Current Genetic Survey and Potential Gene-Targeting Therapeutics for Neuromuscular Diseases

 , ,
, ,  and
and
Abstract
1. Introduction
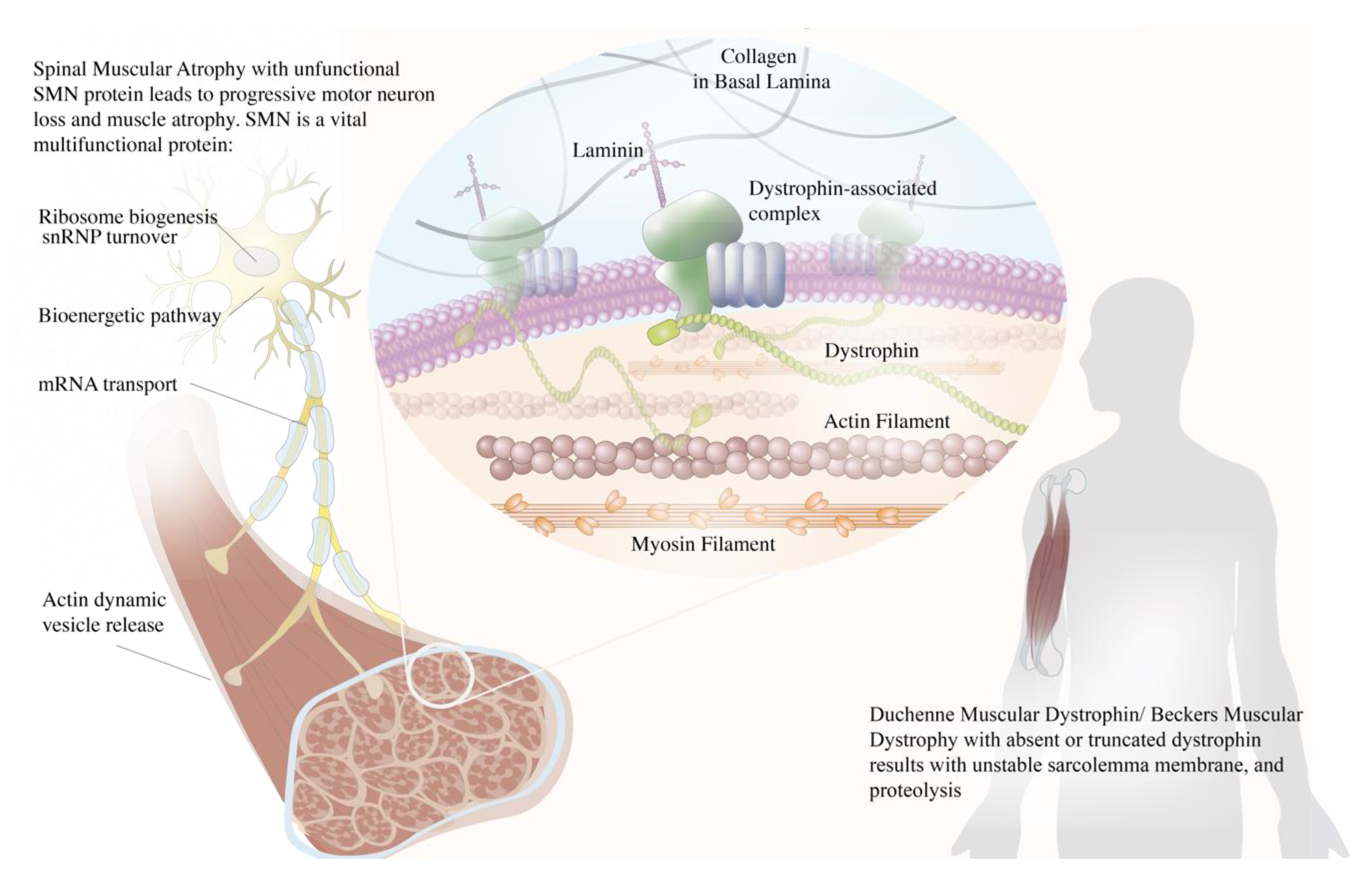
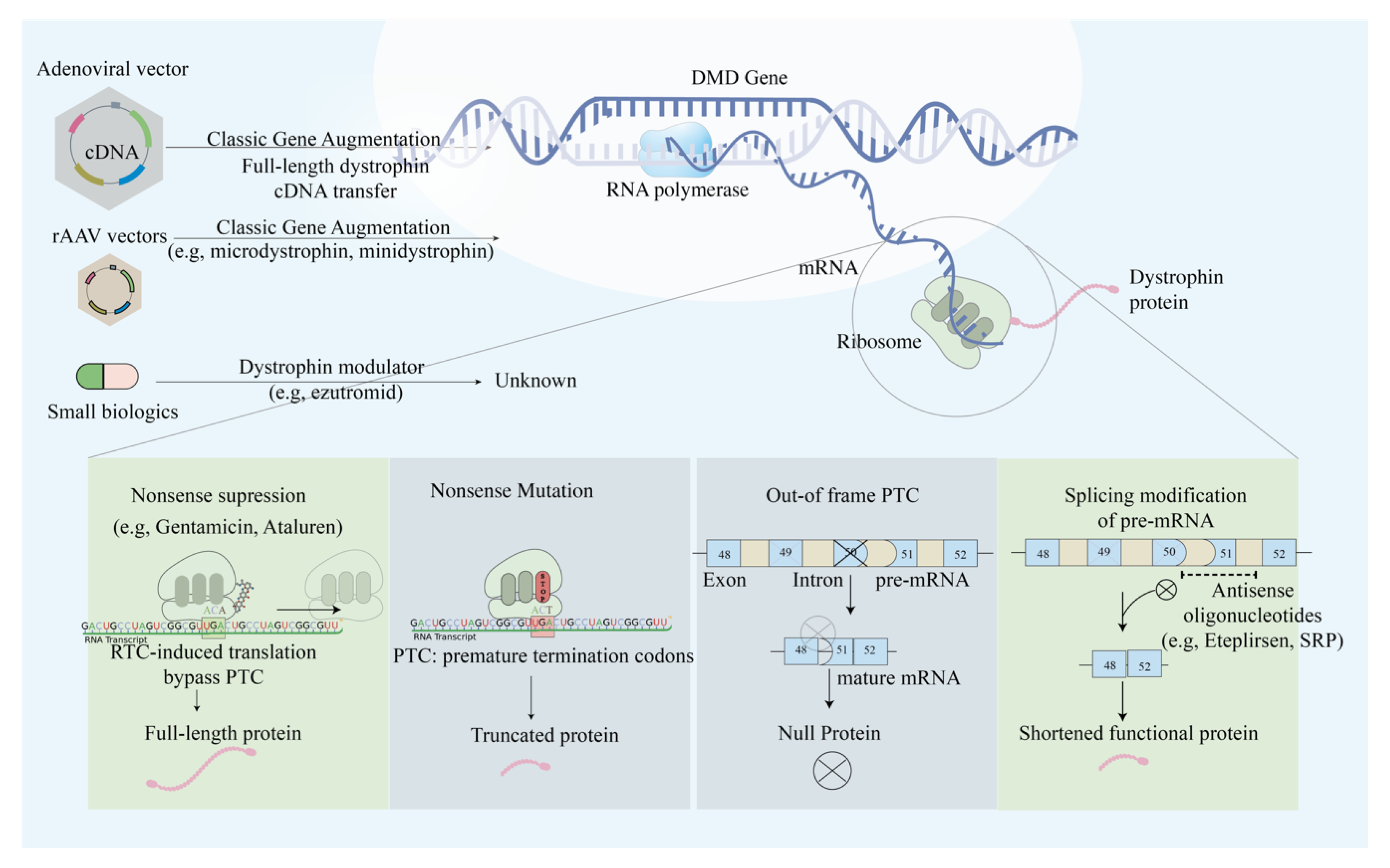
2. Duchenne Muscular Dystrophy (DMD) and Becker Muscular Dystrophy (BMD)
2.1. Single Exon Skipping
2.2. Multi-Exon Skipping
2.3. Mutation Suppression by Readthrough of Stop Codons
2.4. AAV-Based Mini- and Micro-Dystrophin Gene Transfer
2.5. Anti-Myostatin Therapy
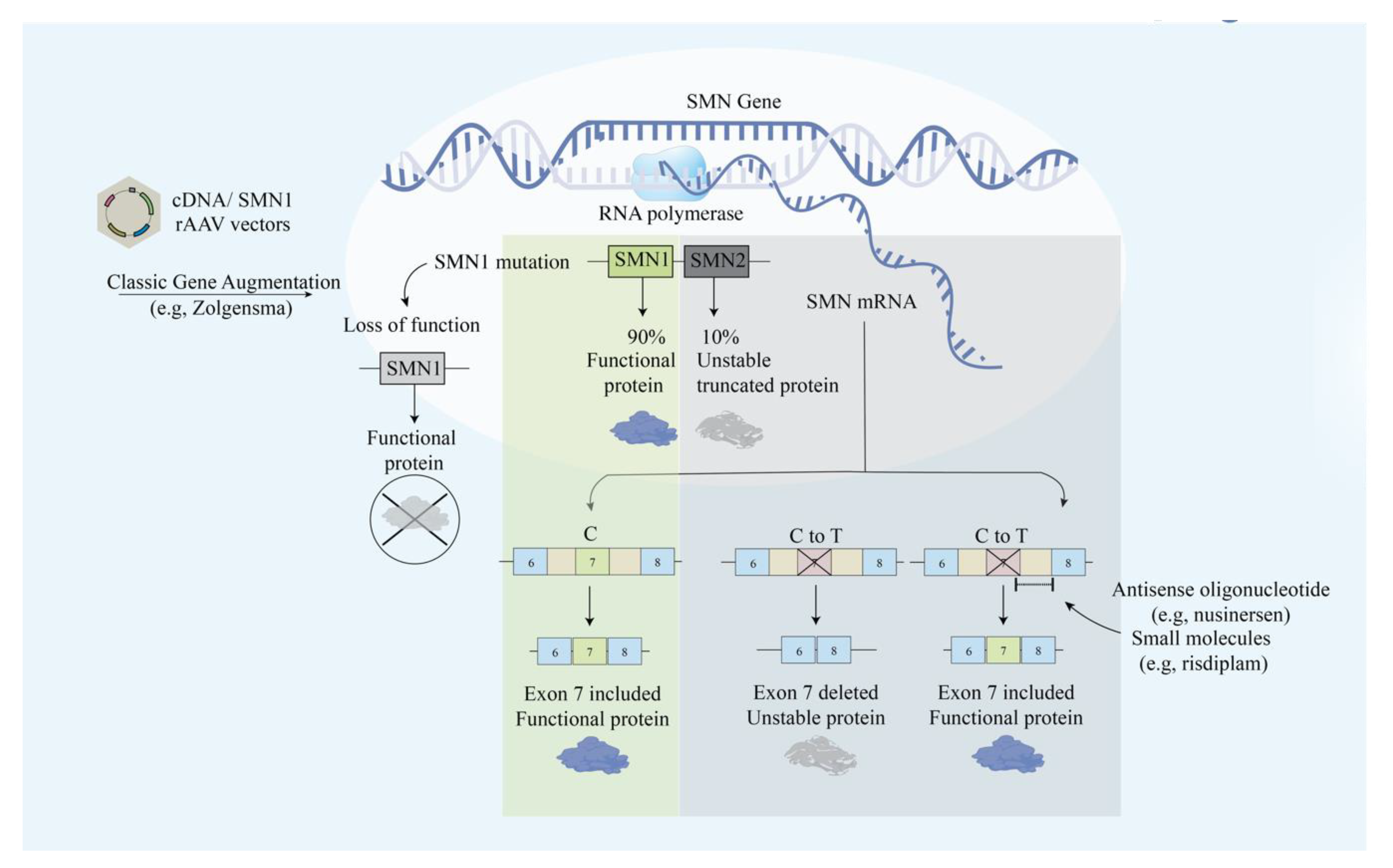
3. Spinal Muscular Atrophy (SMA)
3.1. Modification of SMN2 Alternative Splicing
3.2. AAV-SMN
4. X-Linked Myotubular Myopathy (XLMTM)
4.1. AAV-MTM1
4.2. Antisense Oligonucleotides
5. Limb-Girdle Muscular Dystrophy Type 2C (LGMD2C)
5.1. Multi-Exon Skipping
5.2. AAV-γ-SGC
6. Facioscapulohumeral Muscular Dystrophy (FSHD)
6.1. RNA Interference through AAV Vector
6.2. Antisense Oligonucleotides (AOs) and Phosphorodiamidate Morpholino Oligomers (PMOs)
6.2.1. Modulation of PITX1
6.2.2. Pre-mRNA Level
7. Discussion
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bellayou, H.; Hamzi, K.; Rafai, M.A.; Karkouri, M.; Slassi, I.; Azeddoug, H.; Nadifi, S. Duchenne and Becker muscular dystrophy: Contribution of a molecular and immunohistochemical analysis in diagnosis in Morocco. J. Biomed. Biotechnol. 2009, 2009, 325210. [Google Scholar] [CrossRef]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Athanasopoulos, T.; Graham, I.R.; Foster, H.; Dickson, G. Recombinant adeno-associated viral (rAAV) vectors as therapeutic tools for Duchenne muscular dystrophy (DMD). Gene Ther. 2004, 11 (Suppl. 1), S109–S121. [Google Scholar] [CrossRef][Green Version]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [PubMed]
- Zhu, J.F.; Liu, H.H.; Zhou, T.; Tian, L. Novel mutation in exon 56 of the dystrophin gene in a child with Duchenne muscular dystrophy. Int. J. Mol. Med. 2013, 32, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Ciafaloni, E.; Kumar, A.; Liu, K.; Pandya, S.; Westfield, C.; Fox, D.J.; Caspers Conway, K.M.; Cunniff, C.; Mathews, K.; West, N.; et al. Age at onset of first signs or symptoms predicts age at loss of ambulation in Duchenne and Becker Muscular Dystrophy: Data from the MD STARnet. J. Pediatr. Rehabil. Med. 2016, 9, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Werneck, L.C.; Lorenzoni, P.J.; Ducci, R.D.; Fustes, O.H.; Kay, C.S.K.; Scola, R.H. Duchenne muscular dystrophy: An historical treatment review. Arq. Neuropsiquiatr. 2019, 77, 579–589. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Straub, V.; Hemmings, R.; Haas, M.; Schlosser-Weber, G.; Stoyanova-Beninska, V.; Mercuri, E.; Muntoni, F.; Sepodes, B.; Vroom, E.; et al. Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Perspective on the Outstanding Issues. Nucleic Acid Ther. 2017, 27, 251–259. [Google Scholar] [CrossRef]
- Echigoya, Y.; Lim, K.R.Q.; Nakamura, A.; Yokota, T. Multiple Exon Skipping in the Duchenne Muscular Dystrophy Hot Spots: Prospects and Challenges. J. Pers. Med. 2018, 8, 41. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Janson, A.A.; Kaman, W.E.; Bremmer-Bout, M.; den Dunnen, J.T.; Baas, F.; van Ommen, G.J.; van Deutekom, J.C. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum. Mol. Genet. 2003, 12, 907–914. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Bronson, A.; Levin, A.A.; Takeda, S.; Yokota, T.; Baudy, A.R.; Connor, E.M. Restoring dystrophin expression in duchenne muscular dystrophy muscle progress in exon skipping and stop codon read through. Am. J. Pathol. 2011, 179, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Aupy, P.; Goyenvalle, A. Exon-skipping advances for Duchenne muscular dystrophy. Hum. Mol. Genet. 2018, 27, R163–R172. [Google Scholar] [CrossRef] [PubMed]
- van Deutekom, J.C.; Bremmer-Bout, M.; Janson, A.A.; Ginjaar, I.B.; Baas, F.; den Dunnen, J.T.; van Ommen, G.J. Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum. Mol. Genet. 2001, 10, 1547–1554. [Google Scholar] [CrossRef]
- Sharp, P.S.; Bye-a-Jee, H.; Wells, D.J. Physiological characterization of muscle strength with variable levels of dystrophin restoration in mdx mice following local antisense therapy. Mol. Ther. 2011, 19, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Babic, D.; Rocic, B.; Granic, M.; Skrabalo, Z. Decreased free insulin levels during euglycemic hyperinsulinemic clamp in non-insulin dependent diabetics on insulin therapy. Horm. Metab. Res. 1991, 23, 565–566. [Google Scholar] [CrossRef]
- Goemans, N.; Mercuri, E.; Belousova, E.; Komaki, H.; Dubrovsky, A.; McDonald, C.M.; Kraus, J.E.; Lourbakos, A.; Lin, Z.; Campion, G.; et al. A randomized placebo-controlled phase 3 trial of an antisense oligonucleotide, drisapersen, in Duchenne muscular dystrophy. Neuromuscul. Disord. 2018, 28, 4–15. [Google Scholar] [CrossRef]
- Echigoya, Y.; Yokota, T. Skipping multiple exons of dystrophin transcripts using cocktail antisense oligonucleotides. Nucleic Acid Ther. 2014, 24, 57–68. [Google Scholar] [CrossRef]
- Beroud, C.; Tuffery-Giraud, S.; Matsuo, M.; Hamroun, D.; Humbertclaude, V.; Monnier, N.; Moizard, M.P.; Voelckel, M.A.; Calemard, L.M.; Boisseau, P.; et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum. Mutat. 2007, 28, 196–202. [Google Scholar] [CrossRef]
- Taglia, A.; Petillo, R.; D’Ambrosio, P.; Picillo, E.; Torella, A.; Orsini, C.; Ergoli, M.; Scutifero, M.; Passamano, L.; Palladino, A.; et al. Clinical features of patients with dystrophinopathy sharing the 45-55 exon deletion of DMD gene. Acta Myol. 2015, 34, 9–13. [Google Scholar] [PubMed]
- Nakamura, A.; Shiba, N.; Miyazaki, D.; Nishizawa, H.; Inaba, Y.; Fueki, N.; Maruyama, R.; Echigoya, Y.; Yokota, T. Comparison of the phenotypes of patients harboring in-frame deletions starting at exon 45 in the Duchenne muscular dystrophy gene indicates potential for the development of exon skipping therapy. J. Hum. Genet. 2017, 62, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Echigoya, Y.; Duddy, W.; Saito, T.; Aoki, Y.; Takeda, S.; Yokota, T. Antisense PMO cocktails effectively skip dystrophin exons 45-55 in myotubes transdifferentiated from DMD patient fibroblasts. PLoS ONE 2018, 13, e0197084. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Lim, K.R.Q.; Melo, D.; Bao, B.; Trieu, N.; Mizobe, Y.; Maruyama, R.; Mamchaoui, K.; Tanihata, J.; Aoki, Y.; et al. Exons 45-55 Skipping Using Mutation-Tailored Cocktails of Antisense Morpholinos in the DMD Gene. Mol. Ther. 2019, 27, 2005–2017. [Google Scholar] [CrossRef] [PubMed]
- Aslesh, T.; Maruyama, R.; Yokota, T. Skipping Multiple Exons to Treat DMD-Promises and Challenges. Biomedicines 2018, 6, 1. [Google Scholar] [CrossRef]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of more than 7000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- Bushby, K.; Finkel, R.; Wong, B.; Barohn, R.; Campbell, C.; Comi, G.P.; Connolly, A.M.; Day, J.W.; Flanigan, K.M.; Goemans, N.; et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014, 50, 477–487. [Google Scholar] [CrossRef]
- Barton-Davis, E.R.; Cordier, L.; Shoturma, D.I.; Leland, S.E.; Sweeney, H.L. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Investig. 1999, 104, 375–381. [Google Scholar] [CrossRef]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef]
- Finkel, R.S.; Flanigan, K.M.; Wong, B.; Bonnemann, C.; Sampson, J.; Sweeney, H.L.; Reha, A.; Northcutt, V.J.; Elfring, G.; Barth, J.; et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS ONE 2013, 8, e81302. [Google Scholar] [CrossRef]
- Labella, M.H.; Narayan, A.J.; McCormick, C.M.; Desjardins, C.D.; Masten, A.S. Risk and Adversity, Parenting Quality, and Children’s Social-Emotional Adjustment in Families Experiencing Homelessness. Child Dev. 2019, 90, 227–244. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- Campbell, C.; Barohn, R.J.; Bertini, E.; Chabrol, B.; Comi, G.P.; Darras, B.T.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Iannaccone, S.T.; et al. Meta-analyses of ataluren randomized controlled trials in nonsense mutation Duchenne muscular dystrophy. J. Comp. Eff. Res. 2020, 9, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Li, M.; Berger, S.; Meilak, M.; Rientjes, J.; Currie, P.D. Effect of Ataluren on dystrophin mutations. J. Cell Mol. Med. 2020, 24, 6680–6689. [Google Scholar] [CrossRef]
- Jarmin, S.; Kymalainen, H.; Popplewell, L.; Dickson, G. New developments in the use of gene therapy to treat Duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2014, 14, 209–230. [Google Scholar] [CrossRef]
- Crudele, J.M.; Chamberlain, J.S. AAV-based gene therapies for the muscular dystrophies. Hum. Mol. Genet. 2019, 28, R102–R107. [Google Scholar] [CrossRef]
- Duan, D. Dystrophin Gene Replacement and Gene Repair Therapy for Duchenne Muscular Dystrophy in 2016: An Interview. Hum. Gene. Ther. Clin. Dev. 2016, 27, 9–18. [Google Scholar] [CrossRef]
- Gregorevic, P.; Allen, J.M.; Minami, E.; Blankinship, M.J.; Haraguchi, M.; Meuse, L.; Finn, E.; Adams, M.E.; Froehner, S.C.; Murry, C.E.; et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med. 2006, 12, 787–789. [Google Scholar] [CrossRef]
- Wang, Z.; Kuhr, C.S.; Allen, J.M.; Blankinship, M.; Gregorevic, P.; Chamberlain, J.S.; Tapscott, S.J.; Storb, R. Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol. Ther. 2007, 15, 1160–1166. [Google Scholar] [CrossRef]
- Koo, T.; Okada, T.; Athanasopoulos, T.; Foster, H.; Takeda, S.; Dickson, G. Long-term functional adeno-associated virus-microdystrophin expression in the dystrophic CXMDj dog. J. Gene. Med. 2011, 13, 497–506. [Google Scholar] [CrossRef]
- Zhang, Y.; Duan, D. Novel mini-dystrophin gene dual adeno-associated virus vectors restore neuronal nitric oxide synthase expression at the sarcolemma. Hum. Gene. Ther. 2012, 23, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Pan, X.; Hakim, C.H.; Yang, H.T.; Yue, Y.; Zhang, K.; Terjung, R.L.; Duan, D. Microdystrophin ameliorates muscular dystrophy in the canine model of duchenne muscular dystrophy. Mol. Ther. 2013, 21, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yue, Y.; Li, L.; Hakim, C.H.; Zhang, K.; Thomas, G.D.; Duan, D. Dual AAV therapy ameliorates exercise-induced muscle injury and functional ischemia in murine models of Duchenne muscular dystrophy. Hum. Mol. Genet. 2013, 22, 3720–3729. [Google Scholar] [CrossRef] [PubMed]
- Le Guiner, C.; Montus, M.; Servais, L.; Cherel, Y.; Francois, V.; Thibaud, J.L.; Wary, C.; Matot, B.; Larcher, T.; Guigand, L.; et al. Forelimb treatment in a large cohort of dystrophic dogs supports delivery of a recombinant AAV for exon skipping in Duchenne patients. Mol. Ther. 2014, 22, 1923–1935. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.E.; McPhee, S.W.; Li, C.; Gray, S.J.; Samulski, J.J.; Camp, A.S.; Li, J.; Wang, B.; Monahan, P.E.; Rabinowitz, J.E.; et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol. Ther. 2012, 20, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. JAMA Neurol. 2020, 77, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Kodippili, K.; Hakim, C.H.; Pan, X.; Yang, H.T.; Yue, Y.; Zhang, Y.; Shin, J.H.; Yang, N.N.; Duan, D. Dual AAV Gene Therapy for Duchenne Muscular Dystrophy with a 7-kb Mini-Dystrophin Gene in the Canine Model. Hum. Gene. Ther. 2018, 29, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Chen, X.; Samulski, R.J.; Hirsch, M.L.; Li, C. Inhibition of antigen presentation during AAV gene therapy using virus peptides. Hum. Mol. Genet. 2018, 27, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Hakim, C.H.; Clement, N.; Wasala, L.P.; Yang, H.T.; Yue, Y.; Zhang, K.; Kodippili, K.; Adamson-Small, L.; Pan, X.; Schneider, J.S.; et al. Micro-dystrophin AAV Vectors Made by Transient Transfection and Herpesvirus System Are Equally Potent in Treating mdx Mouse Muscle Disease. Mol. Ther. Methods Clin. Dev. 2020, 18, 664–678. [Google Scholar] [CrossRef]
- Bogdanovich, S.; Krag, T.O.; Barton, E.R.; Morris, L.D.; Whittemore, L.A.; Ahima, R.S.; Khurana, T.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature 2002, 420, 418–421. [Google Scholar] [CrossRef]
- Wagner, K.R.; McPherron, A.C.; Winik, N.; Lee, S.J. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann. Neurol. 2002, 52, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Dumonceaux, J.; Marie, S.; Beley, C.; Trollet, C.; Vignaud, A.; Ferry, A.; Butler-Browne, G.; Garcia, L. Combination of myostatin pathway interference and dystrophin rescue enhances tetanic and specific force in dystrophic mdx mice. Mol. Ther. 2010, 18, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Fleckenstein, J.L.; Amato, A.A.; Barohn, R.J.; Bushby, K.; Escolar, D.M.; Flanigan, K.M.; Pestronk, A.; Tawil, R.; Wolfe, G.I.; et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann. Neurol. 2008, 63, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; McMillan, H.J.; Mah, J.K.; Tarnopolsky, M.; Selby, K.; McClure, T.; Wilson, D.M.; Sherman, M.L.; Escolar, D.; Attie, K.M. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial. Muscle Nerve 2017, 55, 458–464. [Google Scholar] [CrossRef]
- Mendell, J.R.; Sahenk, Z.; Malik, V.; Gomez, A.M.; Flanigan, K.M.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Meadows, E.; Lewis, S.; et al. A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol. Ther. 2015, 23, 192–201. [Google Scholar] [CrossRef]
- Mariot, V.; Joubert, R.; Hourde, C.; Feasson, L.; Hanna, M.; Muntoni, F.; Maisonobe, T.; Servais, L.; Bogni, C.; Le Panse, R.; et al. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat. Commun. 2017, 8, 1859. [Google Scholar] [CrossRef]
- Mariot, V.; Le Guiner, C.; Barthelemy, I.; Montus, M.; Blot, S.; Torelli, S.; Morgan, J.; Muntoni, F.; Voit, T.; Dumonceaux, J. Myostatin Is a Quantifiable Biomarker for Monitoring Pharmaco-gene Therapy in Duchenne Muscular Dystrophy. Mol. Ther. Methods Clin. Dev. 2020, 18, 415–421. [Google Scholar] [CrossRef]
- Rodino-Klapac, L.R.; Janssen, P.M.; Shontz, K.M.; Canan, B.; Montgomery, C.L.; Griffin, D.; Heller, K.; Schmelzer, L.; Handy, C.; Clark, K.R.; et al. Micro-dystrophin and follistatin co-delivery restores muscle function in aged DMD model. Hum. Mol. Genet. 2013, 22, 4929–4937. [Google Scholar] [CrossRef]
- Vasileva, S.; Popkhristova, E. Morphologic and histochemical changes in gingiva of children and youth with diabetes mellitus. Stomatologiia (Sofiia) 1975, 57, 181–187. [Google Scholar]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef]
- Lorson, M.A.; Lorson, C.L. SMN-inducing compounds for the treatment of spinal muscular atrophy. Future Med. Chem. 2012, 4, 2067–2084. [Google Scholar] [CrossRef] [PubMed]
- Palacino, J.; Swalley, S.E.; Song, C.; Cheung, A.K.; Shu, L.; Zhang, X.; Van Hoosear, M.; Shin, Y.; Chin, D.N.; Keller, C.G.; et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol. 2015, 11, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.N.; Singh, N.N. Mechanism of Splicing Regulation of Spinal Muscular Atrophy Genes. Adv. Neurobiol. 2018, 20, 31–61. [Google Scholar] [PubMed]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef]
- Messina, S.; Sframeli, M. New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. J. Clin. Med. 2020, 9, 2222. [Google Scholar] [CrossRef]
- Walter, M.C.; Wenninger, S.; Thiele, S.; Stauber, J.; Hiebeler, M.; Greckl, E.; Stahl, K.; Pechmann, A.; Lochmuller, H.; Kirschner, J.; et al. Safety and Treatment Effects of Nusinersen in Longstanding Adult 5q-SMA Type 3—A Prospective Observational Study. J. Neuromuscul. Dis. 2019, 6, 453–465. [Google Scholar] [CrossRef]
- Al-Zaidy, S.A.; Kolb, S.J.; Lowes, L.; Alfano, L.N.; Shell, R.; Church, K.R.; Nagendran, S.; Sproule, D.M.; Feltner, D.E.; Wells, C.; et al. AVXS-101 (Onasemnogene Abeparvovec) for SMA1: Comparative Study with a Prospective Natural History Cohort. J. Neuromuscul. Dis. 2019, 6, 307–317. [Google Scholar] [CrossRef]
- Matesanz, S.E.; Curry, C.; Gross, B.; Rubin, A.I.; Linn, R.; Yum, S.W.; Kichula, E.A. Clinical Course in a Patient With Spinal Muscular Atrophy Type 0 Treated With Nusinersen and Onasemnogene Abeparvovec. J. Child. Neurol. 2020, 35, 717–723. [Google Scholar] [CrossRef]
- Cheung, A.K.; Hurley, B.; Kerrigan, R.; Shu, L.; Chin, D.N.; Shen, Y.; O’Brien, G.; Sung, M.J.; Hou, Y.; Axford, J.; et al. Discovery of Small Molecule Splicing Modulators of Survival Motor Neuron-2 (SMN2) for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 11021–11036. [Google Scholar] [CrossRef]
- Shorrock, H.K.; Gillingwater, T.H.; Groen, E.J.N. Overview of Current Drugs and Molecules in Development for Spinal Muscular Atrophy Therapy. Drugs 2018, 78, 293–305. [Google Scholar] [CrossRef]
- Kletzl, H.; Marquet, A.; Gunther, A.; Tang, W.; Heuberger, J.; Groeneveld, G.J.; Birkhoff, W.; Mercuri, E.; Lochmuller, H.; Wood, C.; et al. The oral splicing modifier RG7800 increases full length survival of motor neuron 2 mRNA and survival of motor neuron protein: Results from trials in healthy adults and patients with spinal muscular atrophy. Neuromuscul. Disord. 2019, 29, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 ( SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef] [PubMed]
- Sturm, S.; Gunther, A.; Jaber, B.; Jordan, P.; Al Kotbi, N.; Parkar, N.; Cleary, Y.; Frances, N.; Bergauer, T.; Heinig, K.; et al. A phase 1 healthy male volunteer single escalating dose study of the pharmacokinetics and pharmacodynamics of risdiplam (RG7916, RO7034067), a SMN2 splicing modifier. Br. J. Clin. Pharmacol. 2019, 85, 181–193. [Google Scholar] [CrossRef]
- Rashnonejad, A.; Amini Chermahini, G.; Gunduz, C.; Onay, H.; Aykut, A.; Durmaz, B.; Baka, M.; Su, Q.; Gao, G.; Ozkinay, F. Fetal Gene Therapy Using a Single Injection of Recombinant AAV9 Rescued SMA Phenotype in Mice. Mol. Ther. 2019, 27, 2123–2133. [Google Scholar] [CrossRef] [PubMed]
- Boido, M.; Vercelli, A. Neuromuscular Junctions as Key Contributors and Therapeutic Targets in Spinal Muscular Atrophy. Front. Neuroanat. 2016, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Kaifer, K.A.; Villalon, E.; Smith, C.E.; Simon, M.E.; Marquez, J.; Hopkins, A.E.; Morcos, T.I.; Lorson, C.L. AAV9-DOK7 gene therapy reduces disease severity in Smn(2B/-) SMA model mice. Biochem. Biophys. Res. Commun. 2020, 530, 107–114. [Google Scholar] [CrossRef]
- Dowling, J.J.; Lawlor, M.W.; Das, S. X-Linked Myotubular Myopathy, in GeneReviews((R)); Pagon, R.A., Adam, M.P., Ardinger, H.H., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Gonorazky, H.D.; Bonnemann, C.G.; Dowling, J.J. The genetics of congenital myopathies. Handb. Clin. Neurol. 2018, 148, 549–564. [Google Scholar]
- Graham, R.J.; Muntoni, F.; Hughes, I.; Yum, S.W.; Kuntz, N.L.; Yang, M.L.; Byrne, B.J.; Prasad, S.; Alvarez, R.; Genetti, C.A.; et al. Mortality and respiratory support in X-linked myotubular myopathy: A RECENSUS retrospective analysis. Arch. Dis. Child. 2020, 105, 332–338. [Google Scholar] [CrossRef]
- Lawlor, M.W.; Beggs, A.H.; Buj-Bello, A.; Childers, M.K.; Dowling, J.J.; James, E.S.; Meng, H.; Moore, S.A.; Prasad, S.; Schoser, B.; et al. Skeletal Muscle Pathology in X-Linked Myotubular Myopathy: Review With Cross-Species Comparisons. J. Neuropathol. Exp. Neurol. 2016, 75, 102–110. [Google Scholar] [CrossRef]
- Tanner, S.M.; Laporte, J.; Guiraud-Chaumeil, C.; Liechti-Gallati, S. Confirmation of prenatal diagnosis results of X-linked recessive myotubular myopathy by mutational screening, and description of three new mutations in the MTM1 gene. Hum. Mutat. 1998, 11, 62–68. [Google Scholar] [CrossRef]
- Buj-Bello, A.; Fougerousse, F.; Schwab, Y.; Messaddeq, N.; Spehner, D.; Pierson, C.R.; Durand, M.; Kretz, C.; Danos, O.; Douar, A.M.; et al. AAV-mediated intramuscular delivery of myotubularin corrects the myotubular myopathy phenotype in targeted murine muscle and suggests a function in plasma membrane homeostasis. Hum. Mol. Genet. 2008, 17, 2132–2143. [Google Scholar] [CrossRef] [PubMed]
- Childers, M.K.; Joubert, R.; Poulard, K.; Moal, C.; Grange, R.W.; Doering, J.A.; Lawlor, M.W.; Rider, B.E.; Jamet, T.; Daniele, N.; et al. Gene therapy prolongs survival and restores function in murine and canine models of myotubular myopathy. Sci. Transl. Med. 2014, 6, 220ra10. [Google Scholar] [CrossRef] [PubMed]
- Elverman, M.; Goddard, M.A.; Mack, D.; Snyder, J.M.; Lawlor, M.W.; Meng, H.; Beggs, A.H.; Buj-Bello, A.; Poulard, K.; Marsh, A.P.; et al. Long-term effects of systemic gene therapy in a canine model of myotubular myopathy. Muscle Nerve 2017, 56, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Jamett, A.M.; Momboisse, F.; Haro-Acuna, V.; Bevilacqua, J.A.; Caviedes, P.; Cardenas, A.M. Dynamin-2 function and dysfunction along the secretory pathway. Front. Endocrinol. 2013, 4, 126. [Google Scholar] [CrossRef] [PubMed]
- Cowling, B.S.; Chevremont, T.; Prokic, I.; Kretz, C.; Ferry, A.; Coirault, C.; Koutsopoulos, O.; Laugel, V.; Romero, N.B.; Laporte, J. Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J. Clin. Investig. 2014, 124, 1350–1363. [Google Scholar] [CrossRef]
- Buono, S.; Ross, J.A.; Tasfaout, H.; Levy, Y.; Kretz, C.; Tayefeh, L.; Matson, J.; Guo, S.; Kessler, P.; Monia, B.P.; et al. Reducing dynamin 2 (DNM2) rescues DNM2-related dominant centronuclear myopathy. Proc. Natl. Acad. Sci. USA 2018, 115, 11066–11071. [Google Scholar] [CrossRef]
- Tasfaout, H.; Buono, S.; Guo, S.; Kretz, C.; Messaddeq, N.; Booten, S.; Greenlee, S.; Monia, B.P.; Cowling, B.S.; Laporte, J. Antisense oligonucleotide-mediated Dnm2 knockdown prevents and reverts myotubular myopathy in mice. Nat. Commun. 2017, 8, 15661. [Google Scholar] [CrossRef]
- Ozawa, E.; Mizuno, Y.; Hagiwara, Y.; Sasaoka, T.; Yoshida, M. Molecular and cell biology of the sarcoglycan complex. Muscle Nerve 2005, 32, 563–576. [Google Scholar] [CrossRef]
- Rhoades, K.L.; Golub, S.H.; Economou, J.S. The regulation of the human tumor necrosis factor alpha promoter region in macrophage, T cell, and B cell lines. J. Biol. Chem. 1992, 267, 22102–22107. [Google Scholar]
- Wyatt, E.J.; Demonbreun, A.R.; Kim, E.Y.; Puckelwartz, M.J.; Vo, A.H.; Dellefave-Castillo, L.M.; Gao, Q.Q.; Vainzof, M.; Pavanello, R.C.M.; Zatz, M.; et al. Efficient exon skipping of SGCG mutations mediated by phosphorodiamidate morpholino oligomers. JCI Insight 2018, 3, e99357. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; Wyatt, E.J.; Fallon, K.S.; Oosterbaan, C.C.; Page, P.G.; Hadhazy, M.; Quattrocelli, M.; Barefield, D.Y.; McNally, E.M. A gene-edited mouse model of limb-girdle muscular dystrophy 2C for testing exon skipping. Dis. Model. Mech. 2019, 13. [Google Scholar] [CrossRef]
- Herson, S.; Hentati, F.; Rigolet, A.; Behin, A.; Romero, N.B.; Leturcq, F.; Laforet, P.; Maisonobe, T.; Amouri, R.; Haddad, H.; et al. A phase I trial of adeno-associated virus serotype 1-gamma-sarcoglycan gene therapy for limb girdle muscular dystrophy type 2C. Brain 2012, 135 Pt 2, 483–492. [Google Scholar] [CrossRef]
- Israeli, D.; Cosette, J.; Corre, G.; Amor, F.; Poupiot, J.; Stockholm, D.; Montus, M.; Gjata, B.; Richard, I. An AAV-SGCG Dose-Response Study in a gamma-Sarcoglycanopathy Mouse Model in the Context of Mechanical Stress. Mol. Ther. Methods Clin. Dev. 2019, 13, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Deenen, J.C.; Horlings, C.G.; Verschuuren, J.J.; Verbeek, A.L.; van Engelen, B.G. The Epidemiology of Neuromuscular Disorders: A Comprehensive Overview of the Literature. J. Neuromuscul. Dis. 2015, 2, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Tawil, R.; Kissel, J.T.; Heatwole, C.; Pandya, S.; Gronseth, G.; Benatar, M.; Guideline Development, Department; Implementation Subcommittee of the American Academy of Neurology; Practice Issues Review Panel of the American Association of Neurology; Electrodiagnostic Medicine. Evidence-based guideline summary: Evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology 2015, 85, 357–364. [Google Scholar] [PubMed]
- Statland, J.M.; Tawil, R. Facioscapulohumeral Muscular Dystrophy. Continuum 2016, 22, 1916–1931. [Google Scholar]
- Lutz, K.L.; Holte, L.; Kliethermes, S.A.; Stephan, C.; Mathews, K.D. Clinical and genetic features of hearing loss in facioscapulohumeral muscular dystrophy. Neurology 2013, 81, 1374–1377. [Google Scholar] [CrossRef]
- Dandapat, A.; Perrin, B.J.; Cabelka, C.; Razzoli, M.; Ervasti, J.M.; Bartolomucci, A.; Lowe, D.A.; Kyba, M. High Frequency Hearing Loss and Hyperactivity in DUX4 Transgenic Mice. PLoS ONE 2016, 11, e0151467. [Google Scholar] [CrossRef]
- Osborne, R.J.; Welle, S.; Venance, S.L.; Thornton, C.A.; Tawil, R. Expression profile of FSHD supports a link between retinal vasculopathy and muscular dystrophy. Neurology 2007, 68, 569–577. [Google Scholar] [CrossRef]
- Padberg, G.W.; Brouwer, O.F.; de Keizer, R.J.; Dijkman, G.; Wijmenga, C.; Grote, J.J.; Frants, R.R. On the significance of retinal vascular disease and hearing loss in facioscapulohumeral muscular dystrophy. Muscle Nerve 1995, 18 (Suppl. 13), S73–S80. [Google Scholar] [CrossRef]
- Statland, J.M.; Sacconi, S.; Farmakidis, C.; Donlin-Smith, C.M.; Chung, M.; Tawil, R. Coats syndrome in facioscapulohumeral dystrophy type 1: Frequency and D4Z4 contraction size. Neurology 2013, 80, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Gabriels, J.; Beckers, M.C.; Ding, H.; De Vriese, A.; Plaisance, S.; van der Maarel, S.M.; Padberg, G.W.; Frants, R.R.; Hewitt, J.E.; Collen, D.; et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 1999, 236, 25–32. [Google Scholar] [CrossRef]
- Kowaljow, V.; Marcowycz, A.; Ansseau, E.; Conde, C.B.; Sauvage, S.; Matteotti, C.; Arias, C.; Corona, E.D.; Nunez, N.G.; Leo, O.; et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul. Disord. 2007, 17, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Snider, L.; Asawachaicharn, A.; Tyler, A.E.; Geng, L.N.; Petek, L.M.; Maves, L.; Miller, D.G.; Lemmers, R.J.; Winokur, S.T.; Tawil, R.; et al. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: New candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum. Mol. Genet. 2009, 18, 2414–2430. [Google Scholar] [CrossRef]
- Wijmenga, C.; Hewitt, J.E.; Sandkuijl, L.A.; Clark, L.N.; Wright, T.J.; Dauwerse, H.G.; Gruter, A.M.; Hofker, M.H.; Moerer, P.; Williamson, R.; et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat. Genet. 1992, 2, 26–30. [Google Scholar] [CrossRef]
- van Deutekom, J.C.; Wijmenga, C.; van Tienhoven, E.A.; Gruter, A.M.; Hewitt, J.E.; Padberg, G.W.; van Ommen, G.J.; Hofker, M.H.; Frants, R.R. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 1993, 2, 2037–2042. [Google Scholar] [CrossRef]
- Lemmers, R.J.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.; Amell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012, 44, 1370–1374. [Google Scholar] [CrossRef]
- van den Boogaard, M.L.; Lemmers, R.; Balog, J.; Wohlgemuth, M.; Auranen, M.; Mitsuhashi, S.; van der Vliet, P.J.; Straasheijm, K.R.; van den Akker, R.F.P.; Kriek, M.; et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016, 98, 1020–1029. [Google Scholar] [CrossRef]
- Lemmers, R.J.; de Kievit, P.; Sandkuijl, L.; Padberg, G.W.; van Ommen, G.J.; Frants, R.R.; van der Maarel, S.M. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat. Genet. 2002, 32, 235–236. [Google Scholar] [CrossRef]
- Lemmers, R.J.; Wohlgemuth, M.; van der Gaag, K.J.; van der Vliet, P.J.; van Teijlingen, C.M.; de Knijff, P.; Padberg, G.W.; Frants, R.R.; van der Maarel, S.M. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2007, 81, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Lemmers, R.J.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camano, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Dixit, M.; Ansseau, E.; Tassin, A.; Winokur, S.; Shi, R.; Qian, H.; Sauvage, S.; Matteotti, C.; van Acker, A.M.; Leo, O.; et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl. Acad. Sci. USA 2007, 104, 18157–18162. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.N.; Cabotage, J.; Shi, R.; Dixit, M.; Sutherland, M.; Liu, J.; Muger, S.; Harper, S.Q.; Nagaraju, K.; Chen, Y.W. Conditional over-expression of PITX1 causes skeletal muscle dystrophy in mice. Biol. Open 2012, 1, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Tassin, A.; Laoudj-Chenivesse, D.; Vanderplanck, C.; Barro, M.; Charron, S.; Ansseau, E.; Chen, Y.W.; Mercier, J.; Coppee, F.; Belayew, A. DUX4 expression in FSHD muscle cells: How could such a rare protein cause a myopathy? J. Cell. Mol. Med. 2013, 17, 76–89. [Google Scholar] [CrossRef]
- Oliva, J.; Galasinski, S.; Richey, A.; Campbell, A.E.; Meyers, M.J.; Modi, N.; Zhong, J.W.; Tawil, R.; Tapscott, S.J.; Sverdrup, F.M. Clinically Advanced p38 Inhibitors Suppress DUX4 Expression in Cellular and Animal Models of Facioscapulohumeral Muscular Dystrophy. J. Pharmacol. Exp. Ther. 2019, 370, 219–230. [Google Scholar] [CrossRef]
- Rojas, L.A.; Valentine, E.; Accorsi, A.; Maglio, J.; Shen, N.; Robertson, A.; Kazmirski, S.; Rahl, P.; Tawil, R.; Cadavid, D.; et al. p38alpha Regulates Expression of DUX4 in a Model of Facioscapulohumeral Muscular Dystrophy. J. Pharmacol. Exp. Ther. 2020, 374, 489–498. [Google Scholar] [CrossRef]
- DeSimone, A.M.; Leszyk, J.; Wagner, K.; Emerson, C.P., Jr. Identification of the hyaluronic acid pathway as a therapeutic target for facioscapulohumeral muscular dystrophy. Sci. Adv. 2019, 5, eaaw7099. [Google Scholar] [CrossRef]
- Ciszewski, L.; Lu-Nguyen, N.; Slater, A.; Brennan, A.; Williams, H.E.L.; Dickson, G.; Searle, M.S.; Popplewell, L. G-quadruplex ligands mediate downregulation of DUX4 expression. Nucleic Acids Res. 2020, 48, 4179–4194. [Google Scholar] [CrossRef]
- Hamel, J.; Tawil, R. Facioscapulohumeral Muscular Dystrophy: Update on Pathogenesis and Future Treatments. Neurotherapeutics 2018, 15, 863–871. [Google Scholar] [CrossRef]
- Wallace, L.M.; Garwick-Coppens, S.E.; Tupler, R.; Harper, S.Q. RNA interference improves myopathic phenotypes in mice over-expressing FSHD region gene 1 (FRG1). Mol. Ther. 2011, 19, 2048–2054. [Google Scholar] [CrossRef]
- Wallace, L.M.; Liu, J.; Domire, J.S.; Garwick-Coppens, S.E.; Guckes, S.M.; Mendell, J.R.; Flanigan, K.M.; Harper, S.Q. RNA interference inhibits DUX4-induced muscle toxicity in vivo: Implications for a targeted FSHD therapy. Mol. Ther. 2012, 20, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Bortolanza, S.; Nonis, A.; Sanvito, F.; Maciotta, S.; Sitia, G.; Wei, J.; Torrente, Y.; Di Serio, C.; Chamberlain, J.R.; Gabellini, D. AAV6-mediated systemic shRNA delivery reverses disease in a mouse model of facioscapulohumeral muscular dystrophy. Mol. Ther. 2011, 19, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- Wallace, L.M.; Saad, N.Y.; Pyne, N.K.; Fowler, A.M.; Eidahl, J.O.; Domire, J.S.; Griffin, D.A.; Herman, A.C.; Sahenk, Z.; Rodino-Klapac, L.R.; et al. Pre-clinical Safety and Off-Target Studies to Support Translation of AAV-Mediated RNAi Therapy for FSHD. Mol. Ther. Methods Clin. Dev. 2018, 8, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.N.; Lee, Y.C.; Yokota, T.; Chen, Y.W. Morpholino treatment improves muscle function and pathology of Pitx1 transgenic mice. Mol. Ther. 2014, 22, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; King, O.D.; Zhang, Y.; Clayton, N.P.; Spencer, C.; Wentworth, B.M.; Emerson, C.P., Jr.; Wagner, K.R. Morpholino-mediated Knockdown of DUX4 Toward Facioscapulohumeral Muscular Dystrophy Therapeutics. Mol. Ther. 2016, 24, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Ansseau, E.; Vanderplanck, C.; Wauters, A.; Harper, S.Q.; Coppee, F.; Belayew, A. Antisense Oligonucleotides Used to Target the DUX4 mRNA as Therapeutic Approaches in FaciosScapuloHumeral Muscular Dystrophy (FSHD). Genes 2017, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Bittel, A.; Maruyama, R.; Echigoya, Y.; Nguyen, Q.; Huang, Y.; Dzierlega, K.; Zhang, A.; Chen, Y.W.; Yokota, T. DUX4 Transcript Knockdown with Antisense 2′-O-Methoxyethyl Gapmers for the Treatment of Facioscapulohumeral Muscular Dystrophy. Mol. Ther. 2020. [Google Scholar] [CrossRef]
- Grieger, J.C.; Samulski, R.J. Packaging capacity of adeno-associated virus serotypes: Impact of larger genomes on infectivity and postentry steps. J. Virol. 2005, 79, 9933–9944. [Google Scholar] [CrossRef]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef]
- Long, C.; McAnally, J.R.; Shelton, J.M.; Mireault, A.A.; Bassel-Duby, R.; Olson, E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 2014, 345, 1184–1188. [Google Scholar] [CrossRef]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015, 6, 6244. [Google Scholar] [CrossRef]
- Xu, L.; Park, K.H.; Zhao, L.; Xu, J.; El Refaey, M.; Gao, Y.; Zhu, H.; Ma, J.; Han, R. CRISPR-mediated Genome Editing Restores Dystrophin Expression and Function in mdx Mice. Mol. Ther. 2016, 24, 564–569. [Google Scholar] [CrossRef]
- Aoki, Y.; Nakamura, A.; Yokota, T.; Saito, T.; Okazawa, H.; Nagata, T.; Takeda, S. In-frame dystrophin following exon 51-skipping improves muscle pathology and function in the exon 52-deficient mdx mouse. Mol. Ther. 2010, 18, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Arechavala-Gomeza, V.; Graham, I.R.; Popplewell, L.J.; Adams, A.M.; Aartsma-Rus, A.; Kinali, M.; Morgan, J.E.; van Deutekom, J.C.; Wilton, S.D.; Dickson, G.; et al. Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscle. Hum. Gene. Ther. 2007, 18, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Magri, F.; Govoni, A.; D’Angelo, M.G.; Del Bo, R.; Ghezzi, S.; Sandra, G.; Turconi, A.C.; Sciacco, M.; Ciscato, P.; Bordoni, A.; et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J. Neurol. 2011, 258, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Kimura, E.; Mori-Yoshimura, M.; Komaki, H.; Matsuda, Y.; Goto, K.; Hayashi, Y.K.; Nishino, I.; Takeda, S.; Kawai, M. Characteristics of Japanese Duchenne and Becker muscular dystrophy patients in a novel Japanese national registry of muscular dystrophy (Remudy). Orphanet J. Rare Dis. 2013, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Van Deutekom, J.C.; Fokkema, I.F.; Van Ommen, G.J.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef]
- Mendell, J.R.; Rodino-Klapac, L.R. Duchenne muscular dystrophy: CRISPR/Cas9 treatment. Cell Res. 2016, 26, 513–514. [Google Scholar] [CrossRef]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef]
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.R.; Massey, C.; Shelton, J.M.; et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef]
- Ifuku, M.; Iwabuchi, K.A.; Tanaka, M.; Lung, M.S.Y.; Hotta, A. Restoration of Dystrophin Protein Expression by Exon Skipping Utilizing CRISPR-Cas9 in Myoblasts Derived from DMD Patient iPS Cells. Methods Mol. Biol. 2018, 1828, 191–217. [Google Scholar] [PubMed]
- Brescia, M.; Janssen, J.M.; Liu, J.; Goncalves, M. High-Capacity Adenoviral Vectors Permit Robust and Versatile Testing of DMD Gene Repair Tools and Strategies in Human Cells. Cells 2020, 9, 869. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.L.; Li, H.; Rodriguez-Caycedo, C.; Mireault, A.A.; Huang, J.; Shelton, J.M.; McAnally, J.R.; Amoasii, L.; Mammen, P.P.A.; Bassel-Duby, R.; et al. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019, 5, eaav4324. [Google Scholar] [CrossRef] [PubMed]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; January, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef]
- Kemaladewi, D.U.; Cohn, R.D. Exon Snipping in Duchenne Muscular Dystrophy. Trends. Mol. Med. 2016, 22, 187–189. [Google Scholar] [CrossRef]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef]
- Iyombe-Engembe, J.P.; Ouellet, D.L.; Barbeau, X.; Rousseau, J.; Chapdelaine, P.; Lague, P.; Tremblay, J.P. Efficient Restoration of the Dystrophin Gene Reading Frame and Protein Structure in DMD Myoblasts Using the CinDel Method. Mol. Ther. Nucleic Acids 2016, 5, e283. [Google Scholar] [CrossRef]
- Duchene, B.; Iyombe-Engembe, J.P.; Rousseau, J.; Tremblay, J.P.; Ouellet, D.L. From gRNA Identification to the Restoration of Dystrophin Expression: A Dystrophin Gene Correction Strategy for Duchenne Muscular Dystrophy Mutations Using the CRISPR-Induced Deletion Method. Methods Mol. Biol. 2018, 1687, 267–283. [Google Scholar]
- Amoasii, L.; Long, C.; Li, H.; Mireault, A.A.; Shelton, J.M.; Sanchez-Ortiz, E.; McAnally, J.R.; Bhattacharyya, S.; Schmidt, F.; Grimm, D.; et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017, 9, eaan8081. [Google Scholar] [CrossRef]
- Min, Y.L.; Chemello, F.; Li, H.; Rodriguez-Caycedo, C.; Sanchez-Ortiz, E.; Mireault, A.A.; McAnally, J.R.; Shelton, J.M.; Zhang, Y.; Bassel-Duby, R.; et al. Correction of Three Prominent Mutations in Mouse and Human Models of Duchenne Muscular Dystrophy by Single-Cut Genome Editing. Mol. Ther. 2020, 28, 2044–2055. [Google Scholar] [CrossRef]
- Maggio, I.; Liu, J.; Janssen, J.M.; Chen, X.; Goncalves, M.A. Adenoviral vectors encoding CRISPR/Cas9 multiplexes rescue dystrophin synthesis in unselected populations of DMD muscle cells. Sci. Rep. 2016, 6, 37051. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, A.; Murry, C.E. Imprecision Medicine: A One-Size-Fits-Many Approach for Muscle Dystrophy. Cell Stem Cell 2016, 18, 423–424. [Google Scholar] [CrossRef] [PubMed]
- Wojtal, D.; Kemaladewi, D.U.; Malam, Z.; Abdullah, S.; Wong, T.W.; Hyatt, E.; Baghestani, Z.; Pereira, S.; Stavropoulos, J.; Mouly, V.; et al. Spell Checking Nature: Versatility of CRISPR/Cas9 for Developing Treatments for Inherited Disorders. Am. J. Hum. Genet. 2016, 98, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, A.; Duguez, S.; Moiani, A.; Izmiryan, A.; Barbon, E.; Martin, S.; Mamchaoui, K.; Mouly, V.; Bernardi, F.; Mavilio, F.; et al. Correction of the Exon 2 Duplication in DMD Myoblasts by a Single CRISPR/Cas9 System. Mol. Ther. Nucleic Acids 2017, 7, 11–19. [Google Scholar] [CrossRef]
- Valetdinova, K.R.; Ovechkina, V.S.; Zakian, S.M. Methods for Correction of the Single-Nucleotide Substitution c.840C>T in Exon 7 of the SMN2 Gene. Biochemistry (Mosc) 2019, 84, 1074–1084. [Google Scholar] [CrossRef]
- Zhou, M.; Hu, Z.; Qiu, L.; Zhou, T.; Feng, M.; Hu, Q.; Zeng, B.; Li, Z.; Sun, Q.; Wu, Y.; et al. Seamless Genetic Conversion of SMN2 to SMN1 via CRISPR/Cpf1 and Single-Stranded Oligodeoxynucleotides in Spinal Muscular Atrophy Patient-Specific Induced Pluripotent Stem Cells. Hum. Gene. Ther. 2018, 29, 1252–1263. [Google Scholar] [CrossRef]
- Du, M.; Jillette, N.; Zhu, J.J.; Li, S.; Cheng, A.W. CRISPR artificial splicing factors. Nat. Commun. 2020, 11, 2973. [Google Scholar] [CrossRef]
- Selvaraj, S.; Dhoke, N.R.; Kiley, J.; Mateos-Aierdi, A.J.; Tungtur, S.; Mondragon-Gonzalez, R.; Killeen, G.; Oliveira, V.K.P.; Lopez de Munain, A.; Perlingeiro, R.C.R. Gene Correction of LGMD2A Patient-Specific iPSCs for the Development of Targeted Autologous Cell Therapy. Mol. Ther. 2019, 27, 2147–2157. [Google Scholar] [CrossRef]
- Iyer, S.; Suresh, S.; Guo, D.; Daman, K.; Chen, J.C.J.; Liu, P.; Zieger, M.; Luk, K.; Roscoe, B.P.; Mueller, C.; et al. Precise therapeutic gene correction by a simple nuclease-induced double-stranded break. Nature 2019, 568, 561–565. [Google Scholar] [CrossRef]
- Turan, S.; Farruggio, A.P.; Srifa, W.; Day, J.W.; Calos, M.P. Precise Correction of Disease Mutations in Induced Pluripotent Stem Cells Derived From Patients With Limb Girdle Muscular Dystrophy. Mol. Ther. 2016, 24, 685–696. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Class | Phase | Enrolled # | Therapeutics | Route | End Date | Start Date | Trial ID |
|---|---|---|---|---|---|---|---|
| Exon Skipping | 1/2 | 7 | Eteplirsen (AVI-4658/Exondys-51) | IM | December 2008 | October 2007 | NCT00159250 |
| 1/2 | 19 | Eteplirsen (AVI-4658/Exondys-51) | IV | December 2010 | January 2009 | NCT00844597 | |
| 2 | 12 | Eteplirsen(AVI-4658/Exondys-51) | IV | June 2012 | July 2011 | NCT01396239 | |
| 2 | 6 | Casimersen (skip exon45), Eteplirsen (skip exon51), Golodirsen (skip exon53) | IV | September 2021 | February 2020 | NCT04179409 | |
| 2 | 53 | Drisapersen (GSK2402968) | SC | September 2012 | September 2010 | NCT01153932 | |
| 2 | 51 | Drisapersen (GSK2402968) | SC | May 2013 | October 2011 | NCT01462292 | |
| 3 | 186 | Drisapersen (GSK2402968) | SC | June 2013 | December 2010 | NCT01254019 | |
| AAV gene transfer | 1 | 2 | SRP-9001 (rAAVrh74.MHCK.microdystrophin) | IM | September 2017 | March 2015 | NCT02376816 |
| 1 | 6 | d3990 (rAAV2.5-CMV-minidystrophin) | IM | March 2009 | March 2006 | NCT00428935 | |
| 1 | 15 | d3990 (rAAV2.5-CMV-minidystrophin) | IV | August 2021 | January 2018 | NCT03362502 | |
| 1//2 | 4 | PF-06939926 (AAV9- muscle specific promoter- minidystrophin) | IV | March 2020 | January 2018 | NCT03375164 | |
| 1//2 | 6 | SRP-9001 (rAAVrh74.MCK.microDystrophin) | IV | November 2020 | November 2017 | NCT03333590 | |
| 1//2 | 3 | rAAVrh74.MCK.GALGT2 | IM | November 2017 | January 2015 | NCT02354781 | |
| 1//2 | 3 | AAV1-F344 (rAAV1.CMV.huFollistin344) | IV | January 2023 | January 2020 | NCT04240314 | |
| 1//2 | 16 | scAAV9.U7.ACCA (skip exon2) | IV | March 2023 | December 2017 | NCT03368742 | |
| 2 | 41 | SGT-001 (AAV9- muscle specific promoter- microdystrophin) | IM | October 2022 | December 2018 | NCT03769116 | |
| 3 | 99 | SRP-9001 (rAAVrh74.MHCK.microdystrophin) | IV | June 2022 | July 2020 | NCT03368742 | |
| 1 | 2 | PF-06939926 (AAV9- muscle specific promoter- minidystrophin) | IM | September 2017 | March 2015 | NCT02376816 | |
| 1 | 6 | SRP-9001 (rAAVrh74.MHCK.microDystrophin) | IM | March 2009 | March 2006 | NCT00428935 | |
| 1 | 15 | d3990 (rAAV2.5-CMV-minidystrophin) | IV | August 2021 | January 2018 | NCT03362502 | |
| 1//2 | 4 | d3990 (rAAV2.5-CMV-minidystrophin) | IV | March 2020 | January 2018 | NCT03375164 | |
| 1//2 | 6 | PF-06939926 (AAV9- muscle specific promoter- minidystrophin) | IV | November 2020 | November 2017 | NCT03333590 | |
| Nonsense Suppression | 2 | 6 | Ataluren (PTC124) | PO | March 2010 | January 2010 | NCT01009294 |
| 2 | 14 | Ataluren (PTC124) | PO | February 2018 | June 2016 | NCT02819557 | |
| 2 | 36 | Ataluren (PTC124) | PO | May 2010 | July 2008 | NCT00759876 | |
| 3 | 95 | Ataluren (PTC124) | PO | January 2018 | May 2012 | NCT01557400 | |
| 2 | 173 | Ataluren (PTC124) | PO | May 2010 | January 2009 | NCT00847379 | |
| 2 | 174 | Ataluren (PTC124) | PO | December 2009 | February 2008 | NCT00592553 | |
| 2 | 21 | Arbekacin sulfate (NPC-14) | PO | October 2015 | August 2013 | NCT01918384 | |
| 2 | 38 | Ataluren (PTC124) | PO | May 2007 | December 2005 | NCT00264888 | |
| 3 | 230 | Ataluren (PTC124) | PO | August 2015 | March 2013 | NCT01826487 | |
| 2 | 6 | Ataluren (PTC124) | PO | March 2010 | January 2010 | NCT01009294 | |
| 2 | 14 | Ataluren (PTC124) | PO | February 2018 | June 2016 | NCT02819557 | |
| 2 | 36 | Ataluren (PTC124) | PO | May 2010 | July 2008 | NCT00759876 |
| Class | Phase | Enrolled # | Therapeutics | Route | End Date | Start Date | Trial ID |
|---|---|---|---|---|---|---|---|
| AAV gene transfer | 1 | 15 | AAV1-F344 (rAAV1.CMV.huFollistin344) | IM | October 2017 | January 2012 | NCT01519349 |
| Nonsense suppression | 2 | 6 | Ataluren (PTC124) | PO | March 2010 | January 2010 | NCT01009294 |
| 2 | 173 | Ataluren (PTC124) | PO | May 2010 | January 2010 | NCT00847379 |
| Class | Phase | Enrolled # | Therapeutics | Route | End Date | Start Date | Trial ID |
|---|---|---|---|---|---|---|---|
| Alternative splicing | 1 | 33 | Evrysdi (Risdiplam) | PO | August 2016 | January 2016 | NCT02633709 |
| 1 | 33 | Evrysdi (Risdiplam) | PO | August 2016 | January 2016 | NCT02633709 | |
| 2 | 25 | Evrysdi (Risdiplam) | PO | June 2021 | August 2019 | NCT03779334 | |
| 2/3 | 62 | Evrysdi (Risdiplam) | PO | November 2023 | December 2016 | NCT02913482 | |
| 1/2 | 34 | Spinraza (Nusinersen) | IT | January 2015 | October 2012 | NCT01703988 | |
| 3 | 122 | Spinraza (Nusinersen) | IT | August 2023 | November 2015 | NCT02193074 | |
| 3 | 292 | Spinraza (Nusinersen) | IT | November 2016 | August 2014 | NCT02594124 | |
| 1/2 | 40 | Branaplam (LMI070; NVS-SM1) | PO | July 2020 | April, 2015 | NCT02268552 | |
| 1 | 9 | RG7800 (RO6885247) | PO | July 2015 | November 2014 | NCT02240355 | |
| AAV Gene Therapy | 3 | 2 | Zolgensma (Onasemnogene Abeparvovec-xioi; AVXS-101) | IV | June 2021 | May 2019 | NCT03837184 |
| 3 | 22 | Zolgensma (Onasemnogene Abeparvovec-xioi; AVXS-101) | IV | November 2019 | October 2017 | NCT03306277 | |
| 3 | 30 | Zolgensma (Onasemnogene Abeparvovec-xioi; AVXS-101) | IV | June 2021 | April, 2018 | NCT03505099 | |
| 3 | 33 | Zolgensma (Onasemnogene Abeparvovec-xioi; AVXS-101) | IV | September 2020 | August 2018 | NCT03461289 | |
| 3 | 15 | Zolgensma (Onasemnogene Abeparvovec-xioi; AVXS-101) | CVC | December 2017 | May 2014 | NCT02122952 | |
| 4 | 308 | Zolgensma (Onasemnogene Abeparvovec-xioi; AVXS-101) | IV | December 2035 | February 2020 | NCT04042025 |
| Class | Phase | Enrolled # | Therapeutics | Route | End Date | Start Date | Trial ID |
|---|---|---|---|---|---|---|---|
| AAV Gene Transfer | 1/2 | 24 | AT132 (AAV8-DES-hMTM1) | IV | March 2020 | June 2017 | NCT03199469 |
| Antisense Oligonucleotide | 1/2 | 18 | DYN101 (IONIS-DNM2–2.5Rx) | IV | April 2020 | January 2020 | NCT04033159 |
| Class | Phase | Enrolled # | Therapeutics | Route | End Date | Start Date | Trial ID |
|---|---|---|---|---|---|---|---|
| AAV gene transfer | 1 | 9 | AAV1-γ-sarcoglycan | IM | June 2010 | November 2006 | NCT01344798 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiu, W.; Hsun, Y.-H.; Chang, K.-J.; Yarmishyn, A.A.; Hsiao, Y.-J.; Chien, Y.; Chien, C.-S.; Ma, C.; Yang, Y.-P.; Tsai, P.-H.; et al. Current Genetic Survey and Potential Gene-Targeting Therapeutics for Neuromuscular Diseases. Int. J. Mol. Sci. 2020, 21, 9589. https://doi.org/10.3390/ijms21249589
Chiu W, Hsun Y-H, Chang K-J, Yarmishyn AA, Hsiao Y-J, Chien Y, Chien C-S, Ma C, Yang Y-P, Tsai P-H, et al. Current Genetic Survey and Potential Gene-Targeting Therapeutics for Neuromuscular Diseases. International Journal of Molecular Sciences. 2020; 21(24):9589. https://doi.org/10.3390/ijms21249589
Chicago/Turabian StyleChiu, Wei, Ya-Hsin Hsun, Kao-Jung Chang, Aliaksandr A. Yarmishyn, Yu-Jer Hsiao, Yueh Chien, Chian-Shiu Chien, Chun Ma, Yi-Ping Yang, Ping-Hsing Tsai, and et al. 2020. "Current Genetic Survey and Potential Gene-Targeting Therapeutics for Neuromuscular Diseases" International Journal of Molecular Sciences 21, no. 24: 9589. https://doi.org/10.3390/ijms21249589
APA StyleChiu, W., Hsun, Y.-H., Chang, K.-J., Yarmishyn, A. A., Hsiao, Y.-J., Chien, Y., Chien, C.-S., Ma, C., Yang, Y.-P., Tsai, P.-H., Chiou, S.-H., Lin, T.-Y., & Cheng, H.-M. (2020). Current Genetic Survey and Potential Gene-Targeting Therapeutics for Neuromuscular Diseases. International Journal of Molecular Sciences, 21(24), 9589. https://doi.org/10.3390/ijms21249589