Cyclic Peptide Mimotopes for the Detection of Serum Anti–ATIC Autoantibody Biomarker in Hepato-Cellular Carcinoma

Abstract
:1. Introduction
2. Results
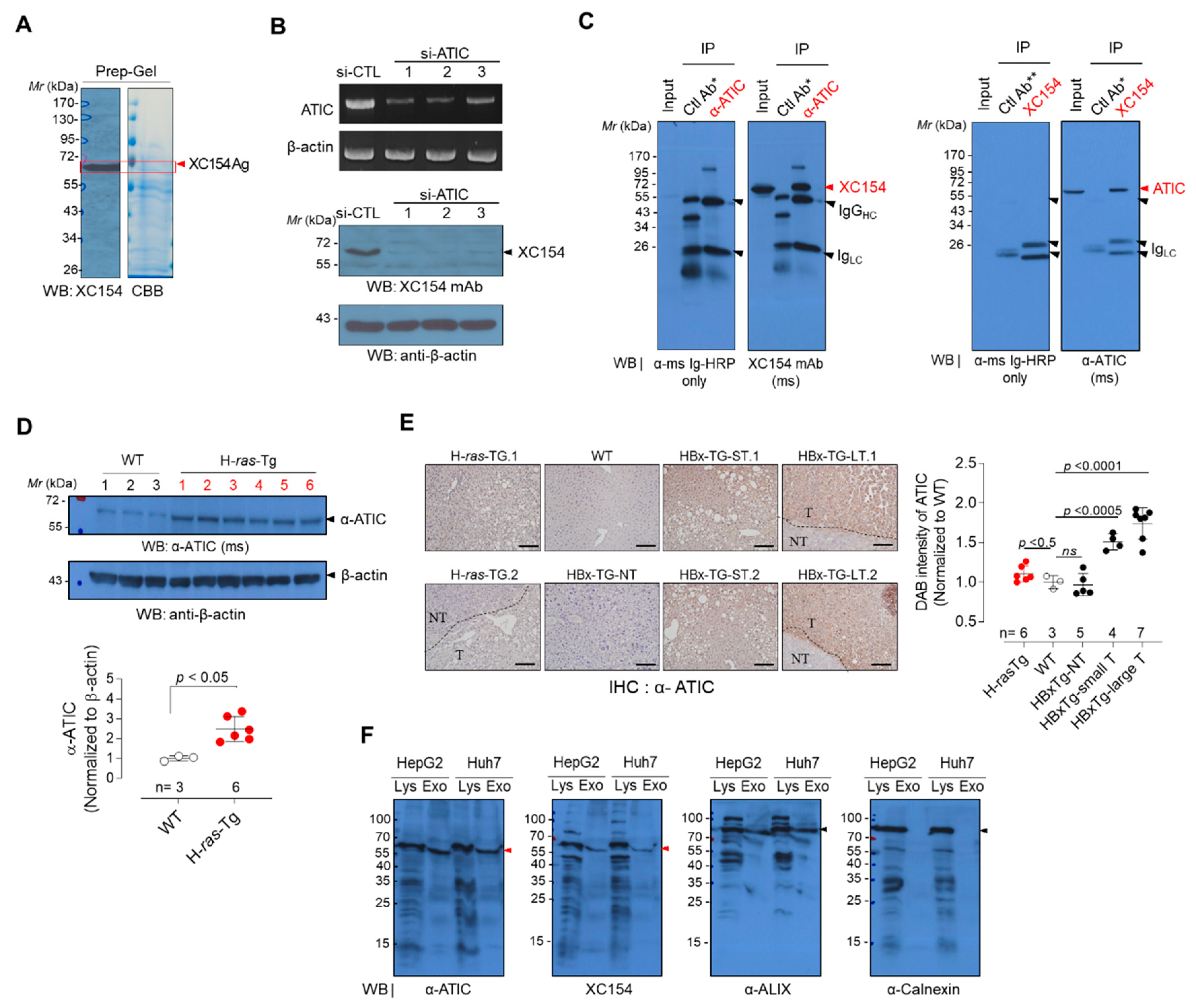
2.1. A Tumor-Associated Autoantibody in the HBx-tg HCC Model Mouse Showed the Elevation of Its Target Antigen in Human HCC Tissues
2.2. The Target Antigen of Tumor-Associated Autoantibody XC154 Was Identified as ATIC, a Bifunctional Purine Biosynthesis Protein
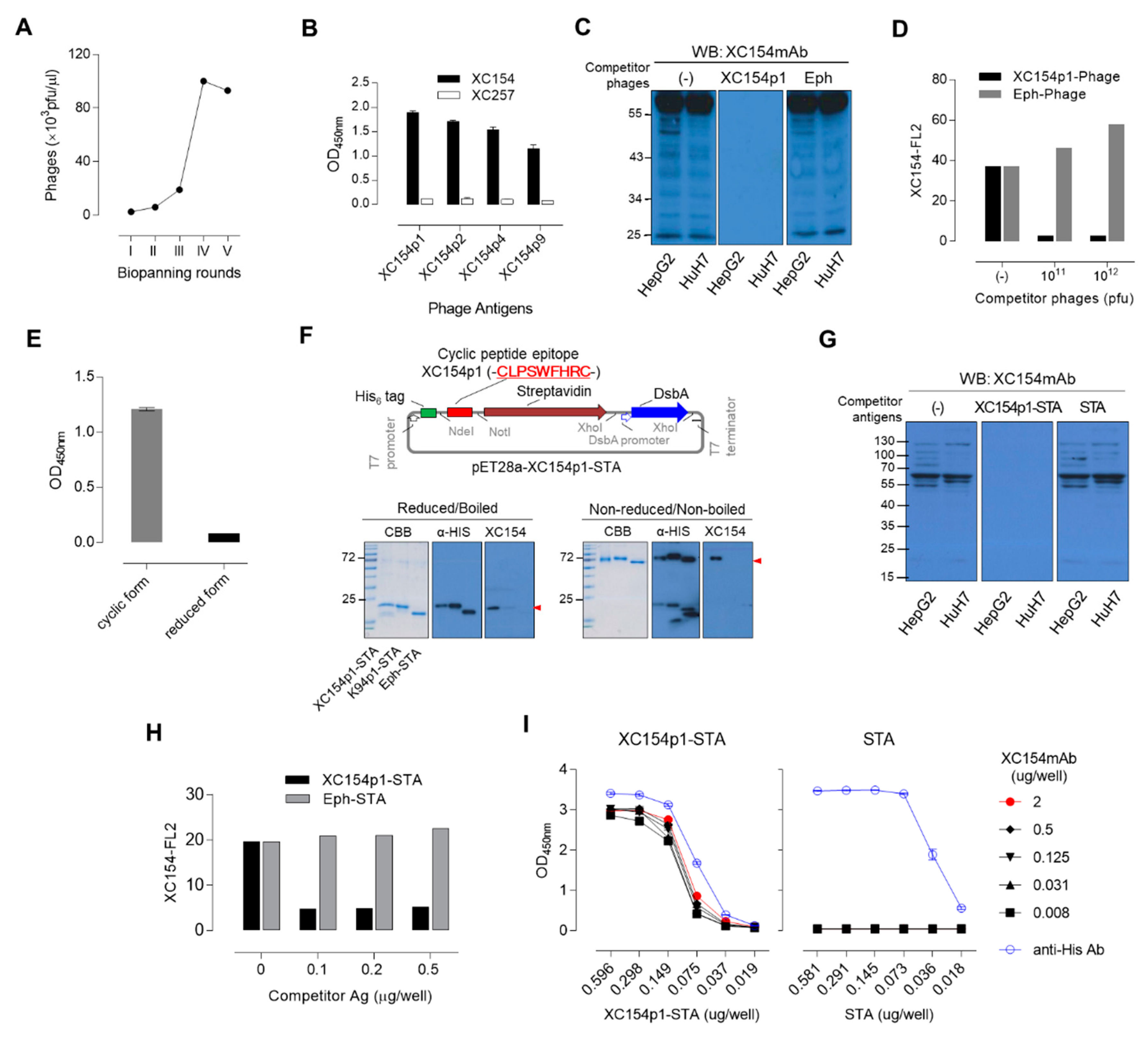
2.3. Specific Mimotopes Against the XC154 Antibody Screened from a Phage Display Random Cyclic Heptapeptide Library Was Selected as a Detection Antigen for Serum Anti-ATIC Autoantibody
2.4. Human Serum Enzyme-Linked Immunosorbent assay (ELISA) to Detect Autoantibodies Against ATIC Using Specific Mimotope, XC154p1
2.5. The Simultaneous Detection of AFP and Anti-ATIC Autoantibody in Patient Serum Improved HCC Diagnosis Accuracy
3. Discussion
4. Materials and Methods
4.1. Tumor-Associated Monoclonal Autoantibody, XC154
4.2. Tumor Cell Lines and Serum Samples
4.3. Western Blot Analysis and Immunoprecipitation
4.4. Immunofluorescence
4.5. Immunohistochemistry
4.6. Identification of XC154 Target Antigen
4.7. Knockdown of ATIC and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.8. Bio-Panning of the XC154-Specific Cyclic Peptide Mimotopes
4.9. ELISA
4.10. Competitive FACS
4.11. Expression and Purification of Recombinant ATIC Protein and XC154p1 Epitope Display Streptavidin (XC154p1-STA)
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- United Nations Development Programme. Human Development Report 2016: Human Development for Everyone; United Nations Development Programme: New York, NY, USA, 2016. [Google Scholar]
- Zaenker, P.; Gray, E.S.; Ziman, M.R. Autoantibody Production in Cancer—The Humoral Immune Response toward Autologous Antigens in Cancer Patients. Autoimmun. Rev. 2016, 15, 477–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauf, F.; Anderson, K.S.; LaBaer, J. Autoantibodies in early detection of breast cancer. Cancer Epidemiol. Biom. Prev. 2020, 29, 2475–2485. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lee, J.R.; Hao, S.; Ling, X.B.; Brooks, J.D.; Wang, S.X.; Gambhir, S.S. Improved detection of prostate cancer using a magneto-nanosensor assay for serum circulating autoantibodies. PLoS ONE 2019, 14, e0221051. [Google Scholar]
- Wang, X.; Yu, J.; Sreekumar, A.; Varambally, S.; Shen, R.; Giacherio, D.; Mehra, R.; Montie, J.E.; Pienta, K.J.; Sanda, M.G.; et al. Autoantibody signatures in prostate cancer. N. Engl. J. Med. 2005, 353, 1224–1235. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Zeng, N.; Yang, T.; Wan, C.; Chen, L.; Shen, Y.; Wen, F. Diagnostic Value of Autoantibodies in Lung Cancer: A Systematic Review and Meta-Analysis. Cell Physiol. Biochem. 2018, 51, 2631–2646. [Google Scholar] [CrossRef]
- Wang, H.; Li, X.; Zhou, D.; Huang, J. Autoantibodies as biomarkers for colorectal cancer: A systematic review, meta-analysis, and bioinformatics analysis. Int. J. Biol. Markers 2019, 34, 334–347. [Google Scholar] [CrossRef] [Green Version]
- Elias, K.M.; Guo, J.; Bast, R.C., Jr. Early Detection of Ovarian Cancer. Hematol. Oncol. Clin. N. Am. 2018, 32, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Fortner, R.T.; Damms-Machado, A.; Kaaks, R. Systematic review: Tumor-associated antigen autoantibodies and ovarian cancer early detection. Gynecol. Oncol. 2017, 147, 465–480. [Google Scholar] [CrossRef]
- Koziol, J.A.; Imai, H.; Dai, L.; Zhang, J.; Tan, E.M. Early detection of hepatocellular carcinoma using autoantibody profiles from a panel of tumor-associated antigens. Cancer Immunol. Immunother. 2018, 67, 835–841. [Google Scholar] [CrossRef]
- Núñez, C. Blood-based protein biomarkers in breast cancer. Clin. Chim. Acta 2019, 490, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Hanash, S.M.; Ostrin, E.J.; Fahrmann, J.F. Blood based biomarkers beyond genomics for lung cancer screening. Transl. Lung Cancer Res. 2018, 7, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Kalniņa, Z.; Meistere, I.; Kikuste, I.; Tolmanis, I.; Zayakin, P.; Linē, A. Emerging blood-based biomarkers for detection of gastric cancer. World J. Gastroenterol. 2015, 21, 11636–11653. [Google Scholar]
- Ludwig, N.; Whiteside, T.L.; Reichert, T.E. Challenges in Exosome Isolation and Analysis in Health and Disease. Int. J. Mol. Sci. 2019, 20, 4684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macdonald, I.K.; Parsy-Kowalska, C.B.; Chapman, C.J. Autoantibodies: Opportunities for Early Cancer Detection. Trends Cancer 2017, 3, 198–213. [Google Scholar] [PubMed]
- Royahem, J.; Conrad, K.; Frey, M.; Melhom, J.; Frank, K.H. Autoantibodies Predictive Parameters of Tumor Development. In Pathogenic and Diagnostic Relevance of Autoantibodies; Konrad, K., Humboldt, R.L., Meurer, M., Shoenfeld, Y., Tan, E.M., Eds.; Pabst: Berlin, Germany, 1988; pp. 412–414. [Google Scholar]
- Kobayashi, M.; Katayama, H.; Fahrmann, J.F.; Hanash, S.M. Development of autoantibody signatures for common cancers. Semin Immunol. 2020, 47, 101388. [Google Scholar] [CrossRef] [PubMed]
- Heo, C.K.; Hwang, H.M.; Lee, H.J.; Kwak, S.S.; Yoo, J.S.; Yu, D.Y.; Lim, K.J.; Lee, S.; Cho, E.W. Serum anti-EIF3A autoantibody as a potential diagnostic marker for hepatocellular carcinoma. Sci. Rep. 2019, 9, 11059. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.Y.; Moon, H.B.; Son, J.K.; Jeong, S.; Yu, S.L.; Yoon, H.; Han, Y.M.; Lee, C.S.; Park, J.S.; Lee, C.H.; et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J. Hepatol. 1999, 31, 123–132. [Google Scholar] [CrossRef]
- Wang, A.G.; Moon, H.B.; Lee, M.R.; Hwang, C.Y.; Kwon, K.S.; Yu, S.L.; Kim, Y.S.; Kim, M.; Kim, J.M.; Kim, S.K.; et al. Gender-dependent hepatic alterations in H-ras12V transgenic mice. J. Hepatol. 2005, 43, 836–844. [Google Scholar] [CrossRef]
- Hwang, H.M.; Heo, C.K.; Lee, H.J.; Kwak, S.S.; Lim, W.H.; Yoo, J.S.; Yu, D.Y.; Lim, K.J.; Kim, J.Y.; Cho, E.W. Identification of anti-SF3B1 autoantibody as a diagnostic marker in patients with hepatocellular carcinoma. J. Transl. Med. 2018, 16, 177. [Google Scholar]
- Heo, C.K.; Hwang, H.M.; Ruem, A.; Yu, D.Y.; Lee, J.Y.; Yoo, J.S.; Kim, I.G.; Yoo, H.S.; Oh, S.; Ko, J.H.; et al. Identification of a mimotope for circulating anti-cytokeratin 8/18 antibody and its usage for the diagnosis of breast cancer. Int. J. Oncol. 2013, 42, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, C.K.; Woo, M.K.; Yu, D.Y.; Lee, J.Y.; Yoo, J.S.; Yoo, H.S.; Ko, J.H.; Kim, J.M.; Choi, J.Y.; Kim, I.G. Identification of autoantibody against fatty acid synthase in hepatocellular carcinoma mouse model and its application to diagnosis of HCC. Int. J. Oncol. 2010, 36, 1453–1459. [Google Scholar] [PubMed]
- Capello, M.; Vykoukal, J.V.; Katayama, H.; Bantis, L.E.; Wang, H.; Kundnani, D.L.; Aguilar-Bonavides, C.; Aguilar, M.; Tripathi, S.C.; Dhillon, D.S. Exosomes harbor B cell targets in pancreatic adenocarcinoma and exert decoy function against complement-mediated cytotoxicity. Nat. Commun. 2019, 10, 254. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Lippert, D.; Krokhin, O.; Cortens, J.P.; Wilkins, J.A. Defining the membrane proteome of NK cells. J. Mass Spectrom. 2010, 45, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Welton, J.L.; Khanna, S.; Giles, P.J.; Brennan, P.; Brewis, I.A.; Staffurth, J.; Mason, M.D.; Clayton, A. Proteomics analysis of bladder cancer exosomes. Mol. Cell Proteom. 2010, 9, 1324–1338. [Google Scholar] [CrossRef] [Green Version]
- Liang, B.; Peng, P.; Chen, S.; Li, L.; Zhang, M.; Cao, D.; Yang, J.; Li, H.; Gui, T.; Li, X.; et al. Characterization and proteomic analysis of ovarian cancer-derived exosomes. J. Proteom. 2013, 80, 171–182. [Google Scholar] [CrossRef]
- Lazar, I.; Clement, E.; Ducoux-Petit, M.; Denat, L.; Soldan, V.; Dauvillier, S.; Balor, S.; Burlet-Schiltz, O.; Larue, L.; Muller, C.; et al. Proteome characterization of melanoma exosomes reveals a specific signature for metastatic cell lines. Pigm. Cell Melan. Res. 2015, 28, 464–475. [Google Scholar] [CrossRef]
- Riedinger, C.; Mendler, M.; Schlotterer, A.; Fleming, T.; Okun, J.; Hammes, H.P.; Herzig, S.; Nawroth, P.P. High-glucose toxicity is mediated by AICAR-transformylase/IMP cyclohydrolase and mitigated by AMP-activated protein kinase in Caenorhabditis elegans. J. Biol. Chem. 2018, 293, 4845–4859. [Google Scholar] [CrossRef]
- Torres, M.J.; Turell, L.; Botti, H.; Antmann, L.; Carballal, S.; Ferrer-Sueta, G.; Radi, R.; Alvarez, B. Modulation of the reactivity of the thiol of human serum albumin and its sulfenic derivative by fatty acids. Arch. Biochem. Biophys. 2012, 521, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Ren, W.; Huang, X.; Deng, J.; Li, T.; Yin, Y. Potential Mechanisms Connecting Purine Metabolism and Cancer Therapy. Front. Immunol. 2018, 9, 1697. [Google Scholar] [CrossRef] [Green Version]
- Vergis, J.M.; Beardsley, G.P. Catalytic mechanism of the cyclohydrolase activity of human aminoimidazole carboxamide ribonucleotide formyltransferase/inosine monophosphate cyclohydrolase. Biochemistry 2004, 43, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Türeci, O.; Usener, D.; Schneider, S.; Sahin, U. Identification of tumor-associated autoantigens with SEREX. Methods Mol. Med. 2005, 109, 137–154. [Google Scholar] [PubMed]
- He, Y.; Wu, Y.; Mou, Z.; Li, W.; Zou, L.; Fu, T.; Zhang, A.; Xiang, D.; Xiao, H.; Wang, X. Proteomics-based identification of HSP60 as a tumor-associated antigen in colorectal cancer. Proteom. Clin. Appl. 2007, 1, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Hardouin, J.; Lasserre, J.P.; Sylvius, L.; Joubert-Caron, R.; Caron, M. Cancer immunomics: From serological proteome analysis to multiple affinity protein profiling. Ann. N. Y. Acad. Sci. 2007, 1107, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Minciacchi, V.R.; Freeman, M.R.; Di Vizio, D. Extracellular vesicles in cancer: Exosomes, microvesicles and the emerging role of large oncosomes. Semin. Cell Dev. Biol. 2015, 40, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livshits, M.A.; Khomyakova, E.; Evtushenko, E.G.; Lazarev, V.N.; Kulemin, N.A.; Semina, S.E.; Generozov, E.V.; Govorun, V.M. Isolation of exosomes by differential centrifugation: Theoretical analysis of a commonly used protocol. Sci. Rep. 2015, 5, 17319. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Protein Name | Accession Number | Molecular Mass (Da) | Protein Score | No. of Matched Peptide | Sequences | emPAI * |
|---|---|---|---|---|---|---|---|
| ATIC | Bifunctional Purine Biosynthesis Protein PURH | IPI00289499 | 64575 | 1780 | 85 (48) | 38 (26) | 16.34 |
| GLA | Alpha-galactosidase A | IPI00025869 | 48735 | 267 | 10 (6) | 3 (3) | 0.56 |
| KRT1 | Keratin, type II cytoskeletal 1 | IPI00220327 | 65999 | 234 | 13 (10) | 13 (10) | 0.83 |
| AIFM1 | Isoform 1 of Apoptosis-inducing factor 1, mitochondrial | IPI00000690 | 66859 | 216 | 16 (6) | 14 (6) | 0.81 |
| KRT9 | Keratin, type I cytoskeletal 9 | IPI00019359 | 62027 | 206 | 11 (7) | 9 (7) | 0.69 |
| TARS | Threonyl-tRNA synthetase, cytoplasmic | IPI00329633 | 83382 | 197 | 12 (6) | 12 (6) | 0.42 |
| HADHA | Trifunctional enzyme subunit alpha, mitochondrial | IPI00031522 | 82947 | 168 | 5 (4) | 5 (4) | 0.19 |
| ENO1 | Isoform alpha-enolase of Alpha-enolase | IPI00465248 | 47139 | 113 | 3 (3) | 2 (2) | 0.26 |
| FH | Isoform Mitochondrial of Fumarate hydratase, mitochondrial | IPI00296053 | 54602 | 106 | 2 (1) | 2 (1) | 0.07 |
| CS | Citrate synthase, mitochondrial | IPI00025366 | 51680 | 97 | 5 (2) | 3 (1) | 0.23 |
| Phage Antigens | Epitope Amino Acid Sequences (X7) | Frequency ** | ||||||
|---|---|---|---|---|---|---|---|---|
| XC154p1 | L *** | P | S | W | F | H | R | 5/10 |
| XC154p2 | A | P | S | W | L | H | R | 1/10 |
| XC154p3 | D | P | S | G | H | R | A | 1/10 |
| XC154p4 | S | P | S | G | L | F | S | 1/10 |
| XC154p8 | A | P | S | W | F | F | R | 1/10 |
| XC154p9 | T | P | S | W | F | T | R | 1/10 |
| Cohort 1 * | Cohort 2 ** | |||
|---|---|---|---|---|
| Patients (number) | HCC | CHB | LC | HCC |
| Gender | ||||
| male/female, | 114/30 | 43/21 | 40/24 | 56/17 |
| n (%) | (79.2/20.8) | (67.2/32.8) | (62.5/37.5) | (76.7/23.3) |
| Age distribution, yr (Avg ± SD) | 33~83 (55.4 ± 10.3) | 23~67 (49.2 ± 10.1) | 41~78 (54.2 ± 9.08) | 41~80 (58.2 ± 7.69) |
| Serum AFP (ng/mL) | 0.8~83000 | 1.4~252 | 0.8~718.9 | 1.08~9786.3 |
| <CV40/>CV40 | 83/61 | 54/10 | 44/20 | 47/26 |
| n (%) | (57.6/42.4) | (84.4/15.6) | (68.8/31.2) | (64.4/35.6) |
| Viral infection | ||||
| (–)/HBV/HCV | 17/118/9 | 0/64/0 | 4/60/0 | 15/58/0 |
| n (%) | (11.8/81.9/6.3) | (0/100/0) | (6.2/93.8/0) | (20.5/79.5/0) |
| Tumor stage T1/T2/T3/T4 n (%) | 34/53/27/30 (23.6/36.8/18.6/20.8) | n.a. § | n.a. | n.a. |
| Tumor size (cm) T < 2/2 < T < 5/5 < T n (%) | 57/48/39 (39.6/33.3/27.1) | n.a. | n.a. | n.a. |
| Anti-ATIC autoantibody (AU) | 0.01~1.1 | 0~0.416 | 0~0.479 | 0.043~0.539 |
| <CV0.223/>CV0.223, | 55/89 | 48/16 | 38/26 | 26/47 |
| n (%) | (38.2/61.8) | (75/25) | (59.4/40.6) | (35.6/64.4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, C.-K.; Hwang, H.-M.; Lim, W.-H.; Lee, H.-J.; Yoo, J.-S.; Lim, K.-J.; Cho, E.-W. Cyclic Peptide Mimotopes for the Detection of Serum Anti–ATIC Autoantibody Biomarker in Hepato-Cellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 9718. https://doi.org/10.3390/ijms21249718
Heo C-K, Hwang H-M, Lim W-H, Lee H-J, Yoo J-S, Lim K-J, Cho E-W. Cyclic Peptide Mimotopes for the Detection of Serum Anti–ATIC Autoantibody Biomarker in Hepato-Cellular Carcinoma. International Journal of Molecular Sciences. 2020; 21(24):9718. https://doi.org/10.3390/ijms21249718
Chicago/Turabian StyleHeo, Chang-Kyu, Hai-Min Hwang, Won-Hee Lim, Hye-Jung Lee, Jong-Shin Yoo, Kook-Jin Lim, and Eun-Wie Cho. 2020. "Cyclic Peptide Mimotopes for the Detection of Serum Anti–ATIC Autoantibody Biomarker in Hepato-Cellular Carcinoma" International Journal of Molecular Sciences 21, no. 24: 9718. https://doi.org/10.3390/ijms21249718