IL-20 in Acute Kidney Injury: Role in Pathogenesis and Potential as a Therapeutic Target


Abstract
1. Introduction
2. Acute Kidney Injury (AKI)
3. Chronic Kidney Disease (CKD)
4. Diabetic Nephropathy
5. Inflammation and Kidney Disease
6. MCP-1(CCL2)/CCR2
7. IL-8/CXCL8
8. TNF-α
9. IL-1β
10. IL-6
11. Transforming Growth Factor-β (TGF-β)
12. Hypoxia in Kidney Disease
13. IL-20
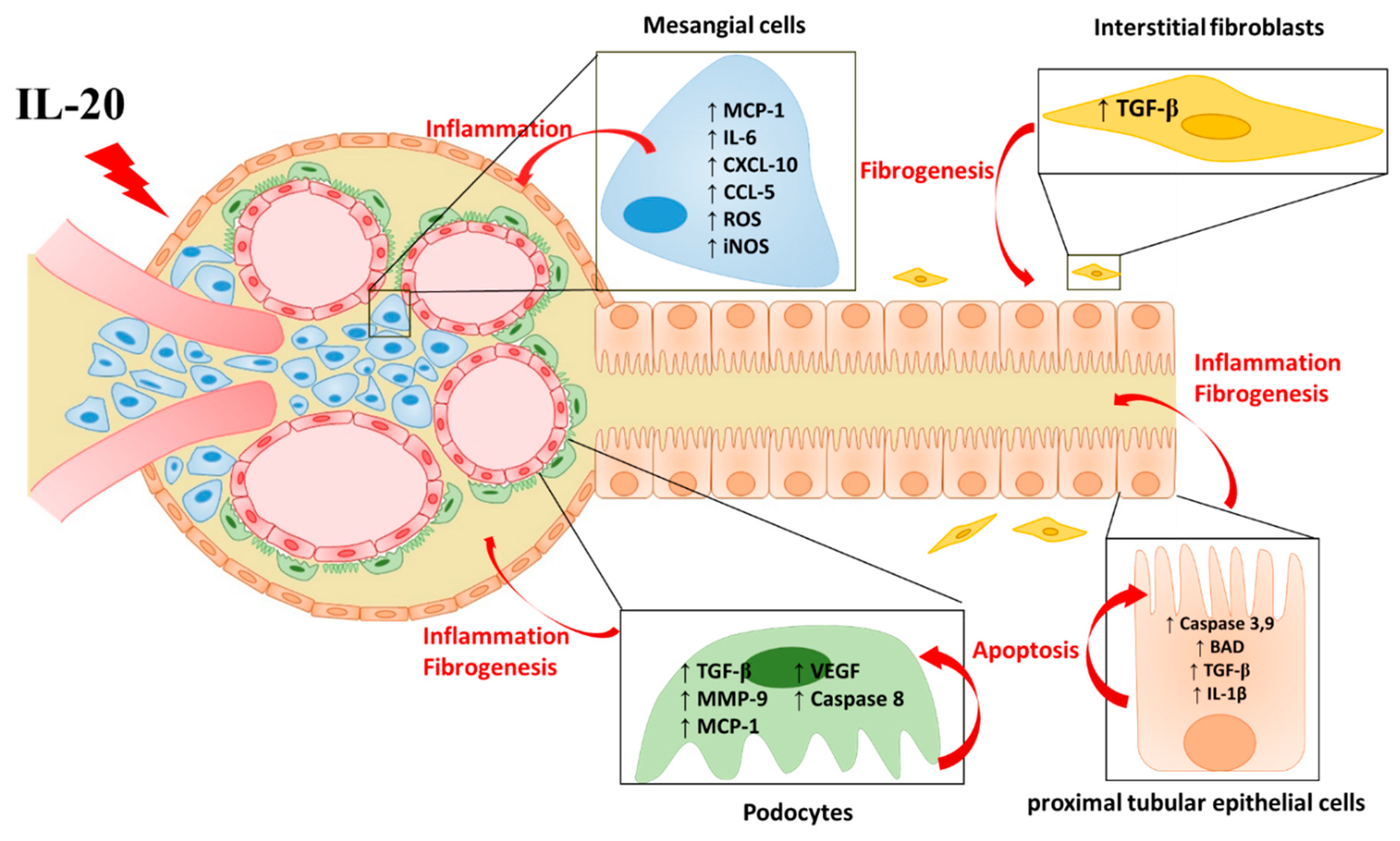
14. IL-20 in AKI
15. IL-20 in CKD
16. IL-20 in DN
17. IL-20 Antibody Therapy in Kidney Disease
18. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef]
- Meng, X.-M.; Tang, P.M.-K.; Li, J.; Lan, H.Y. TGF-β/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82. [Google Scholar] [CrossRef]
- Cooper, M.E. Interaction of metabolic and haemodynamic factors in mediating experimental diabetic nephropathy. Diabetologia 2001, 44, 1957–1972. [Google Scholar] [CrossRef]
- Cao, Z.; Cooper, M.E. Pathogenesis of diabetic nephropathy. J. Diabetes Investig. 2011, 2, 243–247. [Google Scholar] [CrossRef]
- Cooper, M.E. Pathogenesis, prevention, and treatment of diabetic nephropathy. Lancet 1998, 352, 213–219. [Google Scholar] [CrossRef]
- Chang, M.-S.; Hsu, Y.-H. The role of IL-20 in chronic kidney disease and diabetic nephropathy: Pathogenic and therapeutic implications. J. Leukoc. Biol. 2018, 104, 919–923. [Google Scholar] [CrossRef]
- Kes, P.; Basić Jukić, N. Acute kidney injury in the intensive care unit. Bosn. J. Basic Med. Sci. 2010, 10, S8–S12. [Google Scholar] [CrossRef]
- Hamid, S.-A.; Adnan, W.-N.-A.; Naing, N.; Adnan, A. Acute kidney injury in intensive care unit, hospital Universiti Sains Malaysia: A descriptive study. Saudi J. Kidney Dis. Transpl. 2018, 29, 1109–1114. [Google Scholar] [CrossRef]
- Section 2: AKI Definition. Kidney Int. Suppl. (2011) 2012, 2, 19–36. [CrossRef]
- Ysebaert, D.K.; De Greef, K.E.; Vercauteren, S.R.; Ghielli, M.; Verpooten, G.A.; Eyskens, E.J.; De Broe, M.E. Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury. Nephrol. Dial. Transplant. 2000, 15, 1562–1574. [Google Scholar] [CrossRef]
- Berger, K.; Moeller, M.J. Mechanisms of Epithelial Repair and Regeneration After Acute Kidney Injury. Semin. Nephrol. 2014, 34, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.S.; Rouse, M.; Huang, L.; Vergis, A.L.; Reutershan, J.; Cathro, H.P.; Linden, J.; Okusa, M.D. Compartmentalization of neutrophils in the kidney and lung following acute ischemic kidney injury. Kidney Int. 2009, 75, 689–698. [Google Scholar] [CrossRef]
- Forbes, J.M.; Hewitson, T.D.; Becker, G.J.; Jones, C.L. Ischemic acute renal failure: Long-term histology of cell and matrix changes in the rat. Kidney Int. 2000, 57, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.-K.; Sung, S.-A.; Cho, W.-Y.; Go, K.-J.; Kim, H.-K. Macrophages contribute to the initiation of ischaemic acute renal failure in rats. Nephrol. Dial. Transplant. 2006, 21, 1231–1239. [Google Scholar] [CrossRef]
- Kelly, K.J.; Williams, W.W., Jr.; Colvin, R.B.; Meehan, S.M.; Springer, T.A.; Gutierrez-Ramos, J.C.; Bonventre, J.V. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J. Clin. Investig. 1996, 97, 1056–1063. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Yuen, D.A.; Bajwa, A.; Huang, Y.-W.; Sokollik, C.; Huang, L.; Lam, G.Y.; Tole, S.; Liu, G.-Y.; Pan, J.; et al. Slit2 prevents neutrophil recruitment and renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2013, 24, 1274–1287. [Google Scholar] [CrossRef]
- Bikbov, B.; Perico, N.; Remuzzi, G. Disparities in Chronic Kidney Disease Prevalence among Males and Females in 195 Countries: Analysis of the Global Burden of Disease 2016 Study. Nephron 2018, 139, 313–318. [Google Scholar] [CrossRef]
- Kline, J.; Rachoin, J.-S. Acute Kidney Injury and Chronic Kidney Disease: It’s a Two-Way Street. Ren. Fail. 2013, 35, 452–455. [Google Scholar] [CrossRef][Green Version]
- Chawla, L.S.; Kimmel, P.L. Acute kidney injury and chronic kidney disease: An integrated clinical syndrome. Kidney Int. 2012, 82, 516–524. [Google Scholar] [CrossRef]
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [Google Scholar] [CrossRef]
- Ishani, A.; Xue, J.L.; Himmelfarb, J.; Eggers, P.W.; Kimmel, P.L.; Molitoris, B.A.; Collins, A.J. Acute kidney injury increases risk of ESRD among elderly. J. Am. Soc. Nephrol. 2009, 20, 223–228. [Google Scholar] [CrossRef]
- Grgic, I.; Campanholle, G.; Bijol, V.; Wang, C.; Sabbisetti, V.S.; Ichimura, T.; Humphreys, B.D.; Bonventre, J.V. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012, 82, 172–183. [Google Scholar] [CrossRef]
- Pannu, N.; James, M.; Hemmelgarn, B.R.; Dong, J.; Tonelli, M.; Klarenbach, S. Modification of Outcomes After Acute Kidney Injury by the Presence of CKD. Am. J. Kidney Dis. 2011, 58, 206–213. [Google Scholar] [CrossRef]
- Grams, M.E.; Astor, B.C.; Bash, L.D.; Matsushita, K.; Wang, Y.; Coresh, J. Albuminuria and estimated glomerular filtration rate independently associate with acute kidney injury. J. Am. Soc. Nephrol. 2010, 21, 1757–1764. [Google Scholar] [CrossRef]
- James, M.; Hemmelgarn, B.; Wiebe, N.; Pannu, N.; Manns, B.; Klarenbach, S.; Tonelli, M. Glomerular filtration rate, proteinuria, and the incidence and consequences of acute kidney injury: A cohort study. Lancet 2010, 376, 2096–2103. [Google Scholar] [CrossRef]
- Vadivel, N.; Trikudanathan, S.; Singh, A.K. Analgesic nephropathy. Kidney Int. 2007, 72, 517–520. [Google Scholar] [CrossRef]
- Bao, L.; Cunningham, P.N.; Quigg, R.J. Complement in Lupus Nephritis: New Perspectives. Kidney Dis. 2015, 1, 91–99. [Google Scholar] [CrossRef]
- Faurschou, M.; Starklint, H.; Halberg, P.; Jacobsen, S. Prognostic factors in lupus nephritis: Diagnostic and therapeutic delay increases the risk of terminal renal failure. J. Rheumatol. 2006, 33, 1563. [Google Scholar]
- Cheng, Y.; Yang, X.; Zhang, X.; An, Z. Analysis of expression levels of IL-17 and IL-34 and influencing factors for prognosis in patients with lupus nephritis. Exp. Ther. Med. 2019, 17, 2279–2283. [Google Scholar] [CrossRef]
- Tylicki, L.; Rutkowski, B. Hypertensive nephropathy: Pathogenesis, diagnosis and treatment. Pol. Merkur. Lek. Organ Pol. Tow. Lek. 2003, 14, 168–173. [Google Scholar]
- D’ Amico, G. The Commonest Glomerulonephritis in the World: IgA Nephropathy. QJM: An. Int. J. Med. 1987, 64, 709–727. [Google Scholar]
- Shu, D.; Xu, F.; Su, Z.; Zhang, J.; Chen, C.; Zhang, J.; Ding, X.; Lv, Y.; Lin, H.; Huang, P. Risk factors of progressive IgA nephropathy which progress to end stage renal disease within ten years: A case-control study. BMC Nephrol. 2017, 18, 11. [Google Scholar] [CrossRef]
- Lai, K.N. Pathogenesis of IgA nephropathy. Nat. Rev. Nephrol. 2012, 8, 275–283. [Google Scholar] [CrossRef]
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Investig. 2014, 124, 2333–2340. [Google Scholar] [CrossRef]
- Noh, H.; King, G.L. The role of protein kinase C activation in diabetic nephropathy. Kidney Int. 2007, 72, S49–S53. [Google Scholar] [CrossRef]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Böttinger, E.P. Glucose-Induced Reactive Oxygen Species Cause Apoptosis of Podocytes and Podocyte Depletion at the Onset of Diabetic Nephropathy. Diabetes 2006, 55, 225. [Google Scholar] [CrossRef]
- Weil, E.J.; Lemley, K.V.; Mason, C.C.; Yee, B.; Jones, L.I.; Blouch, K.; Lovato, T.; Richardson, M.; Myers, B.D.; Nelson, R.G. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 2012, 82, 1010–1017. [Google Scholar] [CrossRef]
- Lin, J.S.; Susztak, K. Podocytes: The Weakest Link in Diabetic Kidney Disease? Curr. Diabetes Rep. 2016, 16, 45. [Google Scholar] [CrossRef]
- Tashiro, K.; Koyanagi, I.; Saitoh, A.; Shimizu, A.; Shike, T.; Ishiguro, C.; Koizumi, M.; Funabiki, K.; Horikoshi, S.; Shirato, I.; et al. Urinary levels of monocyte chemoattractant protein-1 (MCP-1) and interleukin-8 (IL-8), and renal injuries in patients with type 2 diabetic nephropathy. J. Clin. Lab. Anal. 2002, 16, 1–4. [Google Scholar] [CrossRef]
- Rovin, B.H.; Doe, N.; Tan, L.C. Monocyte chemoattractant protein-1 levels in patients with glomerular disease. Am. J. Kidney Dis. 1996, 27, 640–646. [Google Scholar] [CrossRef]
- Viedt, C.; Dechend, R.; Fei, J.; Hänsch, G.M.; Kreuzer, J.; Orth, S.R. MCP-1 Induces Inflammatory Activation of Human Tubular Epithelial Cells: Involvement of the Transcription Factors, Nuclear Factor-κB and Activating Protein-1. J. Am. Soc. Nephrol. 2002, 13, 1534. [Google Scholar] [CrossRef]
- Giunti, S.; Pinach, S.; Arnaldi, L.; Viberti, G.; Perin, P.C.; Camussi, G.; Gruden, G. The MCP-1/CCR2 system has direct proinflammatory effects in human mesangial cells. Kidney Int. 2006, 69, 856–863. [Google Scholar] [CrossRef]
- Rovin, B.H.; Yoshiumura, T.; Tan, L. Cytokine-induced production of monocyte chemoattractant protein-1 by cultured human mesangial cells. J. Immunol. 1992, 148, 2148. [Google Scholar]
- Prodjosudjadi, W.; Gerritsma, J.S.J.; Klar-Mohamad, N.; Gerritsen, A.F.; Bruijn, J.A.; Daha, M.R.; van Es, L.A. Production and cytokine-mediated regulation of monocyte chemoattractant protein-1 by human proximal tubular epithelial cells. Kidney Int. 1995, 48, 1477–1486. [Google Scholar] [CrossRef]
- Zoja, C.; Wang, J.M.; Bettoni, S.; Sironi, M.; Renzi, D.; Chiaffarino, F.; Abboud, H.E.; Van Damme, J.; Mantovani, A.; Remuzzi, G. Interleukin-1 beta and tumor necrosis factor-alpha induce gene expression and production of leukocyte chemotactic factors, colony-stimulating factors, and interleukin-6 in human mesangial cells. Am. J. Pathol. 1991, 138, 991–1003. [Google Scholar]
- Wu, S.-H.; Lu, C.; Dong, L.; Chen, Z.-Q. Signal transduction involved in CTGF-induced production of chemokines in mesangial cells. Growth Factors 2008, 26, 192–200. [Google Scholar] [CrossRef]
- Ihm, C.G.; Park, J.K.; Hong, S.P.; Lee, T.W.; Cho, B.S.; Kim, M.J.; Cha, D.R.; Ha, H. A High Glucose Concentration Stimulates the Expression of Monocyte Chemotactic Peptide 1 in Human Mesangial Cells. Nephron 1998, 79, 33–37. [Google Scholar] [CrossRef]
- Ha, H.; Yu, M.R.; Choi, Y.J.; Kitamura, M.; Lee, H.B. Role of High Glucose-Induced Nuclear Factor-κB Activation in Monocyte Chemoattractant Protein-1 Expression by Mesangial Cells. J. Am. Soc. Nephrol. 2002, 13, 894. [Google Scholar]
- Banba, N.; Nakamura, T.; Matsumura, M.; Kuroda, H.; Hattori, Y.; Kasai, K. Possible relationship of monocyte chemoattractant protein-1 with diabetic nephropathy. Kidney Int. 2000, 58, 684–690. [Google Scholar] [CrossRef]
- Chow, F.Y.; Nikolic-Paterson, D.J.; Ozols, E.; Atkins, R.C.; Rollin, B.J.; Tesch, G.H. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006, 69, 73–80. [Google Scholar] [CrossRef]
- Wei, M.; Li, Z.; Xiao, L.; Yang, Z. Effects of ROS-relative NF-κB signaling on high glucose-induced TLR4 and MCP-1 expression in podocyte injury. Mol. Immunol. 2015, 68, 261–271. [Google Scholar] [CrossRef]
- Park, J.; Ryu, D.-R.; Li, J.J.; Jung, D.-S.; Kwak, S.-J.; Lee, S.H.; Yoo, T.-H.; Han, S.H.; Lee, J.E.; Kim, D.K.; et al. MCP-1/CCR2 system is involved in high glucose-induced fibronectin and type IV collagen expression in cultured mesangial cells. Am. J. Physiol. Ren. Physiol. 2008, 295, F749–F757. [Google Scholar] [CrossRef]
- Nam, B.Y.; Paeng, J.; Kim, S.H.; Lee, S.H.; Kim, D.H.; Kang, H.-Y.; Li, J.J.; Kwak, S.-J.; Park, J.T.; Yoo, T.-H.; et al. The MCP-1/CCR2 axis in podocytes is involved in apoptosis induced by diabetic conditions. Apoptosis 2012, 17, 1–13. [Google Scholar] [CrossRef]
- Lee, E.Y.; Chung, C.H.; Khoury, C.C.; Yeo, T.K.; Pyagay, P.E.; Wang, A.; Chen, S. The monocyte chemoattractant protein-1/CCR2 loop, inducible by TGF-β, increases podocyte motility and albumin permeability. Am. J. Physiol. Ren. Physiol. 2009, 297, F85–F94. [Google Scholar] [CrossRef]
- Kashyap, S.; Osman, M.; Ferguson, C.M.; Nath, M.C.; Roy, B.; Lien, K.R.; Nath, K.A.; Garovic, V.D.; Lerman, L.O.; Grande, J.P. Ccl2 deficiency protects against chronic renal injury in murine renovascular hypertension. Sci. Rep. 2018, 8, 8598. [Google Scholar] [CrossRef]
- Kitagawa, K.; Wada, T.; Furuichi, K.; Hashimoto, H.; Ishiwata, Y.; Asano, M.; Takeya, M.; Kuziel, W.A.; Matsushima, K.; Mukaida, N.; et al. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am. J. Pathol. 2004, 165, 237–246. [Google Scholar] [CrossRef]
- Kanamori, H.; Matsubara, T.; Mima, A.; Sumi, E.; Nagai, K.; Takahashi, T.; Abe, H.; Iehara, N.; Fukatsu, A.; Okamoto, H.; et al. Inhibition of MCP-1/CCR2 pathway ameliorates the development of diabetic nephropathy. Biochem. Biophys. Res. Commun. 2007, 360, 772–777. [Google Scholar] [CrossRef]
- Furuichi, K.; Wada, T.; Iwata, Y.; Kitagawa, K.; Kobayashi, K.-I.; Hashimoto, H.; Ishiwata, Y.; Asano, M.; Wang, H.; Matsushima, K.; et al. CCR2 Signaling Contributes to Ischemia-Reperfusion Injury in Kidney. J. Am. Soc. Nephrol. 2003, 14, 2503. [Google Scholar] [CrossRef]
- Kashyap, S.; Warner, G.M.; Hartono, S.P.; Boyilla, R.; Knudsen, B.E.; Zubair, A.S.; Lien, K.; Nath, K.A.; Textor, S.C.; Lerman, L.O.; et al. Blockade of CCR2 reduces macrophage influx and development of chronic renal damage in murine renovascular hypertension. Am. J. Physiol. Ren. Physiol. 2015, 310, F372–F384. [Google Scholar] [CrossRef]
- Stroo, I.; Claessen, N.; Teske, G.J.D.; Butter, L.M.; Florquin, S.; Leemans, J.C. Deficiency for the chemokine monocyte chemoattractant protein-1 aggravates tubular damage after renal ischemia/reperfusion injury. PLoS ONE 2015, 10, e0123203. [Google Scholar] [CrossRef]
- Brown, Z.; Strieter, R.M.; Chensue, S.W.; Ceska, M.; Lindley, I.; Neild, G.H.; Kunkel, S.L.; Westwick, J. Cytokine-activated human mesangial cells generate the neutrophil chemoattractant, interleukin 8. Kidney Int. 1991, 40, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Abbott, F.; Ryan, J.J.; Ceska, M.; Matsushima, K.; Sarraf, C.E.; Rees, A.J. Interleukin-1β stimulates human mesangial cells to synthesize and release interleukins-6 and-8. Kidney Int. 1991, 40, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Kusner, D.J.; Luebbers, E.L.; Nowinski, R.J.; Konieczkowski, M.; King, C.H.; Sedor, J.R. Cytokine- and LPS-induced synthesis of interleukin-8 from human mesangial cells. Kidney Int. 1991, 39, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Nord, E.P. IL-8 amplifies CD40/CD154-mediated ICAM-1 production via the CXCR-1 receptor and p38-MAPK pathway in human renal proximal tubule cells. Am. J. Physiol.Ren. Physiol. 2009, 296, F438–F445. [Google Scholar] [CrossRef]
- Gerritsma, J.S.; Hiemstra, P.S.; Gerritsen, A.F.; Prodjosudjadi, W.; Verweij, C.L.; Van Es, L.A.; Daha, M.R. Regulation and production of IL-8 by human proximal tubular epithelial cells in vitro. Clin. Exp. Immunol. 1996, 103, 289–294. [Google Scholar] [CrossRef]
- Wada, T.; Tomosugi, N.; Naito, T.; Yokoyama, H.; Kobayashi, K.; Harada, A.; Mukaida, N.; Matsushima, K. Prevention of proteinuria by the administration of anti-interleukin 8 antibody in experimental acute immune complex-induced glomerulonephritis. J. Exp. Med. 1994, 180, 1135–1140. [Google Scholar] [CrossRef]
- Li, L.; Khan, N.; Li, Q.; Chen, X.; Wei, J.; Wang, B.; Cheng, J.-W.; Gordon, J.; Li, F. G31P, CXCR1/2 inhibitor, with cisplatin inhibits the growth of mice hepatocellular carcinoma and mitigates high-dose cisplatin-induced nephrotoxicity. Oncol. Rep. 2014, 33, 751–757. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, W.; Zhu, H. CXCL8(3–72) K11R/G31P protects against sepsis-induced acute kidney injury via NF-κB and JAK2/STAT3 pathway. Biol. Res. 2019, 52, 29. [Google Scholar] [CrossRef]
- Cui, S.; Zhu, Y.; Du, J.; Khan, M.N.; Wang, B.; Wei, J.; Cheng, J.-W.; Gordon, J.R.; Mu, Y.; Li, F. CXCL8 Antagonist Improves Diabetic Nephropathy in Male Mice With Diabetes and Attenuates High Glucose–Induced Mesangial Injury. Endocrinology 2017, 158, 1671–1684. [Google Scholar] [CrossRef]
- Bertani, T.; Abbate, M.; Zoja, C.; Corna, D.; Perico, N.; Ghezzi, P.; Remuzzi, G. Tumor necrosis factor induces glomerular damage in the rabbit. Am. J. Pathol. 1989, 134, 419–430. [Google Scholar]
- Peralta Soler, A.; Mullin, J.M.; Knudsen, K.A.; Marano, C.W. Tissue remodeling during tumor necrosis factor-induced apoptosis in LLC-PK1 renal epithelial cells. Am. J. Physiol. Ren. Physiol. 1996, 270, F869–F879. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, K.K.; Meldrum, D.R.; Hile, K.L.; Yerkes, E.B.; Ayala, A.; Cain, M.P.; Rink, R.C.; Casale, A.J.; Kaefer, M.A. p38 MAPK mediates renal tubular cell TNF-α production and TNF-α-dependent apoptosis during simulated ischemia. Am. J. Physiol. Cell Physiol. 2001, 281, C563–C570. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-L.; Baysal, K.; Kang, B.; Yang, L.-J.; Williamson, J.R. Correlation between Sustained c-Jun N-terminal Protein Kinase Activation and Apoptosis Induced by Tumor Necrosis Factor-α in Rat Mesangial Cells. J. Biol. Chem. 1998, 273, 4027–4034. [Google Scholar] [CrossRef]
- Lampropoulou, I.-T.; Stangou, M.; Papagianni, A.; Didangelos, T.; Iliadis, F.; Efstratiadis, G. TNF-α and Microalbuminuria in Patients with Type 2 Diabetes Mellitus. J. Diabetes Res. 2014, 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.F.; Mora, C.; Muros, M.; García, J. Urinary tumour necrosis factor-α excretion independently correlates with clinical markers of glomerular and tubulointerstitial injury in type 2 diabetic patients. Nephrol. Dial. Transplant. 2006, 21, 3428–3434. [Google Scholar] [CrossRef]
- McCarthy, E.T.; Sharma, R.; Sharma, M.; Li, J.Z.; Ge, X.L.; Dileepan, K.N.; Savin, V.J. TNF-alpha increases albumin permeability of isolated rat glomeruli through the generation of superoxide. J. Am. Soc. Nephrol. 1998, 9, 433. [Google Scholar]
- Awad, A.S.; You, H.; Gao, T.; Cooper, T.K.; Nedospasov, S.A.; Vacher, J.; Wilkinson, P.F.; Farrell, F.X.; Brian Reeves, W. Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury. Kidney Int. 2015, 88, 722–733. [Google Scholar] [CrossRef]
- Gao, G.; Zhang, B.; Ramesh, G.; Betterly, D.; Tadagavadi, R.K.; Wang, W.; Reeves, W.B. TNF-α mediates increased susceptibility to ischemic AKI in diabetes. Am. J. Physiol. Ren. Physiol. 2013, 304, F515–F521. [Google Scholar] [CrossRef]
- Wang, H.; Li, J.; Gai, Z.; Kullak-Ublick, G.A.; Liu, Z. TNF-α Deficiency Prevents Renal Inflammation and Oxidative Stress in Obese Mice. Kidney Blood Press. Res. 2017, 42, 416–427. [Google Scholar] [CrossRef]
- Karkar, A.M.; Smith, J.; Pusey, C.D. Prevention and treatment of experimental crescentic glomerulonephritis by blocking tumour necrosis factor-α. Nephrol. Dial. Transplant. 2001, 16, 518–524. [Google Scholar] [CrossRef]
- Wen, Y.; Rudemiller, N.P.; Zhang, J.; Robinette, T.; Lu, X.; Ren, J.; Privratsky, J.R.; Nedospasov, S.A.; Crowley, S.D. TNF-α in T lymphocytes attenuates renal injury and fibrosis during nephrotoxic nephritis. Am. J. Physiol. Ren. Physiol. 2019, 318, F107–F116. [Google Scholar] [CrossRef] [PubMed]
- Timoshanko, J.R.; Kitching, A.R.; Iwakura, Y.; Holdsworth, S.R.; Tipping, P.G. Leukocyte-derived interleukin-1beta interacts with renal interleukin-1 receptor I to promote renal tumor necrosis factor and glomerular injury in murine crescentic glomerulonephritis. Am. J. Pathol. 2004, 164, 1967–1977. [Google Scholar] [CrossRef]
- Niemir, Z.I.; Stein, H.; Dworacki, G.; Mundel, P.; Koehl, N.; Koch, B.; Autschbach, F.; Andrassy, K.; Ritz, E.; Waldherr, R.; et al. Podocytes are the major source of IL-1α and IL-1β in human glomerulonephritides. Kidney Int. 1997, 52, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.-J. Of Inflammasomes and Alarmins: IL-1β and IL-1α in Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 2564. [Google Scholar]
- Miao, N.-j.; Xie, H.-y.; Xu, D.; Yin, J.-y.; Wang, Y.-z.; Wang, B.; Yin, F.; Zhou, Z.-l.; Cheng, Q.; Chen, P.-P.; et al. Caspase-11 promotes renal fibrosis by stimulating IL-1β maturation via activating caspase-1. Acta Pharmacol. Sin. 2019, 40, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.-L.; Hua, K.-F.; Chen, A.; Wei, C.-W.; Chen, W.-S.; Wu, C.-Y.; Chu, C.-L.; Yu, Y.-L.; Lo, C.-W.; Ka, S.-M. NLRP3 inflammasome: Pathogenic role and potential therapeutic target for IgA nephropathy. Sci. Rep. 2017, 7, 41123. [Google Scholar] [CrossRef]
- Chow, F.Y.; Nikolic-Paterson, D.J.; Atkins, R.C.; Tesch, G.H. Macrophages in streptozotocin-induced diabetic nephropathy: Potential role in renal fibrosis. Nephrol. Dial. Transplant. 2004, 19, 2987–2996. [Google Scholar] [CrossRef]
- Vesey, D.A.; Cheung, C.; Cuttle, L.; Endre, Z.; Gobe, G.; Johnson, D.W. Interleukin-1β stimulates human renal fibroblast proliferation and matrix protein production by means of a transforming growth factor-β-dependent mechanism. J. Lab. Clin. Med. 2002, 140, 342–350. [Google Scholar] [CrossRef]
- Lovett, D.H.; Ryan, J.L.; Sterzel, R.B. Stimulation of rat mesangial cell proliferation by macrophage interleukin 1. J. Immunol. 1983, 131, 2830. [Google Scholar]
- Lonnemann, G.; Engler-Blum, G.; Müller, G.A.; Koch, K.M.; Dubarello, C.A. Cytokines in human renal interstitial fibrosis. II. Intrinsic interleukin (IL)-1 synthesis and IL-1-dependent production of IL-6 and IL-8 by cultured kidney fibroblasts. Kidney Int. 1995, 47, 845–854. [Google Scholar] [CrossRef][Green Version]
- Robson, R.L.; Westwick, J.; Brown, Z. Interleukin-1-induced IL-8 and IL-6 gene expression and production in human mesangial cells is differentially regulated by cAMP. Kidney Int. 1995, 48, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Han, W.; Song, S.; Du, Y.; Liu, C.; Chen, N.; Wu, H.; Shi, Y.; Duan, H. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol. Cell. Endocrinol. 2018, 478, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Torbohm, I.; Berger, B.; Schönermark, M.; von Kempis, J.; Rother, K.; Hänsch, G.M. Modulation of collagen synthesis in human glomerular epithelial cells by interleukin 1. Clin. Exp. Immunol. 1989, 75, 427–431. [Google Scholar] [PubMed]
- Fan, J.-M.; Huang, X.-R.; Ng, Y.-Y.; Nikolic-Paterson, D.J.; Mu, W.; Atkins, R.C.; Lan, H.Y. Interleukin-1 induces tubular epithelial-myofibroblast transdifferentiation through a transforming growth factor-β1-dependent mechanism in vitro. Am. J. Kidney Dis. 2001, 37, 820–831. [Google Scholar] [CrossRef]
- Zhang, M.; Tang, J.; Li, X. Interleukin-1β-Induced Transdifferentiation of Renal Proximal Tubular Cells Is Mediated by Activation of JNK and p38 MAPK. Nephron Exp. Nephrol. 2005, 99, e68–e76. [Google Scholar] [CrossRef]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.D.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef]
- Timoshanko, J.R.; Kitching, A.R.; Iwakura, Y.; Holdsworth, S.R.; Tipping, P.G. Contributions of IL-1β and IL-1α to Crescentic Glomerulonephritis in Mice. J. Am. Soc. Nephrol. 2004, 15, 910. [Google Scholar] [CrossRef]
- Lei, Y.; Devarapu, S.K.; Motrapu, M.; Cohen, C.D.; Lindenmeyer, M.T.; Moll, S.; Kumar, S.V.; Anders, H.-J. Interleukin-1β Inhibition for Chronic Kidney Disease in Obese Mice With Type 2 Diabetes. Front. Immunol. 2019, 10, 1223. [Google Scholar] [CrossRef]
- Zhang, Z.; Shao, X.; Jiang, N.; Mou, S.; Gu, L.; Li, S.; Lin, Q.; He, Y.; Zhang, M.; Zhou, W.; et al. Caspase-11-mediated tubular epithelial pyroptosis underlies contrast-induced acute kidney injury. Cell Death Dis. 2018, 9, 983. [Google Scholar] [CrossRef]
- Lorenz, G.; Darisipudi, M.N.; Anders, H.-J. Canonical and non-canonical effects of the NLRP3 inflammasome in kidney inflammation and fibrosis. Nephrol. Dial. Transplant. 2013, 29, 41–48. [Google Scholar] [CrossRef]
- Chung, S.D.; Lai, T.Y.; Chien, C.T.; Yu, H.J. Activating Nrf-2 signaling depresses unilateral ureteral obstruction-evoked mitochondrial stress-related autophagy, apoptosis and pyroptosis in kidney. PLoS ONE 2012, 7, e47299. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-R.; Yao, F.-H.; Zhang, J.-G.; Ji, Z.-Y.; Li, K.-L.; Zhan, J.; Tong, Y.-N.; Lin, L.-R.; He, Y.-N. Ischemia-reperfusion induces renal tubule pyroptosis via the CHOP-caspase-11 pathway. Am. J. Physiol. Ren. Physiol. 2013, 306, F75–F84. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Lee, D.W.; Ravichandran, K.; O Keys, D.; Akcay, A.; Nguyen, Q.; He, Z.; Jani, A.; Ljubanovic, D.; Edelstein, C.L. NLRP3 inflammasome knockout mice are protected against ischemic but not cisplatin-induced acute kidney injury. J. Pharm. Exp. Ther. 2013, 346, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Vilaysane, A.; Chun, J.; Seamone, M.E.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.A.; Tschopp, J.; Trpkov, K.; et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. 2010, 21, 1732–1744. [Google Scholar] [CrossRef] [PubMed]
- Hutton, H.L.; Ooi, J.D.; Holdsworth, S.R.; Kitching, A.R. The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology 2016, 21, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Craft, M.L.; Wang, P.; Wyburn, K.R.; Chen, G.; Ma, J.; Hambly, B.; Chadban, S.J. IL-18 contributes to renal damage after ischemia-reperfusion. J. Am. Soc. Nephrol. 2008, 19, 2331–2341. [Google Scholar] [CrossRef]
- Bani-Hani, A.H.; Leslie, J.A.; Asanuma, H.; Dinarello, C.A.; Campbell, M.T.; Meldrum, D.R.; Zhang, H.; Hile, K.; Meldrum, K.K. IL-18 neutralization ameliorates obstruction-induced epithelial–mesenchymal transition and renal fibrosis. Kidney Int. 2009, 76, 500–511. [Google Scholar] [CrossRef]
- Kitching, A.R.; Turner, A.L.; Wilson, G.R.A.; Semple, T.; Odobasic, D.; Timoshanko, J.R.; O’Sullivan, K.M.; Tipping, P.G.; Takeda, K.; Akira, S.; et al. IL-12p40 and IL-18 in Crescentic Glomerulonephritis: IL-12p40 is the Key Th1-Defining Cytokine Chain, Whereas IL-18 Promotes Local Inflammation and Leukocyte Recruitment. J. Am. Soc. Nephrol. 2005, 16, 2023. [Google Scholar] [CrossRef]
- Moutabarrik, A.; Nakanishi, I.; Ishibashi, M. Interleukin-6 and Interleukin-6 Receptor are Expressed by Cultured Glomerular Epithelial Cells. Scand. J. Immunol. 1994, 40, 181–186. [Google Scholar] [CrossRef]
- Boswell, R.N.; Yard, B.A.; Schrama, E.; van Es, L.A.; Daha, M.R.; van der Woude, F.J. Interleukin 6 production by human proximal tubular epithelial cells in vitro: Analysis of the effects of interleukin-1α (IL-1α) and other cytokines. Nephrol. Dial. Transplant. 1994, 9, 599–606. [Google Scholar] [CrossRef]
- van den Dobbelsteen, M.E.A.; van der Woude, F.J.; Schroeijers, W.E.M.; van Es, L.A.; Daha, M.R. Soluble aggregates of IgG and immune complexes enhance IL-6 production by renal mesangial cells. Kidney Int. 1993, 43, 544–553. [Google Scholar] [CrossRef]
- Ruef, C.; Budde, K.; Lacy, J.; Northemann, W.; Baumann, M.; Sterzel, R.B.; Coleman, D.L. Interleukin 6 is an autocrine growth factor for mesangial cells. Kidney Int. 1990, 38, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Lei, C.-T.; Zhang, C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [PubMed]
- Suematsu, S.; Matsuda, T.; Aozasa, K.; Akira, S.; Nakano, N.; Ohno, S.; Miyazaki, J.; Yamamura, K.; Hirano, T.; Kishimoto, T. IgG1 plasmacytosis in interleukin 6 transgenic mice. Proc. Natl. Acad. Sci. USA 1989, 86, 7547–7751. [Google Scholar] [CrossRef] [PubMed]
- Coletta, I.; Soldo, L.; Polentarutti, N.; Mancini, F.; Guglielmotti, A.; Pinza, M.; Mantovani, A.; Milanese, C. Selective Induction of MCP-1 in Human Mesangial Cells by the IL-6/sIL-6R Complex. Nephron Exp. Nephrol. 2000, 8, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.I.; Park, S.H. Sequential signaling cascade of IL-6 and PGC-1α is involved in high glucose-induced podocyte loss and growth arrest. Biochem. Biophys. Res. Commun. 2013, 435, 702–707. [Google Scholar] [CrossRef]
- Nechemia-Arbely, Y.; Barkan, D.; Pizov, G.; Shriki, A.; Rose-John, S.; Galun, E.; Axelrod, J.H. IL-6/IL-6R Axis Plays a Critical Role in Acute Kidney Injury. J. Am. Soc. Nephrol. 2008, 19, 1106. [Google Scholar] [CrossRef]
- Patel, N.S.A.; Chatterjee, P.K.; Di Paola, R.; Mazzon, E.; Britti, D.; De Sarro, A.; Cuzzocrea, S.; Thiemermann, C. Endogenous Interleukin-6 Enhances the Renal Injury, Dysfunction, and Inflammation Caused by Ischemia/Reperfusion. J. Pharmacol. Exp. Ther. 2005, 312, 1170–1178. [Google Scholar] [CrossRef]
- Chen, W.; Yuan, H.; Cao, W.; Wang, T.; Chen, W.; Yu, H.; Fu, Y.; Jiang, B.; Zhou, H.; Guo, H.; et al. Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics 2019, 9, 3980–3991. [Google Scholar] [CrossRef]
- Karkar, A.M.; Smith, J.; Tam, F.W.K.; Pusey, C.D.; Rees, A.J. Abrogation of glomerular injury in nephrotoxic nephritis by continuous infusion of interleukin-6. Kidney Int. 1997, 52, 1313–1320. [Google Scholar] [CrossRef]
- Strutz, F.; Zeisberg, M.; Renziehausen, A.; Raschke, B.; Becker, V.; Van Kooten, C.; Müller, G.A. TGF-β1 induces proliferation in human renal fibroblasts via induction of basic fibroblast growth factor (FGF-2). Kidney Int. 2001, 59, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am. J. Pathol. 2001, 159, 1465–1475. [Google Scholar] [CrossRef]
- Chung, S.; Overstreet, J.M.; Li, Y.; Wang, Y.; Niu, A.; Wang, S.; Fan, X.; Sasaki, K.; Jin, G.-N.; Khodo, S.N.; et al. TGF-β promotes fibrosis after severe acute kidney injury by enhancing renal macrophage infiltration. JCI Insight 2018, 3, e123563. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, M.; Bitzer, M.; Roberts, I.S.; Kopp, J.B.; ten Dijke, P.; Mundel, P.; Böttinger, E.P. Apoptosis in podocytes induced by TGF-beta and Smad7. J. Clin. Investig. 2001, 108, 807–816. [Google Scholar] [CrossRef]
- Das, R.; Xu, S.; Nguyen, T.T.; Quan, X.; Choi, S.-K.; Kim, S.-J.; Lee, E.Y.; Cha, S.-K.; Park, K.-S. Transforming Growth Factor β1-induced Apoptosis in Podocytes via the Extracellular Signal-regulated Kinase-Mammalian Target of Rapamycin Complex 1-NADPH Oxidase 4 Axis. J. Biol. Chem. 2015, 290, 30830–30842. [Google Scholar] [CrossRef]
- Ziyadeh, F.N.; Hoffman, B.B.; Han, D.C.; Iglesias-De La Cruz, M.C.; Hong, S.W.; Isono, M.; Chen, S.; McGowan, T.A.; Sharma, K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 8015–8020. [Google Scholar] [CrossRef]
- Gewin, L.; Vadivelu, S.; Neelisetty, S.; Srichai, M.B.; Paueksakon, P.; Pozzi, A.; Harris, R.C.; Zent, R. Deleting the TGF-β receptor attenuates acute proximal tubule injury. J. Am. Soc. Nephrol. 2012, 23, 2001–2011. [Google Scholar] [CrossRef]
- Kulkarni, A.B.; Ward, J.M.; Yaswen, L.; Mackall, C.L.; Bauer, S.R.; Huh, C.G.; Gress, R.E.; Karlsson, S. Transforming growth factor-beta 1 null mice. An animal model for inflammatory disorders. Am. J. Pathol. 1995, 146, 264–275. [Google Scholar]
- Fujimoto, M.; Maezawa, Y.; Yokote, K.; Joh, K.; Kobayashi, K.; Kawamura, H.; Nishimura, M.; Roberts, A.B.; Saito, Y.; Mori, S. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem. Biophys. Res. Commun. 2003, 305, 1002–1007. [Google Scholar] [CrossRef]
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494. [Google Scholar] [CrossRef]
- Lan, H.Y.; Mu, W.; Tomita, N.; Huang, X.R.; Li, J.H.; Zhu, H.-J.; Morishita, R.; Johnson, R.J. Inhibition of Renal Fibrosis by Gene Transfer of Inducible Smad7 Using Ultrasound-Microbubble System in Rat UUO Model. J. Am. Soc. Nephrol. 2003, 14, 1535. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.-C.; Wang, W.; Huang, X.R.; Fu, P.; Chen, T.-H.; Sheikh-Hamad, D.; Lan, H.Y. Ultrasound-microbubble-mediated gene transfer of inducible Smad7 blocks transforming growth factor-beta signaling and fibrosis in rat remnant kidney. Am. J. Pathol. 2005, 166, 761–771. [Google Scholar] [CrossRef]
- Ka, S.M.; Yeh, Y.C.; Huang, X.R.; Chao, T.K.; Hung, Y.J.; Yu, C.P.; Lin, T.J.; Wu, C.C.; Lan, H.Y.; Chen, A. Kidney-targeting Smad7 gene transfer inhibits renal TGF-β/MAD homologue (SMAD) and nuclear factor κB (NF-κB) signalling pathways, and improves diabetic nephropathy in mice. Diabetologia 2012, 55, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Huang, X.R.; Wang, W.; Li, J.H.; Heuchel, R.L.; Chung, A.C.K.; Lan, H.Y. The protective role of Smad7 in diabetic kidney disease: Mechanism and therapeutic potential. Diabetes 2011, 60, 590–601. [Google Scholar] [CrossRef]
- Nangaku, M. Chronic Hypoxia and Tubulointerstitial Injury: A Final Common Pathway to End-Stage Renal Failure. J. Am. Soc. Nephrol. 2006, 17, 17. [Google Scholar] [CrossRef]
- Tanaka, S.; Tanaka, T.; Nangaku, M. Hypoxia as a key player in the AKI-to-CKD transition. Am. J. Physiol. Ren. Physiol. 2014, 307, F1187–F1195. [Google Scholar] [CrossRef]
- Fine, L.G.; Bandyopadhay, D.; Norman, J.T. Is there a common mechanism for the progression of different types of renal diseases other than proteinuria? Towards the unifying theme of chronic hypoxia. Kidney Int. 2000, 57, S22–S26. [Google Scholar] [CrossRef]
- Singh, P.; Ricksten, S.-E.; Bragadottir, G.; Redfors, B.; Nordquist, L. Renal oxygenation and haemodynamics in acute kidney injury and chronic kidney disease. Clin. Exp. Pharm. Physiol. 2013, 40, 138–147. [Google Scholar] [CrossRef]
- Sada, K.; Nishikawa, T.; Kukidome, D.; Yoshinaga, T.; Kajihara, N.; Sonoda, K.; Senokuchi, T.; Motoshima, H.; Matsumura, T.; Araki, E. Hyperglycemia Induces Cellular Hypoxia through Production of Mitochondrial ROS Followed by Suppression of Aquaporin-1. PLoS ONE 2016, 11, e0158619. [Google Scholar] [CrossRef]
- Basile, D.P.; Friedrich, J.L.; Spahic, J.; Knipe, N.; Mang, H.; Leonard, E.C.; Changizi-Ashtiyani, S.; Bacallao, R.L.; Molitoris, B.A.; Sutton, T.A. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am. J. Physiol. Ren. Physiol. 2010, 300, F721–F733. [Google Scholar] [CrossRef]
- Basile, D.P.; Donohoe, D.; Roethe, K.; Osborn, J.L. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am. J. Physiol. Ren. Physiol. 2001, 281, F887–F899. [Google Scholar] [CrossRef]
- Mazzali, M.; Jefferson, J.A.; Ni, Z.; Vaziri, N.D.; Johnson, R.J. Microvascular and tubulointerstitial injury associated with chronic hypoxia-induced hypertension. Kidney Int. 2003, 63, 2088–2093. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Norman, J.T.; Clark, I.M.; Garcia, P.L. Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int. 2000, 58, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Makino, Y.; Tanaka, T.; Tanaka, H.; Ishizaka, N.; Noiri, E.; Fujita, T.; Nangaku, M. Induction of Renoprotective Gene Expression by Cobalt Ameliorates Ischemic Injury of the Kidney in Rats. J. Am. Soc. Nephrol. 2003, 14, 1825. [Google Scholar] [CrossRef] [PubMed]
- Ohtomo, S.; Nangaku, M.; Izuhara, Y.; Takizawa, S.; Strihou, C.V.Y.D.; Miyata, T. Cobalt ameliorates renal injury in an obese, hypertensive type 2 diabetes rat model. Nephrol. Dial. Transplant. 2007, 23, 1166–1172. [Google Scholar] [CrossRef]
- Brigandi, R.A.; Johnson, B.; Oei, C.; Westerman, M.; Olbina, G.; de Zoysa, J.; Roger, S.D.; Sahay, M.; Cross, N.; McMahon, L.; et al. A Novel Hypoxia-Inducible Factor−Prolyl Hydroxylase Inhibitor (GSK1278863) for Anemia in CKD: A 28-Day, Phase 2A Randomized Trial. Am. J. Kidney Dis. 2016, 67, 861–871. [Google Scholar] [CrossRef]
- Pergola, P.E.; Spinowitz, B.S.; Hartman, C.S.; Maroni, B.J.; Haase, V.H. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney Int. 2016, 90, 1115–1122. [Google Scholar] [CrossRef]
- Provenzano, R.; Besarab, A.; Sun, C.H.; Diamond, S.A.; Durham, J.H.; Cangiano, J.L.; Aiello, J.R.; Novak, J.E.; Lee, T.; Leong, R.; et al. Oral Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor Roxadustat (FG-4592) for the Treatment of Anemia in Patients with CKD. Clin. J. Am. Soc. Nephrol. 2016, 11, 982–991. [Google Scholar] [CrossRef]
- Holdstock, L.; Meadowcroft, A.M.; Maier, R.; Johnson, B.M.; Jones, D.; Rastogi, A.; Zeig, S.; Lepore, J.J.; Cobitz, A.R. Four-Week Studies of Oral Hypoxia-Inducible Factor-Prolyl Hydroxylase Inhibitor GSK1278863 for Treatment of Anemia. J. Am. Soc. Nephrol. 2016, 27, 1234–1244. [Google Scholar] [CrossRef]
- Uchida, T.; Rossignol, F.; Matthay, M.A.; Mounier, R.; Couette, S.; Clottes, E.; Clerici, C. Prolonged Hypoxia Differentially Regulates Hypoxia-inducible Factor (HIF)-1α and HIF-2α Expression in Lung Epithelial Cells: IMPLICATION OF NATURAL ANTISENSE HIF-1α. J. Biol. Chem. 2004, 279, 14871–14878. [Google Scholar] [CrossRef]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.; Heyman, S.N.; Rosen, S.; Shina, A.; Goldfarb, M.; Griethe, W.; Frei, U.; Reinke, P.; Bachmann, S.; Eckardt, K.-U. Up-regulation of HIF in experimental acute renal failure: Evidence for a protective transcriptional response to hypoxia. Kidney Int. 2005, 67, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Ginouvès, A.; Ilc, K.; Macías, N.; Pouysségur, J.; Berra, E. PHDs overactivation during chronic hypoxia “desensitizes” HIFα and protects cells from necrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 4745–4750. [Google Scholar] [CrossRef] [PubMed]
- Marxsen, J.H.; Stengel, P.; Doege, K.; Heikkinen, P.; Jokilehto, T.; Wagner, T.; Jelkmann, W.; Jaakkola, P.; Metzen, E. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. Biochem. J. 2004, 381, 761–767. [Google Scholar] [CrossRef]
- Hsu, Y.-H.; Li, H.-H.; Hsieh, M.-Y.; Liu, M.-F.; Huang, K.-Y.; Chin, L.-S.; Chen, P.-C.; Cheng, H.-H.; Chang, M.-S. Function of interleukin-20 as a proinflammatory molecule in rheumatoid and experimental arthritis. Arthritis Rheum. 2006, 54, 2722–2733. [Google Scholar] [CrossRef]
- Hsu, Y.-H.; Yang, Y.-Y.; Huwang, M.-H.; Weng, Y.-H.; Jou, I.M.; Wu, P.-T.; Lin, T.-Y.; Wu, L.-W.; Chang, M.-S. Anti-IL-20 monoclonal antibody inhibited inflammation and protected against cartilage destruction in murine models of osteoarthritis. PLoS ONE 2017, 12, e0175802. [Google Scholar] [CrossRef]
- Hsu, Y.-H.; Wei, C.-C.; Shieh, D.-B.; Chan, C.-H.; Chang, M.-S. Anti–IL-20 Monoclonal Antibody Alleviates Inflammation in Oral Cancer and Suppresses Tumor Growth. Mol. Cancer Res. 2012, 10, 1430. [Google Scholar] [CrossRef]
- Chen, W.-Y.; Cheng, B.-C.; Jiang, M.-J.; Hsieh, M.-Y.; Chang, M.-S. IL-20 Is Expressed in Atherosclerosis Plaques and Promotes Atherosclerosis in Apolipoprotein E–Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2090–2095. [Google Scholar] [CrossRef]
- Hsieh, M.Y.; Chen, W.Y.; Jiang, M.J.; Cheng, B.C.; Huang, T.Y.; Chang, M.S. Interleukin-20 promotes angiogenesis in a direct and indirect manner. Genes Immun. 2006, 7, 234–242. [Google Scholar] [CrossRef]
- Gong, W.; Wang, X.; Zhang, Y.; Hao, J.; Xing, C.; Chu, Q.; Wang, G.; Zhao, J.; Wang, J.; Dong, Q.; et al. Interleukin-20 Promotes Airway Remodeling in Asthma. Inflammation 2014, 37, 2099–2105. [Google Scholar] [CrossRef]
- Li, H.H.; Hsu, Y.H.; Wei, C.C.; Lee, P.T.; Chen, W.C.; Chang, M.S. Interleukin-20 induced cell death in renal epithelial cells and was associated with acute renal failure. Genes Immun. 2008, 9, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-Y.; Chang, M.-S. IL-20 Is Regulated by Hypoxia-Inducible Factor and Up-Regulated after Experimental Ischemic Stroke. J. Immunol. 2009, 182, 5003. [Google Scholar] [CrossRef]
- Chiu, Y.-S.; Hsing, C.-H.; Li, C.-F.; Lee, C.-Y.; Hsu, Y.-H.; Chang, M.-S. Anti-IL-20 monoclonal antibody inhibited tumor growth in hepatocellular carcinoma. Sci. Rep. 2017, 7, 17609. [Google Scholar] [CrossRef]
- Chiu, Y.-S.; Wei, C.-C.; Lin, Y.-J.; Hsu, Y.-H.; Chang, M.-S. IL-20 and IL-20R1 antibodies protect against liver fibrosis. Hepatology 2014, 60, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Bergholdt, R.; Cucak, H.; Rolin, B.C.; Sams, A.; Rosendahl, A. Neutralizing Anti-IL20 Antibody Treatment Significantly Modulates Low Grade Inflammation without Affecting HbA1c in Type 2 Diabetic db/db Mice. PLoS ONE 2015, 10, e0131306. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.-C.; Li, H.-H.; Hsu, Y.-H.; Hsing, C.-H.; Sung, J.-M.; Chang, M.-S. Interleukin-20 targets renal cells and is associated with chronic kidney disease. Biochem. Biophys. Res. Commun. 2008, 374, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-H.; Li, H.-H.; Sung, J.-M.; Chen, W.-Y.; Hou, Y.-C.; Weng, Y.-H.; Lai, W.-T.; Wu, C.-H.; Chang, M.-S. Interleukin-20 targets podocytes and is upregulated in experimental murine diabetic nephropathy. Exp. Mol. Med. 2017, 49, e310. [Google Scholar] [CrossRef]
- Li, H.-H.; Cheng, H.-H.; Sun, K.-H.; Wei, C.-C.; Li, C.-F.; Chen, W.-C.; Wu, W.-M.; Chang, M.-S. Interleukin-20 targets renal mesangial cells and is associated with lupus nephritis. Clin. Immunol. 2008, 129, 277–285. [Google Scholar] [CrossRef]
- Wei, C.-C.; Chen, W.-Y.; Wang, Y.-C.; Chen, P.-J.; Lee, J.Y.-Y.; Wong, T.-W.; Chen, W.C.; Wu, J.-C.; Chen, G.-y.; Chang, M.-S.; et al. Detection of IL-20 and its receptors on psoriatic skin. Clin. Immunol. 2005, 117, 65–72. [Google Scholar] [CrossRef]
- Stenderup, K.; Rosada, C.; Worsaae, A.; Clausen, J.T.; Norman Dam, T. Interleukin-20 as a Target in Psoriasis Treatment. Ann. N.Y. Acad. Sci. 2007, 1110, 368–381. [Google Scholar] [CrossRef]
- Kragstrup, T.W.; Andersen, M.N.; Schiøttz-Christensen, B.; Jurik, A.G.; Hvid, M.; Deleuran, B. Increased interleukin (IL)-20 and IL-24 target osteoblasts and synovial monocytes in spondyloarthritis. Clin. Exp. Immunol. 2017, 189, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-H.; Chen, W.-Y.; Chan, C.-H.; Wu, C.-H.; Sun, Z.-J.; Chang, M.-S. Anti-IL-20 monoclonal antibody inhibits the differentiation of osteoclasts and protects against osteoporotic bone loss. J. Exp. Med. 2011, 208, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-Y.; Lin, R.M.; Chen, W.-Y.; Lee, C.-L.; Yan, J.; Chang, M.-S. IL20 May Contribute to the Pathogenesis of Human Intervertebral Disc Herniation. Spine 2008, 33, 2034–2040. [Google Scholar] [CrossRef]
- Jha, J.C.; Banal, C.; Chow, B.S.M.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef] [PubMed]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatric Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Manrique, J.; Cravedi, P. Role of monoclonal antibodies in the treatment of immune-mediated glomerular diseases. Nefrol. Publ. Of. de La Soc. Esp. Nefrol. 2014, 34, 388–397. [Google Scholar]
- Hsu, Y.-H.; Chang, M.-S. Interleukin-20 antibody is a potential therapeutic agent for experimental arthritis. Arthritis Rheum. 2010, 62, 3311–3321. [Google Scholar] [CrossRef]
- Kragstrup, T.W.; Andersen, T.; Heftdal, L.D.; Hvid, M.; Gerwien, J.; Sivakumar, P.; Taylor, P.C.; Senolt, L.; Deleuran, B. The IL-20 Cytokine Family in Rheumatoid Arthritis and Spondyloarthritis. Front. Immunol. 2018, 9, 2226. [Google Scholar] [CrossRef]
- Hsu, Y.-H.; Li, H.-H.; Sung, J.-M.; Chen, W.-T.; Hou, Y.-C.; Chang, M.-S. Interleukin-19 mediates tissue damage in murine ischemic acute kidney injury. PLoS ONE 2013, 8, e56028. [Google Scholar] [CrossRef][Green Version]
- Jennings, P.; Crean, D.; Aschauer, L.; Limonciel, A.; Moenks, K.; Kern, G.; Hewitt, P.; Lhotta, K.; Lukas, A.; Wilmes, A.; et al. Interleukin-19 as a translational indicator of renal injury. Arch. Toxicol. 2015, 89, 101–106. [Google Scholar] [CrossRef]
- Li, L.; Jiang, X.-G.; Hu, J.-Y.; Yu, Z.-Q.; Xu, J.-Y.; Liu, F.; Zhao, G.-C.; Zhang, L.; Gu, H.-M.; Zhang, S.-J.; et al. The association between interleukin-19 concentration and diabetic nephropathy. BMC Nephrol. 2017, 18, 65. [Google Scholar] [CrossRef] [PubMed]
- Pap, D.; Sziksz, E.; Kiss, Z.; Rokonay, R.; Veres-Székely, A.; Lippai, R.; Takács, I.M.; Kis, E.; Fekete, A.; Reusz, G.; et al. Microarray Analysis Reveals Increased Expression of Matrix Metalloproteases and Cytokines of Interleukin-20 Subfamily in the Kidneys of Neonate Rats Underwent Unilateral Ureteral Obstruction: A Potential Role of IL-24 in the Regulation of Inflammation and Tissue Remodeling. Kidney Blood Press. Res. 2017, 42, 16–32. [Google Scholar] [PubMed]
- Wang, Z.; Wang, Y.; Chen, Y.; Lv, J. The IL-24 gene protects human umbilical vein endothelial cells against H2O2-induced injury and may be useful as a treatment for cardiovascular disease. Int. J. Mol. Med. 2016, 37, 581–592. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Organ | Diseases | Target Cells | Role | Reference |
|---|---|---|---|---|
| Brain | Ischemic Stroke | Glia-like cells | ↑ Inflammation ↑ Ischemic infarction | [162] |
| Mouth | Oral Cancer | Oral carcinoma cells | ↑ Tumor progression ↑ Inflammation | [157] |
| Airway | Asthma | Lung epithelial cells | ↑ Lung fibrosis | [160] |
| Arterial | Atherosclerosis | Endothelial cells | ↑ Inflammation ↑ Angiogenesis ↑ Atherosclerosis | [158] |
| Liver | Hepatocellular Carcinoma (HCC) | Liver cancer cells | ↑ Tumor progression | [163] |
| Liver Injury | Hepatocytes | ↑ Liver fibrosis ↑ Inflammation | [164] | |
| Pancreas | Type 2 Diabetes | Pancreatic islets | ↑ Inflammation | [165] |
| Kidney | Hgcl2-Induced AKI | Proximal tubular epithelial cells | ↑ Inflammation ↑ Renal fibrosis ↑ Cell death | [161] |
| 5/6 nephrectomy-Induced CKD | Tubular epithelial cells Interstitial fibroblasts | ↑ Renal fibrosis | [166] | |
| STZ-induced DN | Podocytes | ↑ Inflammation ↑ Fibrosis ↓ Renal function | [167] | |
| Lupus Nephritis | Mesangial cells | ↑ Inflammation | [168] | |
| Skin | Psoriasis | Keratinocytes | ↑ Cell proliferation | [169,170] |
| Bone | RA | Synovial fibroblasts Osteoclasts Osteoblasts Chondrocytes | ↑ Inflammation | [155] |
| Spondyloarthritis | Synovial fluid monocytes Synovial fibroblasts Osteoblasts | ↑ Inflammation ↑ Osteoblastogenesis | [171] | |
| Osteoporosis | Osteoclasts Osteoblasts | ↑ Osteoclastogenesis ↓ Osteoblastogenesis | [172] | |
| Osteoarthritis | Synovial fibroblasts Chondrocytes | ↑ Inflammation ↓ Chondrogenesis ↑ Osteoblastogenesis | [156] | |
| Intervertebral Disc (IVD) Herniation | Disc Cells | ↑ Inflammation | [173] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, T.-Y.; Hsu, Y.-H. IL-20 in Acute Kidney Injury: Role in Pathogenesis and Potential as a Therapeutic Target. Int. J. Mol. Sci. 2020, 21, 1009. https://doi.org/10.3390/ijms21031009
Lin T-Y, Hsu Y-H. IL-20 in Acute Kidney Injury: Role in Pathogenesis and Potential as a Therapeutic Target. International Journal of Molecular Sciences. 2020; 21(3):1009. https://doi.org/10.3390/ijms21031009
Chicago/Turabian StyleLin, Tian-Yu, and Yu-Hsiang Hsu. 2020. "IL-20 in Acute Kidney Injury: Role in Pathogenesis and Potential as a Therapeutic Target" International Journal of Molecular Sciences 21, no. 3: 1009. https://doi.org/10.3390/ijms21031009
APA StyleLin, T.-Y., & Hsu, Y.-H. (2020). IL-20 in Acute Kidney Injury: Role in Pathogenesis and Potential as a Therapeutic Target. International Journal of Molecular Sciences, 21(3), 1009. https://doi.org/10.3390/ijms21031009