Abstract
The kidney is innervated by afferent sensory and efferent sympathetic nerve fibers. Norepinephrine (NE) is the primary neurotransmitter for post-ganglionic sympathetic adrenergic nerves, and its signaling, regulated through adrenergic receptors (AR), modulates renal function and pathophysiology under disease conditions. Renal sympathetic overactivity and increased NE level are commonly seen in chronic kidney disease (CKD) and are critical factors in the progression of renal disease. Blockade of sympathetic nerve-derived signaling by renal denervation or AR blockade in clinical and experimental studies demonstrates that renal nerves and its downstream signaling contribute to progression of acute kidney injury (AKI) to CKD and fibrogenesis. This review summarizes our current knowledge of the role of renal sympathetic nerve and adrenergic receptors in AKI, AKI to CKD transition and CKDand provides new insights into the therapeutic potential of intervening in its signaling pathways.
1. Introduction
Acute kidney injury (AKI) is associated with increased mortality and risk of development of chronic kidney disease (CKD) in the long term [1,2]. CKD is characterized by the persistent loss of renal function that frequently occurs over time [1]. Approximately 8–16% of the world’s population have advanced CKD [3], which has added complications of poor quality of life, financial burden, and demand for renal replacement therapy. In addition to renal deficiency, patients with CKD may have comorbidities including diabetes, hypertension, heart diseases, and stroke [1,2]. Despite multiple etiologies, the underlying pathophysiological process of CKD progression is fibrogenesis. Prevention of AKI and its potential progression to CKD is a challenging clinical problem [4]. There is no cure for CKD and no effective treatment for CKD has been developed. Despite being a major unmet medical need, current efforts are restricted to the control of blood pressure (BP) and optimization of renin–angiotensin–aldosterone system (RAAS) blockade. These therapies, at best, may reduce proteinuria, a surrogate marker of renal disease, but they only partially reduce progression of CKD [5].
The kidneys are abundantly innervated by both renal afferent sensory and efferent sympathetic nerves and communicate with the central nervous system via the sensory afferent nerves [6,7]. Increased renal afferent activity directly influences sympathetic outflow to the kidneys via efferent nerves [6,7,8]. The efferent sympathetic nerves are crucial for production of norepinephrine (NE), a key effector of the sympathetic nervous system [9,10]. Epinephrine and NE act by binding to adrenergic receptors (ARs), classified as α1-, α2-, or β-AR [11]. The contribution of sympathetic nervous system to the progression of CKD, a condition commonly characterized by renal sympathetic nerve hyperactivity, is well known [12,13,14]. Increased plasma NE is observed in patients with CKD and is strongly linked with the development of kidney injury [15]. Furthermore, previous studies have found that the infusion of NE into the renal artery causes ischemic renal injury by constricting renal vasculature [16,17,18]. The renal sympathetic nervous system has drawn increasing interest over many years, after the introduction of renal sympathetic denervation into clinical medicine [19,20]. Despite the recognition of the renal sympathetic nerve system as the effector of renal dysfunction in CKD, its role in the progression and development of CKD has not been well defined.
Increased renal sympathetic nerve activity leads to increased renin secretion, increased renal tubular sodium reabsorption, and decreased renal blood flow [6,10,21]. Renal denervation (RDNx) is proposed as a therapeutic strategy used in treatment of resistant hypertension [8,21,22]. RDNx performed surgically or chemically reduces sympathetic nerve activity and has been shown to reduce BP, improve renal function, and attenuate the progression of CKD in experimental models and humans of hypertension and CKD [23,24,25,26,27,28,29,30]. We reported that RDNx prevented the development of tubulointerstitial fibrogenesis and inflammation after unilateral ureteral obstruction (UUO) and kidney ischemia/reperfusion injury (IRI) independent of BP changes [25,31]. In addition, RDNx did not worsen kidney function of patients and renal injured mice—as assessed by glomerular filtration rate (GFR), plasma creatinine, cystatin C, or urea levels, and tubular morphological damages—indicating that it is safe even when CKD is present [25,31,32,33]. Here, we review the progress in our understanding of the molecular mechanisms of NE and ARs signaling in AKI, AKI to CKD transition and CKD.
2. Adrenergic Receptors and Norepinephrine in the Kidney
Norepinephrine (NE) is an organic chemical in the catecholamine family that functions in the body as the main neurotransmitter of sympathetic nerve system [34]. NE released from the nerve terminals, together with co-transmitters, such as ATP and neuropeptide Y, regulate kidney function [35]. NE in the kidney is involved in a number of physiological processes, including control of renal blood flow, glomerular filtration rate, reabsorption of water, sodium, and other ions, and release of renin [6]. We recently found that renal nerve-derived NE signaling via α2-ARs, promotes renal inflammation and interstitial fibrosis [25,31].
ARs, which are G protein coupled receptors, are well known to participate in various physiological renal functions [36,37,38,39]. Localization of ARs has been confirmed using various approaches including in situ hybridization, immunohistochemistry, and competitive binding assays [40,41,42,43]. The presence of adrenergic receptors in nephron segments, including proximal convoluted tubules and cortical and medullary collecting ducts, has previously been demonstrated (Table 1) [40,42,43,44]. In the kidney, α1-AR is expressed in arterioles, whereas α2-AR is expressed predominantly in proximal tubules [45]. α1-AR and α2-AR are responsible for stimulation of renal vasoconstriction and Na+ reabsorption, respectively [45]. β-ARs can be divided into three subtypes: β1-AR, β2-AR, and β3-AR [11] and are expressed in most of the nephron segments, including proximal tubules and distal convoluted tubules [38,43]. The roles of β1- and β2-ARs are well known for regulating renal blood flow, glomerular filtration rate (GFR), sodium and water reabsorption, acid-base balance, and secretion of renin in the kidney [38]. β3-AR is important for regulating renal water and solute reabsorption [38,46]. The renal localization of ARs and their role for the regulation of renal function have been reviewed in detail, largely based on data obtained in experimental animal models [36,37,39,46].

Table 1.
Localization and action of adrenergic receptors in the kidney.
3. Renal Sympathetic Nervous System in AKI and CKD
Acute kidney injury (AKI) is defined as an abrupt or rapid decline in renal filtration function and is the result of combined acting of multiple factors [47,48]. Previous studies suggested that clinical AKI may initiate the onset of progressive renal diseases [49,50,51,52]. AKI can cause end-stage kidney disease (ESKD) directly, and increase the risk of developing CKD. Maladaptive repair after AKI may exceed the capacity of compromised renal reserve, resulting in the functional manifestation of CKD over time [53].
Renal sympathetic nervous system and circulating catecholamines including NE are considered to be involved in the development of the progressive renal tissue injury accompanying AKI [54,55]. In addition, NE injected into renal artery causes ischemic AKI with obstruction of tubule lumens, changes in renal hemodynamics and histological damage in the kidney [16,56,57]. Recent studies reported that renal ischemia is an important primary event leading to increased sympathetic nerve activity [58,59]. Renal venous plasma NE and renal tyrosine hydroxylase expression, which is related to NE synthesis, have been shown to increase after unilateral renal artery occlusion and bilateral renal ischemia/reperfusion (I/R) [58,60,61]. In our previous study, post-ischemic renal NE content increased up to day 16 after unilateral I/R [31]. Other studies reported that plasma and renal NE concentrations increased up to day 3 after bilateral I/R and returned to control level at day 7 [62]. Cisplatin, a widely used chemotherapeutic anti-cancer agent, induces nephrotoxicity and AKI [63]. Cisplatin-induced renal injury causes severe vasoconstriction and tissue damage, which activate renal sympathetic nerves system as shown by the increase in renal venous NE level after cisplatin administration and disrupt normal baroreflex, a reflex mechanism to maintain blood pressure (BP), and control of renal sympathetic nerve activity [64,65,66,67]. Indeed, cisplatin-induced renal failure is accompanied by reduced renal blood flow associated with increased renal vascular resistance [65,68,69].
Renal sympathetic nervous system contributes to pathogenesis of not only ischemic AKI, but also of CKD and hypertensive disease [34,70,71]. In our previous studies, it has been demonstrated that renal nerve-derived NE signaling induces fibrogenesis and inflammation in two different CKD models, UUO and unilateral I/R [25,31]. Sympathetic activity is already elevated in early phases of chronic renal failure [12,71]. These reports clearly demonstrate that enhanced renal sympathetic nerve activity and its consequent effects on NE overflow from nerve endings could play important roles in the development of AKI and its progression to CKD. Sympathetic nervous hyperactivity occurs in the CKD and hypertension, and the level of sympathetic nervous activity is associated with the increase of BP [72]. Increased renal sympathetic nerve activity enhances renin production and release leading to increased angiotensin II (Ang II), the main effector of the renin–angiotensin system (RAS) in hypertensive patients [73,74,75]. NE release mediates vasoconstriction of the renal vasculature, as well as sodium and water reabsorption at renal tubular epithelial cells, and renin release from the juxtaglomerular apparatus, which in turn results in increase of Ang II [76]. Conversely, Ang II can enhance NE release and reduce uptake of NE by acting on adrenergic nerve terminals, resulting in increased sympatho-excitation in the heart, kidney, and vasculature [8,77,78]. It has been proposed that the RAS activates the sympathetic nervous system and angiotensin-converting enzyme inhibition and angiotensin receptor blockade are antiadrenergic [78]. Obesity increases the susceptibility to AKI and subsequent progression to CKD [79,80]. Blockade using doxazosin, an α1-AR antagonist, clonidine, an α2A-AR agonist, and atenolol, a β1-AR antagonist, lowered BP and heart rate in obese dog and human patients [81,82,83]. In addition, RDNx attenuated sodium retention and prevented BP increase in a dog model of high fat diet-induced obesity [84]. There is evidence that angiotensin type 1 receptor interacts with α2A-AR. Genetic inhibition of α2A-AR or pharmacological inhibition of α2-AR lowers Ang II-mediated NE release in kidneys with 5/6 nephrectomy [85].
4. Inactivation of NE-AR Signaling in AKI and CKD
Several studies demonstrated beneficial effects of sympathetic nerve system inhibition on renal function and morphology. Clonidine and moxonidine, α2-AR agonist, and propranolol, β-AR antagonist, are known to have renoprotective actions against post-ischemic AKI [86,87,88]. We previously demonstrated that inhibition of α2-ARs prevents interstitial fibrogenesis after IRI, as indicated by reduced TGF-β1 production, Smad3 phosphorylation, α-SMA expression, and collagen deposition [31]. Other reports have indicated that inhibition of either α1-AR or β-AR protects the kidney against 5/6 nephrectomy-induced injury, and a combinational inhibition of α1-AR and β-AR is more effective in preventing renal injury than the inhibition of α1-AR or β-AR alone [89]. In uninephrectomized rats before ischemia and mice subjected to unilateral ischemia, yohimbine, and atipamezole, both α2-AR blockers and JP-1302, an α2C-AR specific blocker, mitigate I/R-induced renal tubular damage [31,90]. In the cisplatin-induced nephrotoxicity model, it has been reported that α2-AR inhibition with yohimbine and JP-1302 improves renal blood flow, suppresses increased plasma NE level, and ameliorates cisplatin-induced renal dysfunction and histological damages, by suppressing pro-inflammatory cytokine expression such as TNF- α and MCP-1 [91]. RDNx also restores baroreflex sensitivity to normal values, heart rate and BP in cisplatin-induce renal failure [64,92].
In addition, ischemic acute renal failure is ameliorated by RDNx or ganglionic blockade, which blocks the sympathetic nerves, and that effect is accompanied by suppression of elevated renal venous NE level immediately after reperfusion and attenuation of decreased GFR after the I/R [93]. Most studies reported that RDNx provides beneficial effects on BP of patients with stage 3–4 chronic kidney disease [94]. Accumulating evidence suggests that RDNx may help to delay the decline of renal function in chronic kidney disease [95,96]. Pharmacological denervation by DSP-4 also inhibits I/R-induced renal functional and morphological impairments [60]. Renalase is a monoamine oxidase secreted by the renal proximal tubules and degrades catecholamines including NE [97]. Prevented increase in plasma NE levels associated with I/R-induced renal cell death and kidney function impairment [98]. Genetic blockade of renalase increases plasma NE levels and suffers increased renal tubular injury with increased tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein-1 (MCP-1), and MIP-2 after renal I/R. Administration of recombinant renalase suppresses NE overproduction and attenuates ischemic renal damage [98].
Experimental studies demonstrated that renal denervation slowed or delayed the rise in BP in experimental models of hypertension such as spontaneous hypertension, salt-sensitive hypertension, Nω-nitro-L-arginine methyl ester (L-NAME) induced hypertension, Ang II induced hypertension and deoxycorticosterone acetate (DOCA)-salt hypertension [99,100,101,102,103,104,105]. Singh et al. reported that the reduction in BP and albuminuria and improved GFR in sheep with hypertensive CKD, observed at 5 months after catheter-based RDNx, were sustained until 30 months after RDNx, demonstrating the long-term durability of the beneficial effects of RDNx in hypertensive CKD [106]. Although the effect of RDNx is associated with suppressed RAS components expression, it remains to be defined whether renal sympathetic nerves or related signaling control RAS in CKD and hypertension progression through NE and adrenergic receptor.
On the other hand, activation of AR using agonist has protective effect in the kidney. It has been confirmed that dexmedetomidine, a selective α2-AR agonist, exerts notable positive effects in inhibiting sympathetic nerves and plasma NE levels and preventing kidney damage [107,108,109,110]. Dexmedetomidine effectively inhibits sympathetic activity and plays an important role in kidney protection by reducing inflammation, oxidative stress, and apoptosis through activating α2-AR and down-regulating TLR4 expression in I/R-induced [110] and LPS-treated mice [108]. Dexmedetomidine down-regulates the apoptosis of kidney tubular epithelial cells by inhibiting activation of signaling pathways, JAK/STAT and MAPK in I/R-induced rats [109]. Noh et al. have demonstrated that β2AR agonists have an anti-inflammatory action that targets macrophage activation in diabetic rats, especially in the kidney [111]. Overexpression of β2-AR using gene transfer with human β2-AR was effective in preventing endotoxin-induced renal injury with reduced elevation of TNF-α mRNA and leukocyte infiltration into the rat kidney [112].
5. Mechanisms of NE-AR Signaling in AKI and CKD
The mechanisms underlying AKI to CKD progression involves multiple interactions between injured tubules, immune cells, endothelial cells, and fibroblasts [113]. The importance of inflammation in the development and progression of kidney fibrosis is well established [113]. Once the kidney is injured, inflammatory cells infiltrate to the injured site and subsequently precede kidney fibrosis through production and release of profibrotic cytokines and growth factors [114]. These profibrotic cytokines and growth factors contribute to recruitment and activation of myofibroblasts, which cause progressive interstitial fibrosis, leading to CKD [114,115]. Renal efferent sympathetic nerves reportedly mediate renal inflammation through the trafficking and activation renal inflammatory immune cells and releasing inflammatory chemokine and cytokine content [116]. Le Clef et al. reported that the expression level of inflammatory cytokines, TNF-α and interleukin-6 (IL-6), remained higher with longer ischemia times [117]. The activation of α2-AR enhances the progression of dysfunction and inflammatory responses in the kidney [118]. Activation of α2-AR is responsible for upregulation of inflammatory cytokines including TNF-α and IL-6 and enhancement of granulocyte and monocyte recruitment [118]. NE or the α2-AR agonist UK-14304, significantly elevates LPS-induced TNF production from macrophages via α2-AR located on macrophages, while β-adrenergic agonist, isoproterenol, inhibits TNF release from macrophages [119,120,121]. In addition, inhibition of α2C-AR with α2c-AR antagonist JP-1302, after I/R, strongly suppressed cytokines, TNF-α, IL-6, IL-1β, and MCP-1 expression [122]. These increased inflammatory cytokines, IL-1β, IL-18, IL-6, and TNF-α recover at week 2 after RDNx [20]. During kidney interstitial fibrogenesis after UUO and IRI, RDNx attenuates the infiltration of neutrophils and macrophages [25,31]. In addition, it has been reported that RDNx reduces the accumulation of leukocytes, T cells, and both CD4+ and CD8+ T cells in the kidney of Ang II-treated hypertensive mice [116], decreases the activation of monocyte, monocyte-platelet aggregates formation, and plasma levels of monocyte-related cytokines and chemokines, MCP-1, IL-1 β, TNF- α, and IL-12 in human hypertensive patients [123]. NE has been implicated in promoting inflammatory cytokines release and production and myeloid cell recruitment into the injured site [124,125]. We previously reported that increased NE concentration correlated with the levels of inflammatory cytokines and with cell damage, in UUO or the I/R-induced mice model [25,31]. Furthermore, administration of α2-AR antagonists has no effect on NE level but decreases cytokine and chemokine expression after UUO, suggesting that norepinephrine may promote leukocyte recruitment and inflammation [25,31]. Together, these results suggest that following an injury stimulus, NE-AR signaling drives the inflammatory response, which in turn contributes to the development or progression of CKD.
Another potential mechanism for sympathetic nerve system activation in the AKI and CKD model is reduced nitric oxide (NO) availability [77]. In the kidney, NO has numerous physiological roles including the modulation of renal sympathetic nerve activity [126,127]. Ischemic AKI has been shown to alter renal hemodynamics, linked to endothelial cell dysfunction caused by increased reactive oxygen species (ROS) production, leading to decreased NO availability [128]. In addition, renal NO synthase activity is progressively reduced in the chronic renal failure model due to decreasing NO formation [129]. Accumulation of L-arginine analogues, NO synthase inhibitors, in CKD could potentially directly increase sympathetic nervous system (SNS) activity by inhibiting NO production [14]. Inhibition of NO synthesis induces vasoconstriction of the glomerular microvasculature and causes proximal tubular reabsorption to decline [14]. These effects are prevented by prior RDNx or the administration of an α2-ARantagonist [130,131]. Administration of an α2-AR agonist, B-TH933 to the denervated kidney restores both glomerular and tubular responsiveness to NO inhibition [44]. Decreased NOS activity induced by L-NAME, one of the non-selective NO synthase inhibitors, was significantly restored by α1-AR blockers, prazosin, or doxazosin, and attenuation of L-NAME-induced renal injury [132,133,134]. β-blockers, such as carvedilol and nebivolol, are shown to have protective effects via stimulation of NO release with protecting the kidney from 5/6 nephrectomy, gentamycin-induced nephrotoxicity, or hypertensive model [135,136,137].
Kidney tubular cell cycle arrest induced by severe AKI contributes to renal fibrogenesis by releasing profibrotic cytokines such as TGF-β1 and connective tissue growth factor (CTGF) [138]. While cell cycle arrest is normally used as a protective mechanism to avoid cell division during stress and injury, sustained cell cycle arrest at the G2/M phase results in the activation of senescence-associated secretory phenotype (SASP) and c-jun NH2-terminal kinase (JNK) signaling cascade, leading to the synthesis and the secretion of proliferative and profibrotic factors, which induce fibroblast proliferation and collagen deposition in renal interstitium [138,139]. In a previous study, it has been reported that RDNx prevents tubular cell cycle arrest at the G2/M phase observed during fibrogenesis after IRI with decreased number of tubular epithelial cells positive for phosphorylated histone H3, markers of the G2/M and decreased ratio of cyclin B1 to cyclin D1, markers of the G2/M cell cycle arrest [31]. However, in the denervated kidney, administration of NE induces cell cycle arrest at the G2/M phase, activates TGF- β1 signaling and increases α-SMA expression [31]. These suggest that renal nerve-derived NE may contribute to cell cycle arrest, leading to renal fibrogenesis.
The pathophysiology of AKI is generally characterized by a common cascade of cell death-induced inflammation and fibrosis following injury [140]. Inhibition of cell death attenuates renal inflammation and the consequent renal fibrogenesis after kidney injury [141,142,143,144]. Our recent study investigating the role of renal sympathetic nerves in renal fibrogenesis after renal I/R demonstrated that unilateral RDNx at the time of injury, or up to 1-day post-injury, improved histology, decreased pro-inflammatory and pro-fibrotic responses and apoptosis, thereby indicating that renal nerve stimulation is a primary mechanism and renal nerve-derived factors drive epithelial cell cycle arrest and the inflammatory cascade causing interstitial fibrogenesis after ischemia reperfusion injury [31]. NE can induce apoptosis by activating the ROS/JNK signaling pathway [145]. In cultured kidney proximal tubule cells, NE treatment increases poly (ADP-ribose) polymerase 1 (PARP1) and cleaved caspase-3 expression, which is reduced by co-treatment with caspase-3 inhibitor [59]. Consistent with our data, RDNx inhibits renal sympathetic activation, significantly attenuating the expression levels of p53, TNF-α, NF-κB, caspase-2 and -3, and improving apoptosis [146]. On the other hand, activation of α2-AR with dexmedetomidine, α2-AR agonist attenuates renal cell apoptosis and renal tubule impairment by inhibiting mitochondrial apoptosis pathways in the sepsis- or I/R-induced injury model, which may be effective by regulating NE release [147,148,149]. These findings indicate that inhibiting NE-AR signaling may attenuate apoptosis and may represent one of the mechanisms by which NE-AR signaling may prevent or limit progression of renal fibrogenesis at its onset in AKI-induced CKD.
6. Role of Sympathetic Nerves in Other Organs
Heart failure is a disorder in which the heart is unable to pump an adequate supply of blood [150]. SNS overactivity is associated with an increased risk of cardiovascular mortality in patients with renal failure (Table 2) [151]. Sympatho-excitation is a major component of the pathological relation between the kidney and heart in congestive heart failure [150]. It has been reported that the efferent renal nerves are essential to the renal hypoperfusion in congestive heart failure [152]. Furthermore, NE is released from the heart with an impaired NE re-uptake [153,154], thereby producing direct effects on β-AR signal transduction and impaired inotropic stimulation of the heart [155]. RDNx showed improvement of sodium excretion in experimental heart failure [156], increased cardiac output, improved renal blood flow [157], and a down-regulation of angiotensin II type 1 receptors mediating maladaptive responses [158]. Linz et al. have suggested that RDNx attenuates malignant ventricular arrhythmias in pigs with ventricular ischemia [159]. Similarly, RDNx improved ventricular end-diastolic diameter and fractional shortening in rats with heart failure induced by myocardial infarction [160]. It has been reported that α2-ARs play essential roles in the prevention of heart failure [161]. α2-AR subtypes are the presynaptic inhibitory receptor controlling sympathetic NE release [10]. Deletion of α2A-AR and α2C-AR subtypes increased the susceptibility to develop heart failure following chronic pressure overload in vivo [161,162]. In accordance with this, activation of α2A-AR using α2A-AR agonists, clonidine and moxonidine though sympatho-inhibition effect protect the kidney against renal IRI [87,163]. Conversely, in our previous reports, we demonstrated that the activation of the α2-AR accelerates the progression of renal fibrogenesis after renal IRI and UUO, suggesting the activation of α2-AR in this model may trigger inflammatory and fibrotic signaling pathways than sympatho-inhibition to induce fibrogenesis [25,31]. The contrasting results in the heart failure models versus the renal and IRI and UUO models could be due to the fact that in the mouse heart failure model, deletion of sympatho-inhibitory α2-ARs results in increased norepinephrine levels, which elicit typical cardiovascular consequences, including aggressive remodeling of the left ventricle, cardiac hypertrophy, and fibrosis [161,164]. In the renal denervated kidney, administration of exogenous NE can promote leukocyte recruitment, activate the TGF-β1 signaling pathway, and induce G2/M cell cycle arrest [25,31].
Table 2.
Activated sympathetic nerve in other organs.
Hepatorenal syndrome (HRS) is currently defined as the occurrence of AKI, in patients with advanced chronic liver disease [165,166]. Since catecholamines are metabolized by the liver, elevated arterial plasma NE level in cirrhosis might be due to impaired hepatic function (Table 2). With increasing severity of cirrhosis, the sympathetic nerves are progressively activated with increased circulating and renal NE concentrations [167,168,169]. Enhanced sympathetic nervous activity and NE is important in maintaining the level of arterial blood pressure in cirrhosis [170]. The treatment of HRS using α1-AR agonist, midodrine administration is associated with a significant improvement in renal function in HRS patients [171]. Other α2-AR agonist, clonidine, reduces portal pressure, arterial BP, and circulating NE in patients with cirrhosis [172]. On the other hand, in end-stage cirrhosis, β-blockers may reduce survival due to their negative effect on the cardiac compensatory reserve [173,174]. β-blockers are associated with increased mortality in patients with refractory ascites at high risk of paracentesis-induced circulatory dysfunction [173]. For this reason, these studies suggest that β-blockers should be avoided in patients with both cirrhosis and renal failure. Moreover, renal vasoconstriction caused by RAS activation and sympathetic nervous systems is the primary cause of HRS [175]. As mentioned above, Renin, a major RAS component, is produced in the juxtaglomerular cells of the afferent renal arteriole, reduced sodium intake, and increased activity of the sympathetic nervous system [78]. RAS activation is one of the main factors associated with renal vasoconstriction in HRS [176,177]. Thus, sympathetic nervous overactivity plays an important role in chronic liver disease.
7. Conclusions
The role of the sympathetic nervous system in AKI and CKD are discussed through a literature review investigating sympathetic nervous mechanisms [14,39,58,96]. As discussed, various studies have proposed a role for the renal sympathetic nervous system in contributing to renal interstitial inflammation and fibrogenesis in AKI and CKD. The renal sympathetic nerve-derived NE mediates inflammatory and fibrogenic response, and blockade of NE-AR signaling can prevent renal interstitial fibrogenesis following kidney injury (Figure 1). In addition, accumulating evidence suggested that renal denervation can help to delay reduction of renal function in CKD [31,178,179]. However, the NE-AR signaling pathway leading to renal injury in AKI and CKD remains to be defined. Thus, further research is needed to investigate the mechanisms by which renal sympathetic nerve-derived signaling may lead to progressive CKD. Defining the key molecules and understanding their functions may lead to designing of novel therapeutic strategies to prevent the progression of AKI to CKD.
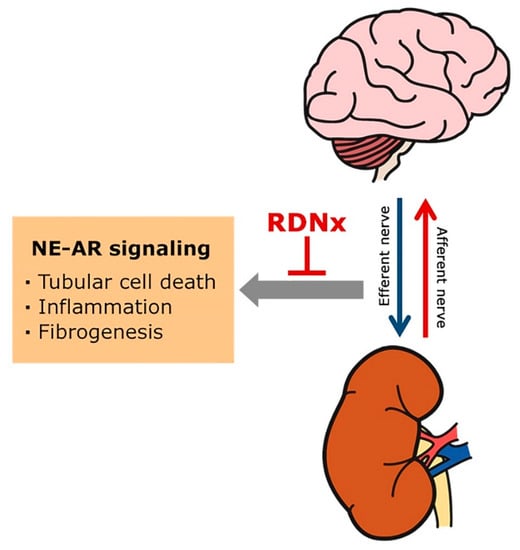
Figure 1.
Renal sympathetic nerve-derived norepinephrine (NE) and adrenergic receptor (AR) in chronic kidney disease development and progression. Sympathetic efferent activation, which is generated in the central nervous system, contributes to sympatho-excitation in the vasculature and renal tubules, and mediate their effects via norepinephrine and adrenergic receptors (ARs). The increased renal sympathetic nerve-derived NE may trigger tubular cell death, renal inflammation, and fibrogenesis progression, leading to CKD. Renal denervation (RDNx) lowers NE in renal tissue after renal injury and has a protective effect against fibrogenesis and CKD progression.
Author Contributions
Conceptualization, writing—Original Draft Preparation, Writing—Review and Editing, M.R.N., H.-S.J., J.K., B.J.P. All authors have read and agree to the published version of the manuscript.
Funding
B.J.P. is supported by NIH grants DK-116987, DK-120533, DK 120846 and American Heart Association (AHA) Grant in Aid 15GRNT25080031. H.S.J. is supported by AHA postdoctoral fellowship Grant 15POST25130003. J.K. is supported by grant (NRF-2019R1F1A1041410) from the National Research Foundation of Korea.
Conflicts of Interest
The authors declare no conflict of interest.
References
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; et al. AKI on CKD: heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef]
- Belayev, L.Y.; Palevsky, P.M. The link between acute kidney injury and chronic kidney disease. Curr. Opin. Nephrol. Hypertens 2014, 23, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Kaushal, G.P.; Shah, S.V. Challenges and advances in the treatment of AKI. J. Am. Soc. Nephrol. 2014, 25, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, T.; Oka, M.; Maesato, K.; Mano, T.; Ikee, R.; Moriya, H.; Kobayashi, S. Pathological regression by angiotensin II type 1 receptor blockade in patients with mesangial proliferative glomerulonephritis. Hypertens Res. 2008, 31, 387–394. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sata, Y.; Head, G.A.; Denton, K.; May, C.N.; Schlaich, M.P. Role of the Sympathetic Nervous System and Its Modulation in Renal Hypertension. Front. Med. 2018, 5, 82. [Google Scholar] [CrossRef]
- Nishi, E.E.; Bergamaschi, C.T.; Campos, R.R. The crosstalk between the kidney and the central nervous system: the role of renal nerves in blood pressure regulation. Exp. Physiol. 2015, 100, 479–484. [Google Scholar] [CrossRef]
- Johns, E.J.; Kopp, U.C.; DiBona, G.F. Neural control of renal function. Compr. Physiol. 2011, 1, 731–767. [Google Scholar] [CrossRef] [PubMed]
- McCorry, L.K. Physiology of the autonomic nervous system. Am. J. Pharm. Educ. 2007, 71, 78. [Google Scholar] [CrossRef]
- Kanagy, N.L. Alpha(2)-adrenergic receptor signalling in hypertension. Clin. Sci. 2005, 109, 431–437. [Google Scholar] [CrossRef]
- Molinoff, P.B. Alpha- and beta-adrenergic receptor subtypes properties, distribution and regulation. Drugs 1984, 28 (Suppl. 2), 1–15. [Google Scholar] [CrossRef]
- Converse, R.L., Jr.; Jacobsen, T.N.; Toto, R.D.; Jost, C.M.; Cosentino, F.; Fouad-Tarazi, F.; Victor, R.G. Sympathetic overactivity in patients with chronic renal failure. N. Engl. J. Med. 1992, 327, 1912–1918. [Google Scholar] [CrossRef] [PubMed]
- Veelken, R.; Vogel, E.M.; Hilgers, K.; Amann, K.; Hartner, A.; Sass, G.; Neuhuber, W.; Tiegs, G. Autonomic renal denervation ameliorates experimental glomerulonephritis. J. Am. Soc. Nephrol. 2008, 19, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Young, B.E.; Fadel, P.J. Sympathetic Overactivity in Chronic Kidney Disease: Consequences and Mechanisms. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Zoccali, C.; Mallamaci, F.; Tripepi, G.; Parlongo, S.; Cutrupi, S.; Benedetto, F.A.; Cataliotti, A.; Malatino, L.S.; Investigators, C. Norepinephrine and concentric hypertrophy in patients with end-stage renal disease. Hypertension 2002, 40, 41–46. [Google Scholar] [CrossRef]
- Cronin, R.E.; Erickson, A.M.; de Torrente, A.; McDonald, K.M.; Schrier, R.W. Norepinephrine-induced acute renal failure: a reversible ischemic model of acute renal failure. Kidney Int. 1978, 14, 187–190. [Google Scholar] [CrossRef]
- Conger, J.D.; Robinette, J.B.; Hammond, W.S. Differences in vascular reactivity in models of ischemic acute renal failure. Kidney Int. 1991, 39, 1087–1097. [Google Scholar] [CrossRef]
- Bellomo, R.; Wan, L.; May, C. Vasoactive drugs and acute kidney injury. Crit. Care Med. 2008, 36, S179–S186. [Google Scholar] [CrossRef]
- Schlaich, M.P.; Sobotka, P.A.; Krum, H.; Lambert, E.; Esler, M.D. Renal sympathetic-nerve ablation for uncontrolled hypertension. N. Engl. J. Med. 2009, 361, 932–934. [Google Scholar] [CrossRef]
- Krum, H.; Schlaich, M.; Whitbourn, R.; Sobotka, P.A.; Sadowski, J.; Bartus, K.; Kapelak, B.; Walton, A.; Sievert, H.; Thambar, S.; et al. Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet 2009, 373, 1275–1281. [Google Scholar] [CrossRef]
- Kannan, A.; Medina, R.I.; Nagajothi, N.; Balamuthusamy, S. Renal sympathetic nervous system and the effects of denervation on renal arteries. World J. Cardiol. 2014, 6, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Mulder, J.; Hokfelt, T.; Knuepfer, M.M.; Kopp, U.C. Renal sensory and sympathetic nerves reinnervate the kidney in a similar time-dependent fashion after renal denervation in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R675–R682. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Denton, K.M. Renal Denervation. Hypertension 2018, 72, 528–536. [Google Scholar] [CrossRef] [PubMed]
- DiBona, G.F.; Esler, M. Translational medicine: the antihypertensive effect of renal denervation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R245–R253. [Google Scholar] [CrossRef]
- Kim, J.; Padanilam, B.J. Renal nerves drive interstitial fibrogenesis in obstructive nephropathy. J. Am. Soc. Nephrol. 2013, 24, 229–242. [Google Scholar] [CrossRef]
- Azizi, M.; Schmieder, R.E.; Mahfoud, F.; Weber, M.A.; Daemen, J.; Davies, J.; Basile, J.; Kirtane, A.J.; Wang, Y.; Lobo, M.D.; et al. Endovascular ultrasound renal denervation to treat hypertension (RADIANCE-HTN SOLO): a multicentre, international, single-blind, randomised, sham-controlled trial. Lancet 2018, 391, 2335–2345. [Google Scholar] [CrossRef]
- Kandzari, D.E.; Bohm, M.; Mahfoud, F.; Townsend, R.R.; Weber, M.A.; Pocock, S.; Tsioufis, K.; Tousoulis, D.; Choi, J.W.; East, C.; et al. Effect of renal denervation on blood pressure in the presence of antihypertensive drugs: 6-month efficacy and safety results from the SPYRAL HTN-ON MED proof-of-concept randomised trial. Lancet 2018, 391, 2346–2355. [Google Scholar] [CrossRef]
- Smithwick, R.H.; Thompson, J.E. Splanchnicectomy for essential hypertension; results in 1,266 cases. J. Am. Med. Assoc. 1953, 152, 1501–1504. [Google Scholar] [CrossRef]
- Azizi, M.; Sapoval, M.; Gosse, P.; Monge, M.; Bobrie, G.; Delsart, P.; Midulla, M.; Mounier-Vehier, C.; Courand, P.Y.; Lantelme, P.; et al. Optimum and stepped care standardised antihypertensive treatment with or without renal denervation for resistant hypertension (DENERHTN): a multicentre, open-label, randomised controlled trial. Lancet 2015, 385, 1957–1965. [Google Scholar] [CrossRef]
- Mahfoud, F.; Ukena, C.; Schmieder, R.E.; Cremers, B.; Rump, L.C.; Vonend, O.; Weil, J.; Schmidt, M.; Hoppe, U.C.; Zeller, T.; et al. Ambulatory blood pressure changes after renal sympathetic denervation in patients with resistant hypertension. Circulation 2013, 128, 132–140. [Google Scholar] [CrossRef]
- Kim, J.; Padanilam, B.J. Renal denervation prevents long-term sequelae of ischemic renal injury. Kidney Int. 2015, 87, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Mahfoud, F.; Bohm, M.; Schmieder, R.; Narkiewicz, K.; Ewen, S.; Ruilope, L.; Schlaich, M.; Williams, B.; Fahy, M.; Mancia, G. Effects of renal denervation on kidney function and long-term outcomes: 3-year follow-up from the Global SYMPLICITY Registry. Eur. Heart J. 2019, 40, 3474–3482. [Google Scholar] [CrossRef] [PubMed]
- Schlaich, M.P.; Bart, B.; Hering, D.; Walton, A.; Marusic, P.; Mahfoud, F.; Bohm, M.; Lambert, E.A.; Krum, H.; Sobotka, P.A.; et al. Feasibility of catheter-based renal nerve ablation and effects on sympathetic nerve activity and blood pressure in patients with end-stage renal disease. Int. J. Cardiol. 2013, 168, 2214–2220. [Google Scholar] [CrossRef] [PubMed]
- Esler, M.; Jennings, G.; Korner, P.; Willett, I.; Dudley, F.; Hasking, G.; Anderson, W.; Lambert, G. Assessment of human sympathetic nervous system activity from measurements of norepinephrine turnover. Hypertension 1988, 11, 3–20. [Google Scholar] [CrossRef]
- Oberhauser, V.; Vonend, O.; Rump, L.C. Neuropeptide Y and ATP interact to control renovascular resistance in the rat. J. Am. Soc. Nephrol. 1999, 10, 1179–1185. [Google Scholar]
- Cotecchia, S. The alpha1-adrenergic receptors: diversity of signaling networks and regulation. J. Recept Signal. Transduct Res. 2010, 30, 410–419. [Google Scholar] [CrossRef]
- Arif, E.; Nihalani, D. Beta2-adrenergic receptor in kidney biology: A current prospective. Nephrology (Carlton) 2019, 24, 497–503. [Google Scholar] [CrossRef]
- Procino, G.; Carmosino, M.; Milano, S.; Dal Monte, M.; Schena, G.; Mastrodonato, M.; Gerbino, A.; Bagnoli, P.; Svelto, M. beta3 adrenergic receptor in the kidney may be a new player in sympathetic regulation of renal function. Kidney Int. 2016, 90, 555–567. [Google Scholar] [CrossRef]
- Jang, H.S.; Kim, J.; Padanilam, B.J. Renal sympathetic nerve activation via alpha2-adrenergic receptors in chronic kidney disease progression. Kidney Res. Clin. Pract. 2019, 38, 6–14. [Google Scholar] [CrossRef]
- Summers, R.J.; Stephenson, J.A.; Kuhar, M.J. Localization of beta adrenoceptor subtypes in rat kidney by light microscopic autoradiography. J. Pharmacol. Exp. Ther. 1985, 232, 561–569. [Google Scholar]
- Summers, R.J.; Kuhar, M.J. Autoradiographic localization of beta-adrenoceptors in rat kidney. Eur. J. Pharmacol. 1983, 91, 305–310. [Google Scholar] [CrossRef]
- Struyker-Boudier, H.A.; Janssen, B.J.; Smits, J.F. Adrenoceptors in the kidney: localization and pharmacology. Clin. Exp. Hypertens A 1987, 9 (Suppl. 1), 135–150. [Google Scholar] [CrossRef]
- Boivin, V.; Jahns, R.; Gambaryan, S.; Ness, W.; Boege, F.; Lohse, M.J. Immunofluorescent imaging of beta 1- and beta 2-adrenergic receptors in rat kidney. Kidney Int. 2001, 59, 515–531. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.C.; Vallon, V. Alpha 2-adrenoceptors determine the response to nitric oxide inhibition in the rat glomerulus and proximal tubule. J. Am. Soc. Nephrol. 1995, 6, 1482–1490. [Google Scholar] [PubMed]
- Snavely, M.D.; Ziegler, M.G.; Insel, P.A. Subtype-selective down-regulation of rat renal cortical alpha- and beta-adrenergic receptors by catecholamines. Endocrinology 1985, 117, 2182–2189. [Google Scholar] [CrossRef] [PubMed]
- Morla, L.; Edwards, A.; Crambert, G. New insights into sodium transport regulation in the distal nephron: Role of G-protein coupled receptors. World J. Biol. Chem. 2016, 7, 44–63. [Google Scholar] [CrossRef]
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [CrossRef]
- Forni, L.G.; Darmon, M.; Ostermann, M.; Oudemans-van Straaten, H.M.; Pettila, V.; Prowle, J.R.; Schetz, M.; Joannidis, M. Renal recovery after acute kidney injury. Intensive Care Med. 2017, 43, 855–866. [Google Scholar] [CrossRef]
- Ishani, A.; Xue, J.L.; Himmelfarb, J.; Eggers, P.W.; Kimmel, P.L.; Molitoris, B.A.; Collins, A.J. Acute kidney injury increases risk of ESRD among elderly. J. Am. Soc. Nephrol. 2009, 20, 223–228. [Google Scholar] [CrossRef]
- Lo, L.J.; Go, A.S.; Chertow, G.M.; McCulloch, C.E.; Fan, D.; Ordonez, J.D.; Hsu, C.Y. Dialysis-requiring acute renal failure increases the risk of progressive chronic kidney disease. Kidney Int. 2009, 76, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Zager, R.A.; Johnson, A.C.; Becker, K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and "end-stage" kidney disease. Am. J. Physiol. Renal Physiol. 2011, 301, F1334–F1345. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Bonventre, J.V.; Mehta, R.; Nangaku, M.; Unwin, R.; Rosner, M.H.; Kellum, J.A.; Ronco, C.; Group, A.X.W. Progression after AKI: Understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. J. Am. Soc. Nephrol. 2016, 27, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Baines, A.D.; DeBold, A.J.; Sonnenberg, H. Natriuretic effect of atrial extract on isolated perfused rat kidney. Can. J. Physiol. Pharmacol. 1983, 61, 1462–1466. [Google Scholar] [CrossRef]
- Iaina, A.; Eliahou, H.E. The sympathetic nervous system in the pathogenesis of acute renal failure. Clin. Exp. Dial. Apheresis 1983, 7, 115–125. [Google Scholar] [CrossRef]
- Bellomo, R.; Giantomasso, D.D. Noradrenaline and the kidney: friends or foes? Crit. Care 2001, 5, 294–298. [Google Scholar] [CrossRef]
- Conger, J.D.; Robinette, J.B.; Guggenheim, S.J. Effect of acetylcholine on the early phase of reversible norepinephrine-induced acute renal failure. Kidney Int. 1981, 19, 399–409. [Google Scholar] [CrossRef]
- Fujii, T.; Kurata, H.; Takaoka, M.; Muraoka, T.; Fujisawa, Y.; Shokoji, T.; Nishiyama, A.; Abe, Y.; Matsumura, Y. The role of renal sympathetic nervous system in the pathogenesis of ischemic acute renal failure. Eur. J. Pharmacol. 2003, 481, 241–248. [Google Scholar] [CrossRef]
- Tanaka, R.; Tsutsui, H.; Ohkita, M.; Takaoka, M.; Yukimura, T.; Matsumura, Y. Sex differences in ischemia/reperfusion-induced acute kidney injury are dependent on the renal sympathetic nervous system. Eur. J. Pharmacol. 2013, 714, 397–404. [Google Scholar] [CrossRef]
- Mutoh, J.; Ohsawa, M.; Hisa, H. Involvement of renal sympathetic nerve activation on the progression of ischemic acute kidney injury in the mouse. J. Pharmacol. Sci. 2014, 125, 415–421. [Google Scholar] [CrossRef]
- Tsutsui, H.; Tanaka, R.; Yamagata, M.; Yukimura, T.; Ohkita, M.; Matsumura, Y. Protective effect of ischemic preconditioning on ischemia/reperfusion-induced acute kidney injury through sympathetic nervous system in rats. Eur. J. Pharmacol. 2013, 718, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Li, A.; Li, J.; Wu, C.; Cui, S.; Zhou, Z.; Liu, Y.; Wilcox, C.S.; Hou, F.F. Reno-Cerebral Reflex Activates the Renin-Angiotensin System, Promoting Oxidative Stress and Renal Damage After Ischemia-Reperfusion Injury. Antioxid Redox Signal. 2017, 27, 415–432. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef] [PubMed]
- Goulding, N.E.; Johns, E.J. Neural regulation of the kidney function in rats with cisplatin induced renal failure. Front. Physiol. 2015, 6, 192. [Google Scholar] [CrossRef]
- Matsushima, H.; Yonemura, K.; Ohishi, K.; Hishida, A. The role of oxygen free radicals in cisplatin-induced acute renal failure in rats. J. Lab. Clin. Med. 1998, 131, 518–526. [Google Scholar] [CrossRef]
- Bagnis, C.; Beaufils, H.; Jacquiaud, C.; Adabra, Y.; Jouanneau, C.; Le Nahour, G.; Jaudon, M.C.; Bourbouze, R.; Jacobs, C.; Deray, G. Erythropoietin enhances recovery after cisplatin-induced acute renal failure in the rat. Nephrol. Dial. Transplant. 2001, 16, 932–938. [Google Scholar] [CrossRef]
- Hye Khan, M.A.; Sattar, M.A.; Abdullah, N.A.; Johns, E.J. Influence of combined hypertension and renal failure on functional alpha(1)-adrenoceptor subtypes in the rat kidney. Br. J. Pharmacol. 2008, 153, 1232–1241. [Google Scholar] [CrossRef]
- Kribben, A.; Edelstein, C.L.; Schrier, R.W. Pathophysiology of acute renal failure. J. Nephrol. 1999, 12 (Suppl. 2), S142–S151. [Google Scholar]
- Yatsu, T.; Aoki, M.; Inagaki, O. Preventive effect of zelandopam, a dopamine D1 receptor agonist, on cisplatin-induced acute renal failure in rats. Eur. J. Pharmacol. 2003, 461, 191–195. [Google Scholar] [CrossRef]
- Hasking, G.J.; Esler, M.D.; Jennings, G.L.; Burton, D.; Johns, J.A.; Korner, P.I. Norepinephrine spillover to plasma in patients with congestive heart failure: evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation 1986, 73, 615–621. [Google Scholar] [CrossRef]
- Hausberg, M.; Kosch, M.; Harmelink, P.; Barenbrock, M.; Hohage, H.; Kisters, K.; Dietl, K.H.; Rahn, K.H. Sympathetic nerve activity in end-stage renal disease. Circulation 2002, 106, 1974–1979. [Google Scholar] [CrossRef] [PubMed]
- Denker, M.G.; Cohen, D.L. Resistant Hypertension and Renal Nerve Denervation. Methodist Debakey Cardiovasc. J. 2015, 11, 240–244. [Google Scholar] [CrossRef] [PubMed]
- DiBona, G.F. Functionally specific renal sympathetic nerve fibers: role in cardiovascular regulation. Am. J. Hypertens 2001, 14, 163S–170S. [Google Scholar] [CrossRef]
- Taddei, S.; Virdis, A.; Abdel-Haq, B.; Giovannetti, R.; Duranti, P.; Arena, A.M.; Favilla, S.; Salvetti, A. Indirect evidence for vascular uptake of circulating renin in hypertensive patients. Hypertension 1993, 21, 852–860. [Google Scholar] [CrossRef][Green Version]
- Campese, V.M.; Mitra, N.; Sandee, D. Hypertension in renal parenchymal disease: why is it so resistant to treatment? Kidney Int. 2006, 69, 967–973. [Google Scholar] [CrossRef]
- DiBona, G.F.; Kopp, U.C. Neural control of renal function. Physiol. Rev. 1997, 77, 75–197. [Google Scholar] [CrossRef]
- Schlaich, M.P.; Socratous, F.; Hennebry, S.; Eikelis, N.; Lambert, E.A.; Straznicky, N.; Esler, M.D.; Lambert, G.W. Sympathetic activation in chronic renal failure. J. Am. Soc. Nephrol. 2009, 20, 933–939. [Google Scholar] [CrossRef]
- Stegbauer, J.; Vonend, O.; Habbel, S.; Quack, I.; Sellin, L.; Gross, V.; Rump, L.C. Angiotensin II modulates renal sympathetic neurotransmission through nitric oxide in AT2 receptor knockout mice. J. Hypertens 2005, 23, 1691–1698. [Google Scholar] [CrossRef]
- Hall, M.E.; do Carmo, J.M.; da Silva, A.A.; Juncos, L.A.; Wang, Z.; Hall, J.E. Obesity, hypertension, and chronic kidney disease. Int. J. Nephrol. Renov. Dis. 2014, 7, 75–88. [Google Scholar] [CrossRef]
- Schiffl, H.; Lang, S.M. Obesity, acute kidney injury and outcome of critical illness. Int. Urol. Nephrol. 2017, 49, 461–466. [Google Scholar] [CrossRef]
- Richards, R.J.; Thakur, V.; Reisin, E. Obesity-related hypertension: its physiological basis and pharmacological approaches to its treatment. J. Hum. Hypertens 1996, 10 (Suppl. 3), S59–S64. [Google Scholar]
- Wofford, M.R.; Anderson, D.C., Jr.; Brown, C.A.; Jones, D.W.; Miller, M.E.; Hall, J.E. Antihypertensive effect of alpha- and beta-adrenergic blockade in obese and lean hypertensive subjects. Am. J. Hypertens 2001, 14, 694–698. [Google Scholar] [CrossRef]
- Rocchini, A.P.; Mao, H.Z.; Babu, K.; Marker, P.; Rocchini, A.J. Clonidine prevents insulin resistance and hypertension in obese dogs. Hypertension 1999, 33, 548–553. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kassab, S.; Kato, T.; Wilkins, F.C.; Chen, R.; Hall, J.E.; Granger, J.P. Renal denervation attenuates the sodium retention and hypertension associated with obesity. Hypertension 1995, 25, 893–897. [Google Scholar] [CrossRef]
- Hoch, H.; Stegbauer, J.; Potthoff, S.A.; Hein, L.; Quack, I.; Rump, L.C.; Vonend, O. Regulation of renal sympathetic neurotransmission by renal alpha(2A)-adrenoceptors is impaired in chronic renal failure. Br. J. Pharmacol. 2011, 163, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Solez, K.; Freshwater, M.F.; Su, C.T. The effect of propranolol on postischemic acute renal failure in the rat. Transplantation 1977, 24, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Solez, K.; Ideura, T.; Silvia, C.B.; Hamilton, B.; Saito, H. Clonidine after renal ischemia to lessen acute renal failure and microvascular damage. Kidney Int. 1980, 18, 309–322. [Google Scholar] [CrossRef]
- Chevalier, R.L.; Finn, W.F. Effects of propranolol on post-ischemic acute renal failure. Nephron 1980, 25, 77–81. [Google Scholar] [CrossRef]
- Amann, K.; Koch, A.; Hofstetter, J.; Gross, M.L.; Haas, C.; Orth, S.R.; Ehmke, H.; Rump, L.C.; Ritz, E. Glomerulosclerosis and progression: effect of subantihypertensive doses of alpha and beta blockers. Kidney Int. 2001, 60, 1309–1323. [Google Scholar] [CrossRef]
- Shimokawa, T.; Tsutsui, H.; Miura, T.; Nishinaka, T.; Terada, T.; Takama, M.; Yoshida, S.; Tanba, T.; Tojo, A.; Yamagata, M.; et al. Renoprotective effect of yohimbine on ischaemia/reperfusion-induced acute kidney injury through alpha2C-adrenoceptors in rats. Eur. J. Pharmacol. 2016, 781, 36–44. [Google Scholar] [CrossRef]
- Salman, I.M.; Ameer, O.Z.; Sattar, M.A.; Abdullah, N.A.; Yam, M.F.; Najim, H.S.; Abdulkarim, M.F.; Abdullah, G.Z.; Kaur, G.; Khan, M.A.; et al. Characterization of renal hemodynamic and structural alterations in rat models of renal impairment: role of renal sympathoexcitation. J. Nephrol. 2011, 24, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Sattar, M.A.; Rathore, H.A.; Abdulla, M.H.; Ud Din Ahmad, F.; Ahmad, A.; Afzal, S.; Abdullah, N.A.; Johns, E.J. Renal denervation restores the baroreflex control of renal sympathetic nerve activity and heart rate in Wistar-Kyoto rats with cisplatin-induced renal failure. Acta Physiol. 2014, 210, 690–700. [Google Scholar] [CrossRef]
- Ogawa, T.; Mimura, Y.; Kaminishi, M. Renal denervation abolishes the protective effects of ischaemic preconditioning on function and haemodynamics in ischaemia-reperfused rat kidneys. Acta Physiol. Scand. 2002, 174, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Hering, D.; Mahfoud, F.; Walton, A.S.; Krum, H.; Lambert, G.W.; Lambert, E.A.; Sobotka, P.A.; Bohm, M.; Cremers, B.; Esler, M.D.; et al. Renal denervation in moderate to severe CKD. J. Am. Soc. Nephrol. 2012, 23, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Schmid, A.; Toennes, S.W.; Ditting, T.; Veelken, R.; Uder, M.; Schmieder, R.E. Central pulse pressure predicts BP reduction after renal denervation in patients with treatment-resistant hypertension. EuroIntervention 2015, 11, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, M.H. Renal nerves and CKD: is renal denervation the answer? J. Am. Soc. Nephrol. 2012, 23, 1132–1135. [Google Scholar] [CrossRef]
- Desir, G. Novel insights into the physiology of renalase and its role in hypertension and heart disease. Pediatr. Nephrol. 2012, 27, 719–725. [Google Scholar] [CrossRef]
- Lee, H.T.; Kim, J.Y.; Kim, M.; Wang, P.; Tang, L.; Baroni, S.; D’Agati, V.D.; Desir, G.V. Renalase protects against ischemic AKI. J. Am. Soc. Nephrol 2013, 24, 445–455. [Google Scholar] [CrossRef]
- Johns, E.J.; Abdulla, M.H. Renal nerves in blood pressure regulation. Curr. Opin. Nephrol. Hypertens 2013, 22, 504–510. [Google Scholar] [CrossRef]
- Lee, J.Y.; Walsh, G.M. Systemic and regional haemodynamic effects of renal denervation in spontaneously hypertensive rats. J. Hypertens 1983, 1, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Krueger, A.D.; Lee, J.Y.; Yang, P.C.; Papaioannou, S.E.; Walsh, G.M. Selective vasodilation produced by renal denervation in adult spontaneously hypertensive rats. Hypertension 1986, 8, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Foss, J.D.; Fink, G.D.; Osborn, J.W. Reversal of genetic salt-sensitive hypertension by targeted sympathetic ablation. Hypertension 2013, 61, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, H.; Nishida, H.; Nomura, G.; Van Vliet, B.N.; Toshima, H. Hypertension induced by nitric oxide synthesis inhibition is renal nerve dependent. Hypertension 1994, 23, 971–975. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jacob, F.; Clark, L.A.; Guzman, P.A.; Osborn, J.W. Role of renal nerves in development of hypertension in DOCA-salt model in rats: a telemetric approach. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1519–H1529. [Google Scholar] [CrossRef] [PubMed]
- Kandlikar, S.S.; Fink, G.D. Splanchnic sympathetic nerves in the development of mild DOCA-salt hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1965–H1973. [Google Scholar] [CrossRef]
- Singh, R.R.; McArdle, Z.M.; Iudica, M.; Easton, L.K.; Booth, L.C.; May, C.N.; Parkington, H.C.; Lombardo, P.; Head, G.A.; Lambert, G.; et al. Sustained Decrease in Blood Pressure and Reduced Anatomical and Functional Reinnervation of Renal Nerves in Hypertensive Sheep 30 Months After Catheter-Based Renal Denervation. Hypertension 2019, 73, 718–727. [Google Scholar] [CrossRef]
- Ma, S.; Evans, R.G.; Iguchi, N.; Tare, M.; Parkington, H.C.; Bellomo, R.; May, C.N.; Lankadeva, Y.R. Sepsis-induced acute kidney injury: A disease of the microcirculation. Microcirculation 2019, 26, e12483. [Google Scholar] [CrossRef]
- Tan, F.; Chen, Y.; Yuan, D.; Gong, C.; Li, X.; Zhou, S. Dexmedetomidine protects against acute kidney injury through downregulating inflammatory reactions in endotoxemia rats. Biomed. Rep. 2015, 3, 365–370. [Google Scholar] [CrossRef]
- Si, Y.; Bao, H.; Han, L.; Shi, H.; Zhang, Y.; Xu, L.; Liu, C.; Wang, J.; Yang, X.; Vohra, A.; et al. Dexmedetomidine protects against renal ischemia and reperfusion injury by inhibiting the JAK/STAT signaling activation. J. Transl. Med. 2013, 11, 141. [Google Scholar] [CrossRef]
- Gu, J.; Sun, P.; Zhao, H.; Watts, H.R.; Sanders, R.D.; Terrando, N.; Xia, P.; Maze, M.; Ma, D. Dexmedetomidine provides renoprotection against ischemia-reperfusion injury in mice. Crit. Care 2011, 15, R153. [Google Scholar] [CrossRef]
- Noh, H.; Yu, M.R.; Kim, H.J.; Lee, J.H.; Park, B.W.; Wu, I.H.; Matsumoto, M.; King, G.L. Beta 2-adrenergic receptor agonists are novel regulators of macrophage activation in diabetic renal and cardiovascular complications. Kidney Int. 2017, 92, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Imaizumi, A.; Yanagawa, Y.; Kohsaka, T.; Johns, E.J. beta(2)-Adrenoceptor activation attenuates endotoxin-induced acute renal failure. J. Am. Soc. Nephrol. 2004, 15, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yanagita, M. Immune cells and inflammation in AKI to CKD progression. Am. J. Physiol. Renal. Physiol. 2018, 315, F1501–F1512. [Google Scholar] [CrossRef] [PubMed]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Kirabo, A.; Wu, J.; Saleh, M.A.; Zhu, L.; Wang, F.; Takahashi, T.; Loperena, R.; Foss, J.D.; Mernaugh, R.L.; et al. Renal Denervation Prevents Immune Cell Activation and Renal Inflammation in Angiotensin II-Induced Hypertension. Circ. Res. 2015, 117, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Le Clef, N.; Verhulst, A.; D’Haese, P.C.; Vervaet, B.A. Unilateral Renal Ischemia-Reperfusion as a Robust Model for Acute to Chronic Kidney Injury in Mice. PLoS ONE 2016, 11, e0152153. [Google Scholar] [CrossRef]
- Padro, C.J.; Sanders, V.M. Neuroendocrine regulation of inflammation. Semin Immunol. 2014, 26, 357–368. [Google Scholar] [CrossRef]
- Severn, A.; Rapson, N.T.; Hunter, C.A.; Liew, F.Y. Regulation of tumor necrosis factor production by adrenaline and beta-adrenergic agonists. J. Immunol. 1992, 148, 3441–3445. [Google Scholar]
- Spengler, R.N.; Allen, R.M.; Remick, D.G.; Strieter, R.M.; Kunkel, S.L. Stimulation of alpha-adrenergic receptor augments the production of macrophage-derived tumor necrosis factor. J. Immunol. 1990, 145, 1430–1434. [Google Scholar]
- Spengler, R.N.; Chensue, S.W.; Giacherio, D.A.; Blenk, N.; Kunkel, S.L. Endogenous norepinephrine regulates tumor necrosis factor-alpha production from macrophages in vitro. J. Immunol. 1994, 152, 3024–3031. [Google Scholar] [PubMed]
- Shimokawa, T.; Tsutsui, H.; Miura, T.; Takama, M.; Hayashi, K.; Nishinaka, T.; Terada, T.; Yoneda, K.; Yamagata, M.; Yukimura, T. Post-treatment with JP-1302 protects against renal ischemia/reperfusion-induced acute kidney injury in rats. J. Pharmacol. Sci. 2019, 139, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Zaldivia, M.T.; Rivera, J.; Hering, D.; Marusic, P.; Sata, Y.; Lim, B.; Eikelis, N.; Lee, R.; Lambert, G.W.; Esler, M.D.; et al. Renal Denervation Reduces Monocyte Activation and Monocyte-Platelet Aggregate Formation: An Anti-Inflammatory Effect Relevant for Cardiovascular Risk. Hypertension 2017, 69, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Szelenyi, J.; Kiss, J.P.; Vizi, E.S. Differential involvement of sympathetic nervous system and immune system in the modulation of TNF-alpha production by alpha2- and beta-adrenoceptors in mice. J. Neuroimmunol. 2000, 103, 34–40. [Google Scholar] [CrossRef]
- Flierl, M.A.; Rittirsch, D.; Nadeau, B.A.; Chen, A.J.; Sarma, J.V.; Zetoune, F.S.; McGuire, S.R.; List, R.P.; Day, D.E.; Hoesel, L.M.; et al. Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature 2007, 449, 721–725. [Google Scholar] [CrossRef]
- Mount, P.F.; Power, D.A. Nitric oxide in the kidney: functions and regulation of synthesis. Acta Physiol. 2006, 187, 433–446. [Google Scholar] [CrossRef]
- Eppel, G.A.; Denton, K.M.; Malpas, S.C.; Evans, R.G. Nitric oxide in responses of regional kidney perfusion to renal nerve stimulation and renal ischaemia. Pflugers Arch. 2003, 447, 205–213. [Google Scholar] [CrossRef]
- Basile, D.P.; Yoder, M.C. Renal endothelial dysfunction in acute kidney ischemia reperfusion injury. Cardiovasc. Hematol. Disord Drug Targets 2014, 14, 3–14. [Google Scholar] [CrossRef]
- Aiello, S.; Noris, M.; Todeschini, M.; Zappella, S.; Foglieni, C.; Benigni, A.; Corna, D.; Zoja, C.; Cavallotti, D.; Remuzzi, G. Renal and systemic nitric oxide synthesis in rats with renal mass reduction. Kidney Int. 1997, 52, 171–181. [Google Scholar] [CrossRef]
- Baylis, C.; Braith, R.; Santmyire, B.R.; Engels, K. Renal nerves do not mediate vasoconstrictor responses to acute nitric oxide synthesis inhibition in conscious rats. J. Am. Soc. Nephrol. 1997, 8, 887–892. [Google Scholar]
- Bruck, H.; Gossl, M.; Spitthover, R.; Schafers, R.F.; Kohnle, M.; Philipp, T.; Wenzel, R.R. The nitric oxide synthase inhibitor L-NMMA potentiates noradrenaline-induced vasoconstriction: effects of the alpha2-receptor antagonist yohimbine. J. Hypertens 2001, 19, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Tojo, A.; Kobayashi, N.; Kimura, K.; Hirata, Y.; Matsuoka, H.; Yagi, S.; Omata, M. Effects of antihypertensive drugs on nitric oxide synthase activity in rat kidney. Kidney Int. Suppl. 1996, 55, S138–S140. [Google Scholar]
- Wangensteen, R.; O’Valle, F.; Del Moral, R.; Vargas, F.; Osuna, A. Chronic alpha1-adrenergic blockade improves hypertension and renal injury in L-NAME and low-renin L-NAME-DOCA hypertensive rats. Med. Sci. Monit 2002, 8, BR378–BR384. [Google Scholar] [PubMed]
- Erley, C.M.; Rebmann, S.; Strobel, U.; Schmidt, T.; Wehrmann, M.; Osswald, H.; Risler, T. Effects of antihypertensive therapy on blood pressure and renal function in rats with hypertension due to chronic blockade of nitric oxide synthesis. Exp. Nephrol. 1995, 3, 293–299. [Google Scholar] [PubMed]
- Van den Branden, C.; Gabriels, M.; Vamecq, J.; Vanden Houte, K.; Verbeelen, D. Carvedilol protects against glomerulosclerosis in rat remnant kidney without general changes in antioxidant enzyme status. A comparative study of two beta-blocking drugs, carvedilol and propanolol. Nephron 1997, 77, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.V.; Shifow, A.A.; Naidu, M.U.; Ratnakar, K.S. Carvedilol: a beta blocker with antioxidant property protects against gentamicin-induced nephrotoxicity in rats. Life Sci. 2000, 66, 2603–2611. [Google Scholar] [CrossRef]
- Barone, F.C.; Nelson, A.H.; Ohlstein, E.H.; Willette, R.N.; Sealey, J.E.; Laragh, J.H.; Campbell, W.G., Jr.; Feuerstein, G.Z. Chronic carvedilol reduces mortality and renal damage in hypertensive stroke-prone rats. J. Pharmacol. Exp. Ther. 1996, 279, 948–955. [Google Scholar]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef]
- Canaud, G.; Bonventre, J.V. Cell cycle arrest and the evolution of chronic kidney disease from acute kidney injury. Nephrol. Dial. Transplant. 2015, 30, 575–583. [Google Scholar] [CrossRef]
- Agarwal, A.; Dong, Z.; Harris, R.; Murray, P.; Parikh, S.M.; Rosner, M.H.; Kellum, J.A.; Ronco, C.; Acute Dialysis Quality Initiative, X.W.G. Cellular and Molecular Mechanisms of AKI. J. Am. Soc. Nephrol. 2016, 27, 1288–1299. [Google Scholar] [CrossRef]
- Daemen, M.A.; van’t Veer, C.; Denecker, G.; Heemskerk, V.H.; Wolfs, T.G.; Clauss, M.; Vandenabeele, P.; Buurman, W.A. Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J. Clin. Invest. 1999, 104, 541–549. [Google Scholar] [CrossRef]
- Jang, H.S.; Padanilam, B.J. Simultaneous deletion of Bax and Bak is required to prevent apoptosis and interstitial fibrosis in obstructive nephropathy. Am. J. Physiol. Renal. Physiol. 2015, 309, F540–F550. [Google Scholar] [CrossRef] [PubMed]
- Homsi, E.; Janino, P.; de Faria, J.B. Role of caspases on cell death, inflammation, and cell cycle in glycerol-induced acute renal failure. Kidney Int. 2006, 69, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Servais, H.; Ortiz, A.; Devuyst, O.; Denamur, S.; Tulkens, P.M.; Mingeot-Leclercq, M.P. Renal cell apoptosis induced by nephrotoxic drugs: cellular and molecular mechanisms and potential approaches to modulation. Apoptosis 2008, 13, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.C.; Yin, S.C.; Chi, C.S.; Hwang, B.; Hsu, S.L. Norepinephrine induces apoptosis in neonatal rat endothelial cells via a ROS-dependent JNK activation pathway. Apoptosis 2006, 11, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Wang, N.; Qian, J.; Bai, L.; Zheng, X.; Hou, G.; Qiu, X.; Yang, B. Renal sympathetic denervation improves myocardial apoptosis in rats with isoproterenol-induced heart failure by downregulation of tumor necrosis factor-alpha and nuclear factor-kappaB. Exp. Ther Med. 2017, 14, 4104–4110. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chi, X.; Wu, S.; Jin, Y.; Yao, H.; Wang, Y.; Xia, Z.; Cai, J. Dexmedetomidine Pretreatment Attenuates Kidney Injury and Oxidative Stress during Orthotopic Autologous Liver Transplantation in Rats. Oxid Med. Cell Longev 2016, 2016, 4675817. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Feng, X.; Hu, X.; Sha, J.; Li, B.; Zhang, H.; Fan, H. Dexmedetomidine Ameliorates Acute Stress-Induced Kidney Injury by Attenuating Oxidative Stress and Apoptosis through Inhibition of the ROS/JNK Signaling Pathway. Oxid Med. Cell Longev 2018, 2018, 4035310. [Google Scholar] [CrossRef]
- Koca, U.; Olguner, C.G.; Ergur, B.U.; Altekin, E.; Tasdogen, A.; Duru, S.; Girgin, P.; Gunduz, K.; Cilaker Micili, S.; Guzeldag, S.; et al. The effects of dexmedetomidine on secondary acute lung and kidney injuries in the rat model of intra-abdominal sepsis. Sci. World J. 2013, 2013, 292687. [Google Scholar] [CrossRef]
- Schiller, A.M.; Pellegrino, P.R.; Zucker, I.H. The renal nerves in chronic heart failure: efferent and afferent mechanisms. Front. Physiol. 2015, 6, 224. [Google Scholar] [CrossRef]
- Park, J. Cardiovascular risk in chronic kidney disease: role of the sympathetic nervous system. Cardiol. Res. Pract 2012, 2012, 319432. [Google Scholar] [CrossRef] [PubMed]
- Kon, V.; Yared, A.; Ichikawa, I. Role of renal sympathetic nerves in mediating hypoperfusion of renal cortical microcirculation in experimental congestive heart failure and acute extracellular fluid volume depletion. J. Clin. Invest. 1985, 76, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Swedberg, K.; Viquerat, C.; Rouleau, J.L.; Roizen, M.; Atherton, B.; Parmley, W.W.; Chatterjee, K. Comparison of myocardial catecholamine balance in chronic congestive heart failure and in angina pectoris without failure. Am. J. Cardiol. 1984, 54, 783–786. [Google Scholar] [CrossRef]
- Bohm, M.; La Rosee, K.; Schwinger, R.H.; Erdmann, E. Evidence for reduction of norepinephrine uptake sites in the failing human heart. J. Am. Coll Cardiol. 1995, 25, 146–153. [Google Scholar] [CrossRef]
- Bohm, M.; Gierschik, P.; Jakobs, K.H.; Pieske, B.; Schnabel, P.; Ungerer, M.; Erdmann, E. Increase of Gi alpha in human hearts with dilated but not ischemic cardiomyopathy. Circulation 1990, 82, 1249–1265. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, D.; Freeman, R.H.; Johnson, R.A.; Simmons, J.C. Effects of renal denervation on postprandial sodium excretion in experimental heart failure. Am. J. Physiol. 1994, 266, R1599–R1604. [Google Scholar] [CrossRef]
- Masaki, H.; Imaizumi, T.; Harasawa, Y.; Takeshita, A. Dynamic arterial baroreflex in rabbits with heart failure induced by rapid pacing. Am. J. Physiol. 1994, 267, H92–H99. [Google Scholar] [CrossRef]
- Clayton, S.C.; Haack, K.K.; Zucker, I.H. Renal denervation modulates angiotensin receptor expression in the renal cortex of rabbits with chronic heart failure. Am. J. Physiol. Renal Physiol. 2011, 300, F31–F39. [Google Scholar] [CrossRef]
- Linz, D.; Wirth, K.; Ukena, C.; Mahfoud, F.; Poss, J.; Linz, B.; Bohm, M.; Neuberger, H.R. Renal denervation suppresses ventricular arrhythmias during acute ventricular ischemia in pigs. Heart Rhythm 2013, 10, 1525–1530. [Google Scholar] [CrossRef]
- Brandt, M.C.; Mahfoud, F.; Reda, S.; Schirmer, S.H.; Erdmann, E.; Bohm, M.; Hoppe, U.C. Renal sympathetic denervation reduces left ventricular hypertrophy and improves cardiac function in patients with resistant hypertension. J. Am. Coll. Cardiol. 2012, 59, 901–909. [Google Scholar] [CrossRef]
- Brede, M.; Wiesmann, F.; Jahns, R.; Hadamek, K.; Arnolt, C.; Neubauer, S.; Lohse, M.J.; Hein, L. Feedback inhibition of catecholamine release by two different alpha2-adrenoceptor subtypes prevents progression of heart failure. Circulation 2002, 106, 2491–2496. [Google Scholar] [CrossRef] [PubMed]
- Brede, M.; Philipp, M.; Knaus, A.; Muthig, V.; Hein, L. alpha2-adrenergic receptor subtypes - novel functions uncovered in gene-targeted mouse models. Biol. Cell 2004, 96, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Sugiura, T.; Hayashi, K.; Ohkita, M.; Takaoka, M.; Yukimura, T.; Matsumura, Y. Moxonidine prevents ischemia/reperfusion-induced renal injury in rats. Eur. J. Pharmacol. 2009, 603, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Gilsbach, R.; Schneider, J.; Lother, A.; Schickinger, S.; Leemhuis, J.; Hein, L. Sympathetic alpha(2)-adrenoceptors prevent cardiac hypertrophy and fibrosis in mice at baseline but not after chronic pressure overload. Cardiovasc. Res. 2010, 86, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.K.; Chan, M.H.; Tai, M.H.; Lam, C.W. Hepatorenal syndrome. Clin. Biochem. Rev. 2007, 28, 11–17. [Google Scholar]
- Moreau, R.; Lebrec, D. Acute renal failure in patients with cirrhosis: perspectives in the age of MELD. Hepatology 2003, 37, 233–243. [Google Scholar] [CrossRef]
- Stadlbauer, V.; Wright, G.A.; Banaji, M.; Mukhopadhya, A.; Mookerjee, R.P.; Moore, K.; Jalan, R. Relationship between activation of the sympathetic nervous system and renal blood flow autoregulation in cirrhosis. Gastroenterology 2008, 134, 111–119. [Google Scholar] [CrossRef]
- Bendtsen, F.; Christensen, N.J.; Sorensen, T.I.; Henriksen, J.H. Effect of oral propranolol administration on azygos, renal and hepatic uptake and output of catecholamines in cirrhosis. Hepatology 1991, 14, 237–243. [Google Scholar] [CrossRef]
- Henriksen, J.H.; Ring-Larsen, H. Hepatorenal disorders: role of the sympathetic nervous system. Semin Liver Dis. 1994, 14, 35–43. [Google Scholar] [CrossRef]
- Arroyo, V.; Guevara, M.; Gines, P. Hepatorenal syndrome in cirrhosis: pathogenesis and treatment. Gastroenterology 2002, 122, 1658–1676. [Google Scholar] [CrossRef]
- Angeli, P.; Volpin, R.; Gerunda, G.; Craighero, R.; Roner, P.; Merenda, R.; Amodio, P.; Sticca, A.; Caregaro, L.; Maffei-Faccioli, A.; et al. Reversal of type 1 hepatorenal syndrome with the administration of midodrine and octreotide. Hepatology 1999, 29, 1690–1697. [Google Scholar] [CrossRef] [PubMed]
- Roulot, D.; Braillon, A.; Gaudin, C.; Ozier, Y.; Girod, C.; Lebrec, D. Mechanisms of a clonidine-induced decrease in portal pressure in normal and cirrhotic conscious rats. Hepatology 1989, 10, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Serste, T.; Melot, C.; Francoz, C.; Durand, F.; Rautou, P.E.; Valla, D.; Moreau, R.; Lebrec, D. Deleterious effects of beta-blockers on survival in patients with cirrhosis and refractory ascites. Hepatology 2010, 52, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Krag, A.; Wiest, R.; Albillos, A.; Gluud, L.L. The window hypothesis: haemodynamic and non-haemodynamic effects of beta-blockers improve survival of patients with cirrhosis during a window in the disease. Gut 2012, 61, 967–969. [Google Scholar] [CrossRef] [PubMed]
- Simoes, E.S.A.C.; Miranda, A.S.; Rocha, N.P.; Teixeira, A.L. Renin angiotensin system in liver diseases: Friend or foe? World J. Gastroenterol. 2017, 23, 3396–3406. [Google Scholar] [CrossRef] [PubMed]
- Grace, J.A.; Herath, C.B.; Mak, K.Y.; Burrell, L.M.; Angus, P.W. Update on new aspects of the renin-angiotensin system in liver disease: clinical implications and new therapeutic options. Clin. Sci. 2012, 123, 225–239. [Google Scholar] [CrossRef]
- Ahmadian, E.; Pennefather, P.S.; Eftekhari, A.; Heidari, R.; Eghbal, M.A. Role of renin-angiotensin system in liver diseases: an outline on the potential therapeutic points of intervention. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 1279–1288. [Google Scholar] [CrossRef]
- Ott, C.; Mahfoud, F.; Schmid, A.; Toennes, S.W.; Ewen, S.; Ditting, T.; Veelken, R.; Ukena, C.; Uder, M.; Bohm, M.; et al. Renal denervation preserves renal function in patients with chronic kidney disease and resistant hypertension. J. Hypertens 2015, 33, 1261–1266. [Google Scholar] [CrossRef]
- Hering, D.; Marusic, P.; Duval, J.; Sata, Y.; Head, G.A.; Denton, K.M.; Burrows, S.; Walton, A.S.; Esler, M.D.; Schlaich, M.P. Effect of renal denervation on kidney function in patients with chronic kidney disease. Int. J. Cardiol. 2017, 232, 93–97. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).