, M.; Stelmaszewska, J.; Slezak, T.; Anisimowicz, S.; Plockinger, U.; Quinkler, M.; Bonomi, M.; Wolczynski, S.; Huhtaniemi, I.; et al. GnRH antagonist treatment of malignant adrenocortical tumors. Endocr. Relat. Cancer 2019, 26, 103–117. [Google Scholar] [CrossRef]
- Lamharzi, N.; Schally, A.V.; Koppán, M. Luteinizing hormone–releasing hormone (LH–RH) antagonist Cetrorelix inhibits growth of DU-145 human androgen-independent prostate carcinoma in nude mice and suppresses the levels and mRNA expression of IGF-II in tumors. Regul. Pept. 1998, 77, 185–192. [Google Scholar] [CrossRef]
- Moretti, R.M.; Marelli, M.M.; van Groeninghen, J.C.; Motta, M.; Limonta, P. Inhibitory activity of luteinizing hormone-releasing hormone on tumor growth and progression. Endocr. Relat. Cancer 2003, 10, 161–167. [Google Scholar] [CrossRef]
- Msaouel, P.; Diamanti, E.; Tzanela, M.; Koutsilieris, M. Luteinising hormone-releasing hormone antagonists in prostate cancer therapy. Expert Opin. Emerg. Drugs 2007, 12, 285–299. [Google Scholar] [CrossRef]
- Festuccia, C.; Dondi, D.; Piccolella, M.; Locatelli, A.; Gravina, G.L.; Tombolini, V.; Motta, M. Ozarelix, a fourth generation GnRH antagonist, induces apoptosis in hormone refractory androgen receptor negative prostate cancer cells modulating expression and activity of death receptors. Prostate 2010, 70, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Martinez-Arguelles, D.B.; Patterson, N.H.; Chaurand, P.; Papadopoulos, V. In search of the molecular mechanisms mediating the inhibitory effect of the GnRH antagonist degarelix on human prostate cell growth. PLoS ONE 2015, 10, e0120670. [Google Scholar] [CrossRef] [PubMed]
- Cucchiara, V.; Yang, J.C.; Liu, C.; Adomat, H.H.; Guns, E.S.T.; Gleave, M.E.; Gao, A.C.; Evans, C.P. GnRH antagonists have direct inhibitory effects on castration-resistant prostate cancer via intracrine androgen and AR-V7 expression. Mol. Cancer Ther. 2019, 18, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Millar, R.P.; Pawson, A.J. Outside-in and inside-out signaling: The new concept that selectivity of ligand binding at the gonadotropin-releasing hormone receptor is modulated by the intracellular environment. Endocrinology 2004, 145, 3590–3593. [Google Scholar] [CrossRef] [PubMed]
- Darby, S.; Stockley, J.; Khan, M.M.; Robson, C.N.; Leung, H.Y.; Gnanapragasam, V.J. Expression of GnRH type II is regulated by the androgen receptor in prostate cancer. Endocr. Relat. Cancer 2007, 14, 613–624. [Google Scholar] [CrossRef]
- Gründker, C.; Schlotawa, L.; Viereck, V.; Eicke, N.; Horst, A.; Kairies, B.; Emons, G. Antiproliferative effects of the GnRH antagonist cetrorelix and of GnRH-II on human endometrial and ovarian cancer cells are not mediated through the GnRH type I receptor. Eur. J. Endocrinol. 2004, 151, 141–149. [Google Scholar] [CrossRef][Green Version]
- Kim, J.W.; Yadav, D.K.; Kim, S.J.; Lee, M.-Y.; Park, J.-M.; Kim, B.S.; Kim, M.-H.; Park, H.-G.; Kang, K.W. Anti-cancer effect of GV1001 for prostate cancer: Function as a ligand of GnRHR. Endocr. Relat. Cancer 2019, 26, 147–162. [Google Scholar] [CrossRef]
- Marelli, M.M.; Moretti, R.M.; Mai, S.; Procacci, P.; Limonta, P. Gonadotropin-releasing hormone agonists reduce the migratory and the invasive behavior of androgen-independent prostate cancer cells by interfering with the activity of IGF-I. Int. J. Oncol. 2007, 30, 261–271. [Google Scholar] [CrossRef][Green Version]
- Dondi, D.; Festuccia, C.; Piccolella, M.; Bologna, M.; Motta, M. GnRH agonists and antagonists decrease the metastatic progression of human prostate cancer cell lines by inhibiting the plasminogen activator system. Oncol. Rep. 2006, 15, 393–400. [Google Scholar] [CrossRef][Green Version]
- Enomoto, M.; Utsumi, M.; Park, M.K. Gonadotropin-releasing hormone induces actin cytoskeleton remodeling and affects cell migration in a cell-type-specific manner in TSU-Pr1 and DU145 cells. Endocrinology 2006, 147, 530–542. [Google Scholar] [CrossRef][Green Version]
- Sica, G.; Angelucci, C.; Lama, G.; Iacopino, F. Leuprorelin acetate affects adhesion molecule expression in human prostate cancer cells. Int. J. Oncol. 2011, 38, 1501–1509. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moretti, R.M.; Mai, S.; Marelli, M.M.; Bani, M.R.; Ghilardi, C.; Giavazzi, R.; Taylor, D.M.; Martini, P.G.V.; Limonta, P. Dual targeting of tumor and endothelial cells by gonadotropin-releasing hormone agonists to reduce melanoma angiogenesis. Endocrinol. 2010, 151, 4643–4653. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.; Wells, A.; Turner, T. Luteinising hormone-releasing hormone analogue reverses the cell adhesion profile of EGFR overexpressing DU-145 human prostate carcinoma subline. Br. J. Cancer 2005, 92, 366–375. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dobkin-Bekman, M.; Naidich, M.; Pawson, A.J.; Millar, R.P.; Seger, R.; Naor, Z. Activation of Mitogen-activated protein kinase (MAPK) by GnRH is cell-context dependent. Mol. Cell. Endocrinol. 2006, 252, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Huang, T.; Chen, W.; Lu, H. GnRH participates in the self-renewal of A549-derived lung cancer stem-like cells through upregulation of the JNK signaling pathway. Oncol. Rep. 2015, 34, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Imai, A.; Horibe, S.; Takagi, H.; Fuseya, T.; Tamaya, T. Signal transduction of GnRH receptor in the reproductive tract tumor. Endocr. J. 1996, 43, 249–260. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maudsley, S. Gonadotropin-releasing hormone (GnRH) antagonists promote proapoptotic signaling in peripheral reproductive tumor cells by activating a Gαi-coupling state of the type I GnRH receptor. Cancer Res. 2004, 64, 7533–7544. [Google Scholar] [CrossRef]
- Grundker, C. Antiproliferative signaling of luteinizing hormone-releasing hormone in human endometrial and ovarian cancer cells through G proteinαI-mediated activation of phosphotyrosine phosphatase. Endocrinology 2001, 142, 2369–2380. [Google Scholar] [CrossRef]
- Imai, A.; Tamaya, T. GnRH receptor and apoptotic signaling. Vitam. Horm. 2000, 59, 1–33. [Google Scholar] [CrossRef]
- Johnson, G.L. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Nadel, G.; Yao, Z.; Ben-Ami, I.; Naor, Z.; Seger, R. Gq-induced apoptosis is mediated by AKT inhibition that leads to PKC-induced JNK activation. Cell. Physiol. Biochem. 2018, 50, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Lawrentschuk, N.; Fernandes, K.; Bell, D.; Barkin, J.; Fleshner, N. Efficacy of a second line luteinizing hormone-releasing hormone agonist after advanced prostate cancer biochemical recurrence. J. Urol. 2011, 185, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Teply, B.A.; Wang, H.; Luber, B.; Sullivan, R.; Rifkind, I.; Bruns, A.; Spitz, A.; DeCarli, M.; Sinibaldi, V.; Pratz, C.F.; et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: An open-label, phase 2, multicohort study. Lancet Oncol. 2018, 19, 76–86. [Google Scholar] [CrossRef]
- Jang, H.S.; Koo, K.C.; Cho, K.S.; Chung, B.H. Survival outcomes of concurrent treatment with docetaxel and androgen deprivation therapy in metastatic castration-resistant prostate cancer. Yonsei Med. J. 2016, 57, 1070–1078. [Google Scholar] [CrossRef]
- Organ, M.; Wood, L.; Wilke, D.; Skedgel, C.; Cheng, T.; North, S.; Thompson, K.; Winch, S.; Rendon, R. Intermittent LHRH therapy in the management of castrate-resistant prostate cancer (CRPCa): Results of a multi-institutional randomized prospective clinical trial. Am. J. Clin. Oncol. 2013, 36, 601–605. [Google Scholar] [CrossRef]
- Merseburger, A.S.; Hammerer, P.; Rozet, F.; Roumeguère, T.; Caffo, O.; da Silva, F.C.; Alcaraz, A. Androgen deprivation therapy in castrate-resistant prostate cancer: How important is GnRH agonist backbone therapy? World J. Urol. 2015, 33, 1079–1085. [Google Scholar] [CrossRef]
- Merseburger, A.S.; Hupe, M.C. An update on triptorelin: Current thinking on androgen deprivation therapy for prostate cancer. Adv. Ther. 2016, 33, 1072–1093. [Google Scholar] [CrossRef]
- Tombal, B.; Cornel, E.B.; Persad, R.; Stari, A.; Veiga, F.G.; Schulman, C. Clinical outcomes and testosterone levels following continuous androgen deprivation in patients with relapsing or locally advanced prostate cancer: A post hoc analysis of the ICELAND study. J. Urol. 2017, 198, 1054–1060. [Google Scholar] [CrossRef]
- Pinthus, J.H. Antagonizing GnRH receptors: A temporary ADT salvage maneuver for prostate cancer patients experiencing PSA failure with GnRH agonist. Can. Urol. Assoc. J. 2020, 14, 42. [Google Scholar] [CrossRef]
- Ben-Josef, E.; Yang, S.Y.; Ji, T.H.; Bidart, J.M.; Garde, S.V.; Chopra, D.P.; Porter, A.T.; Tang, D.G. Hormone-refractory prostate cancer cells express functional follicle-stimulating hormone receptor (FSHR). J. Urol. 1999, 161, 970–976. [Google Scholar] [CrossRef]
- Mariani, S.; Salvatori, L.; Basciani, S.; Arizzi, M.; Franco, G.; Petrangeli, E.; Spera, G.; Gnessi, L. Expression and cellular localization of follicle-stimulating hormone receptor in normal human prostate, benign prostatic hyperplasia and prostate cancer. J. Urol. 2006, 175, 2072–2077. [Google Scholar] [CrossRef]
- Crawford, E.D.; Schally, A.V.; Pinthus, J.H.; Block, N.L.; Rick, F.G.; Garnick, M.B.; Eckel, R.H.; Keane, T.E.; Shore, N.D.; Dahdal, D.N.; et al. The potential role of follicle-stimulating hormone in the cardiovascular, metabolic, skeletal, and cognitive effects associated with androgen deprivation therapy. Urol. Oncol. Semin. Orig. Investig. 2017, 35, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; Tombal, B.; Keane, T.; Boccardo, F.; Miller, K.; Shore, N.; Moul, J.W.; Damber, J.-E.; Collette, L.; Persson, B.-E. FSH suppression and tumour control in patients with prostate cancer during androgen deprivation with a GnRH agonist or antagonist. Scand. J. Urol. 2018, 52, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Abufaraj, M.; Iwata, T.; Kimura, S.; Haddad, A.; Al-Ani, H.; Abusubaih, L.; Moschini, M.; Briganti, A.; Karakiewicz, P.I.; Shariat, S.F. Differential impact of gonadotropin-releasing hormone antagonist versus agonist on clinical safety and oncologic outcomes on patients with metastatic prostate cancer: A meta-analysis of randomized controlled trials. Eur. Urol. 2020, S0302-2838, 30429–30432. [Google Scholar] [CrossRef] [PubMed]
- Boccon-Gibod, L.; van der Meulen, E.; Persson, B.-E. An update on the use of gonadotropin-releasing hormone antagonists in prostate cancer. Ther. Adv. Urol. 2011, 3, 127–140. [Google Scholar] [CrossRef]
- Higano, C.S. Update on cardiovascular and metabolic risk profiles of hormonal agents used in managing advanced prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 912–917. [Google Scholar] [CrossRef]
- Margel, D.; Ber, Y.; Peer, A.; Shavit-Grievink, L.; Pinthus, J.H.; Witberg, G.; Baniel, J.; Kedar, D.; Rosenbaum, E. Cardiac biomarkers in patients with prostate cancer and cardiovascular disease receiving gonadotrophin releasing hormone agonist vs antagonist. Prostate Cancer Prostatic Dis. 2020. [Google Scholar] [CrossRef]
- Perrone, V.; Degli Esposti, L.; Giacomini, E.; Veronesi, C.; Blini, V.; Oderda, M. Cardiovascular risk profile in prostate cancer patients treated with GnRH agonists versus antagonists: An Italian real-world analysis. Ther. Clin. Risk Manag. 2020, 16, 393–401. [Google Scholar] [CrossRef]
- Pham, T.; Sadowski, M.C.; Li, H.; Richard, D.; d’Emden, M.C.; Richard, K. Advances in hormonal therapies for hormone naïve and castration-resistant prostate cancers with or without previous chemotherapy. Exp. Hematol. Oncol. 2015, 5, 1–11. [Google Scholar] [CrossRef]
- Masson-Lecomte, A.; Guy, L.; Pedron, P.; Bruyère, F.; Rouprêt, M.; Nsabimbona, B.; Dahan, M.; Hoffman, P.; Salomon, L.; Vordos, D.; et al. A switch from GnRH agonist to GnRH antagonist in castration-resistant prostate cancer patients leads to a low response rate on PSA. World J. Urol. 2012, 31, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, R.; Kawahara, T.; Miyoshi, Y.; Yao, M.; Chiba, S.; Uemura, H. A case of switching from GnRH agonist to antagonist for castration resistant prostate cancer control. Case Rep. Oncol. 2019, 12, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Atchia, K.S.; Wallis, C.J.; Fleshner, N.; Toren, P. Switching from a gonadotropin-releasing hormone (GnRH) agonist to a GnRH antagonist in prostate cancer patients: A systematic review and meta-analysis. Can. Urol. Assoc. J. 2019, 14, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Uemura, H.; DiBonaventura, M.; Wang, E.; Ledesma, D.A.; Concialdi, K.; Aitoku, Y. The treatment patterns of castration-resistant prostate cancer in Japan, including symptomatic skeletal events and associated treatment and healthcare resource use. Expert Rev. Pharmacoecon. Outcomes Res. 2017, 17, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Schally, A.V. Targeting of cytotoxic luteinizing hormone-releasing hormone analogs to breast, ovarian, endometrial, and prostate cancers. Biol. Reprod. 2005, 73, 851–859. [Google Scholar] [CrossRef]
- Schally, A.V.; Engel, J.B.; Emons, G.; Block, N.L.; Pinski, J. Use of analogs of peptide hormones conjugated to cytotoxic radicals for chemotherapy targeted to receptors on tumors. Curr. Drug Deliv. 2011, 8, 11–25. [Google Scholar] [CrossRef]
- Engel, J.B.; Tinneberg, H.-R.; Rick, F.G.; Berkes, E.; Schally, A.V. Targeting of peptide cytotoxins to LHRH receptors for treatment of cancer. Curr. Drug Targets 2016, 17, 488–494. [Google Scholar] [CrossRef]
- Fodor, K.; Dobos, N.; Schally, A.; Steiber, Z.; Olah, G.; Sipos, E.; Székvölgyi, L.; Halmos, G. The targeted LHRH analog AEZS-108 alters expression of genes related to angiogenesis and development of metastasis in uveal melanoma. Oncotarget 2020, 11, 175–187. [Google Scholar] [CrossRef]
- Letsch, M.; Schally, A.V.; Szepeshazi, K.; Halmos, G.; Nagy, A. Preclinical evaluation of targeted cytotoxic luteinizing hormone-releasing hormone analogue AN-152 in androgen-sensitive and insensitive prostate cancers. Clin. Cancer Res. 2003, 9, 4505–4513. [Google Scholar]
- Engel, J.; Emons, G.; Pinski, J.; Schally, A.V. AEZS-108: A targeted cytotoxic analog of LHRH for the treatment of cancers positive for LHRH receptors. Expert Opin. Investig. Drugs 2012, 21, 891–899. [Google Scholar] [CrossRef]
- Popovics, P.; Schally, A.V.; Szalontay, L.; Block, N.L.; Rick, F.G. Targeted cytotoxic analog of luteinizing hormone-releasing hormone (LHRH), AEZS-108 (AN-152), inhibits the growth of DU-145 human castration-resistant prostate cancer in vivo and in vitro through elevating p21 and ROS levels. Oncotarget 2014, 5, 4567–4578. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, S.; Kotulla, G.; Kaiser, F.; Krauss, W.; Werning, G.; Elsasser, H.P.; Nagy, A.; Schulz, K.D.; Gründker, C.; Schally, A.V.; et al. Receptor mediated antiproliferative effects of the cytotoxic LHRH agonist AN-152 in human ovarian and endometrial cancer cell lines. Int. J. Oncol. 2000, 17, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tsao-Wei, D.D.; Xiong, S.; Groshen, S.; Dorff, T.B.; Quinn, D.I.; Tai, Y.-C.; Engel, J.; Hawes, D.; Schally, A.V.; et al. Phase I, dose-escalation study of the targeted cytotoxic LHRH analog AEZS-108 in patients with castration- and taxane-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 6277–6283. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.S.; Athreya, K.; Liu, S.; Schally, A.V.; Tsao-Wei, D.; Groshen, S.; Quinn, D.I.; Dorff, T.B.; Xiong, S.; Engel, J.; et al. A phase II trial of AEZS-108 in castration- and taxane-resistant prostate cancer. Clin. Genitourin. Cancer 2017, 15, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Karampelas, T.; Argyros, O.; Sayyad, N.; Spyridaki, K.; Pappas, C.; Morgan, K.; Kolios, G.; Millar, R.P.; Liapakis, G.; Tzakos, A.G.; et al. GnRH-gemcitabine conjugates for the treatment of androgen-independent prostate cancer: Pharmacokinetic enhancements combined with targeted drug delivery. Bioconjug. Chem. 2014, 25, 813–823. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Argyros, O.; Karampelas, T.; Asvos, X.; Varela, A.; Sayyad, N.; Papakyriakou, A.; Davos, C.H.; Tzakos, A.G.; Fokas, D.; Tamvakopoulos, C. Peptide-drug conjugate GnRH-sunitinib targets angiogenesis selectively at the site of action to inhibit tumor growth. Cancer Res. 2016, 76, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Lovas, S.; Pályi, I.; Vincze, B.; Horváth, J.; Kovács, M.; Mezö, I.; Tóth, G.; Teplán, I.; Murphy, R.F. Direct anticancer activity of gonadotropin-releasing hormone-III. J. Pept. Res. 1998, 52, 384–389. [Google Scholar] [CrossRef]
- Orban, E.; Mezo, G.; Schlage, P.; Csik, G.; Kulic, Z.; Ansorge, P.; Fellinger, E.; Moller, H.M.; Manea, M. In vitro degradation and antitumor activity of oxime bond-linked daunorubicin-GnRH-III bioconjugates and DNA-binding properties of daunorubicin-amino acid metabolites. Amino Acids 2011, 41, 469–483. [Google Scholar] [CrossRef]
- Schlage, P.; Mezo, G.; Orban, E.; Bosze, S.; Manea, M. Anthracycline-GnRH derivative bioconjugates with different linkages: Synthesis, in vitro drug release and cytostatic effect. J. Control Release 2011, 156, 170–178. [Google Scholar] [CrossRef]
- Manea, M.; Tovari, J.; Tejeda, M.; Schulcz, A.; Kapuvari, B.; Vincze, B.; Mezo, G. In-vivo antitumour effect of daunorubicin-GnRH-III derivative conjugates on colon carcinoma-bearing mice. Anticancer Drugs 2012, 23, 90–97. [Google Scholar] [CrossRef]
- Kapuvari, B.; Hegedus, R.; Schulcz, A.; Manea, M.; Tovari, J.; Gacs, A.; Vincze, B.; Mezo, G. Improved in vivo antitumor effect of a daunorubicin—GnRH-III bioconjugate modified by apoptosis inducing agent butyric acid on colorectal carcinoma bearing mice. Investig. New Drugs 2016, 34, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Manea, M.; Leurs, U.; Orban, E.; Baranyai, Z.; Ohlschlager, P.; Marquardt, A.; Schulcz, A.; Tejeda, M.; Kapuvari, B.; Tovari, J.; et al. Enhanced enzymatic stability and antitumor activity of daunorubicin-GnRH-III bioconjugates modified in position. Bioconjug. Chem. 2011, 22, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Leurs, U.; Mezo, G.; Orban, E.; Ohlschlager, P.; Marquardt, A.; Manea, M. Design, synthesis, in vitro stability and cytostatic effect of multifunctional anticancer drug-bioconjugates containing GnRH-III as a targeting moiety. Biopolymers 2012, 98, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Leurs, U.; Lajko, E.; Mezo, G.; Orban, E.; Ohlschlager, P.; Marquardt, A.; Kohidai, L.; Manea, M. GnRH-III based multifunctional drug delivery systems containing daunorubicin and methotrexate. Eur. J. Med. Chem. 2012, 52, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Schreier, V.N.; Mezo, G.; Orban, E.; Durr, C.; Marquardt, A.; Manea, M. Synthesis, enzymatic stability and in vitro cytostatic effect of Daunorubicin-GnRH-III derivative dimers. Bioorg. Med. Chem. Lett. 2013, 23, 2145–2150. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Biri-Kovacs, B.; Szeder, B.; Buday, L.; Gardi, J.; Szabo, Z.; Halmos, G.; Mezo, G. Enhanced in vitro antitumor activity of GnRH-III-daunorubicin bioconjugates influenced by sequence modification. Pharmaceutics 2018, 10, 223. [Google Scholar] [CrossRef]
- Marelli, M.M.; Manea, M.; Moretti, R.M.; Marzagalli, M.; Limonta, P. Oxime bond-linked daunorubicin-GnRH-III bioconjugates exert antitumor activity in castration-resistant prostate cancer cells via the type I GnRH receptor. Int. J. Oncol. 2014, 46, 243–253. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).