1. Introduction
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is amongst the chronic diseases which most adversely affect quality of life [
1]. Despite this, diagnosis is often a slow process and relies on internationally varying case criteria, in the absence of a gold standard diagnostic biomarker. The most commonly used definitions require the presence of post-exertional malaise for diagnosis, accompanied by combinations of other, variably presenting symptoms [
2,
3,
4]. Dependence on these cumbersome and varying definitions, which likely capture heterogenous patient populations, has presented a longstanding challenge for patients, clinical practice and the substantial body of ME/CFS research [
5]. The identification of robust biomarkers has consequently been one of the most recurring pursuits in the field, since it would allow for more rapid, specific and sensitive diagnoses of patients. While past and ongoing attempts at identifying such a diagnostic solution are numerous, none have yet resulted in a clinically proven biomarker of ME/CFS. It is therefore imperative that new diagnostic tools continue to be sought.
To combat the subjectivity introduced by self-reported symptom scales, objective clinical measures have been explored previously, such as hand grip strength [
6] or orthostatic intolerance and standing difficulty [
7]. These physical measures have been demonstrated to have value in stratifying ME/CFS patients by disease state/severity and in aiding the separation of patients from healthy subjects. Measures of physical ability may, however, be confounded by other conditions also affecting physical ability. Such tools are, therefore, very useful for the clinical characterisation of patients but are not proven specific to this disease. This highlights the unmet need for specific, molecular biomarkers of ME/CFS.
Accordingly, molecular biomarkers have been pursued across multiple areas of research. For example, the role of the immune system in ME/CFS has been studied for decades, raising cytokines as potential biomarkers of disease [
8,
9,
10,
11,
12,
13,
14] whose clinical utility would be aided by their accessibility in blood. Other cytokine studies have utilised cerebrospinal fluid [
15,
16], the acquisition of which is more invasive than drawing blood, and so could be less amenable to a routine diagnostic test. Despite the number of studies, these reports as a whole are largely inconsistent for individual cytokines [
17,
18], with initially promising candidates such as transforming growth factor beta 1 being subsequently refuted [
19]. Therefore, the utility of cytokine measurements such as an ME/CFS diagnostic tool remains uncertain.
By contrast, studies reporting altered levels of many metabolites in the blood of ME/CFS patients [
20,
21,
22,
23,
24,
25,
26,
27] are thought to reflect pathway alterations that are largely consistent, albeit with some discrepancies between studies [
23]. Such metabolite differences have been proposed as potential candidates for diagnostic blood tests [
20,
21,
22,
23,
24]. One strength of a diagnostic test which measures multiple molecular parameters is that the likelihood of other diseases displaying the same biochemical pattern is low. However, the techniques utilised in these studies (mass spectrometry or nuclear magnetic resonance spectroscopy) may be subject to rapid fluctuations with patient diet and activity, as well as requiring strict sample acquisition and handling conditions [
28,
29] and expensive, specialised equipment. These requirements could introduce challenges in a broad clinical setting. Further investigation is therefore warranted, both to address the clinical suitability of blood-based metabolite measurements for diagnosing ME/CFS, and to better understand the remaining inconsistencies.
The ideal molecular biomarkers would be straightforward to sample and assess, and they also exhibit high sensitivity and specificity. For these reasons, the search for biomarkers has extended to investigating the utility of routine blood pathology tests, but the results are either inconsistent [
9,
10] or require further study [
30]. The recent development of a nanoneedle bioarray to measure the electrical impedance of ME/CFS peripheral blood mononuclear cells (PBMCs) in plasma is highly promising, but requires identification of the underlying mechanism and proven specificity for ME/CFS [
31]. Therefore, the need for a simple blood-based biomarker remains currently unfulfilled.
It has long been suspected that mitochondrial dysfunction might play a role in the cytopathology, but the small number of direct investigations of this had produced confusing, contradictory results. We recently reported both specific mitochondrial dysfunction and cellular signalling dysregulation in ME/CFS patient blood-derived lymphoblasts, as well as an associated reduction in the viability in culture of ex vivo, stored PBMCs from patients versus healthy controls [
32]. We now examine the discriminatory utility of these differences as components of a potential, blood-based diagnostic test.
3. Discussion
The diagnosis of ME/CFS currently requires patients to exhibit, for at least six months, the hallmark symptoms of chronic fatigue and post-exertional malaise that cannot be explained by other conditions. For patients, physicians and the health care system, this diagnosis—that depends on exclusion of other illnesses—is a long, frustrating and potentially expensive process. Suitable diagnostic biomarkers have not yet been identified, although a recent report of an altered electrical impedance response to salt stress in patient lymphocytes looks highly promising [
31]. We reported, in an accompanying paper, that ex vivo lymphocytes from ME/CFS patients exhibit an elevated death rate in culture after recovery from frozen storage and that lymphoblastoid cell lines (lymphoblasts) isolated from them exhibit multiple mitochondrial and cellular stress signalling abnormalities. These abnormalities were measured in four different laboratory tests—lymphocyte death rates, lymphoblast mitochondrial membrane potential, lymphoblast mitochondrial respiratory function and lymphoblast TORC1 signalling activity. The mitochondrial respiratory dysfunction included changes in several key measures of mitochondrial activity—reduced mitochondrial membrane potential and lower efficiency of ATP synthesis, increased “proton leak” relative to basal metabolic rate, and elevated maximum rates of respiration and Complex I activity. Our purpose here was to determine if these abnormalities in cells derived from ME/CFS patient blood samples could be used as biomarkers of disease.
The results from the linear discrimination and logistic regression analyses were almost identical for all of the biomarkers we used here. This is not surprising as they are related methods and have produced similar outcomes in other studies [
37]. However, logistic regression is considered to be more robust against departures of the input data from normality, equality of variances and the presence of outliers. To confirm that the proposed biomarkers would be effective in correctly diagnosing ME/CFS in new samples that were not part of the original analysis, we randomly selected 80% of our sample for use as a training set, while the remaining 20% was used as a test data set. Despite the fact that the smaller training set makes the parameter estimates and threshold determination less accurate, very similar results to those obtained with the full data set were obtained in all of our assays and all performed as well with the test data set as with the training set (
Appendix A Table A2). We also trialed and found that support vector machine and neural network models produced very similar outcomes for all of the biomarkers we tested here. These results give confidence that the proposed biomarkers could be usefully deployed for diagnostic purposes.
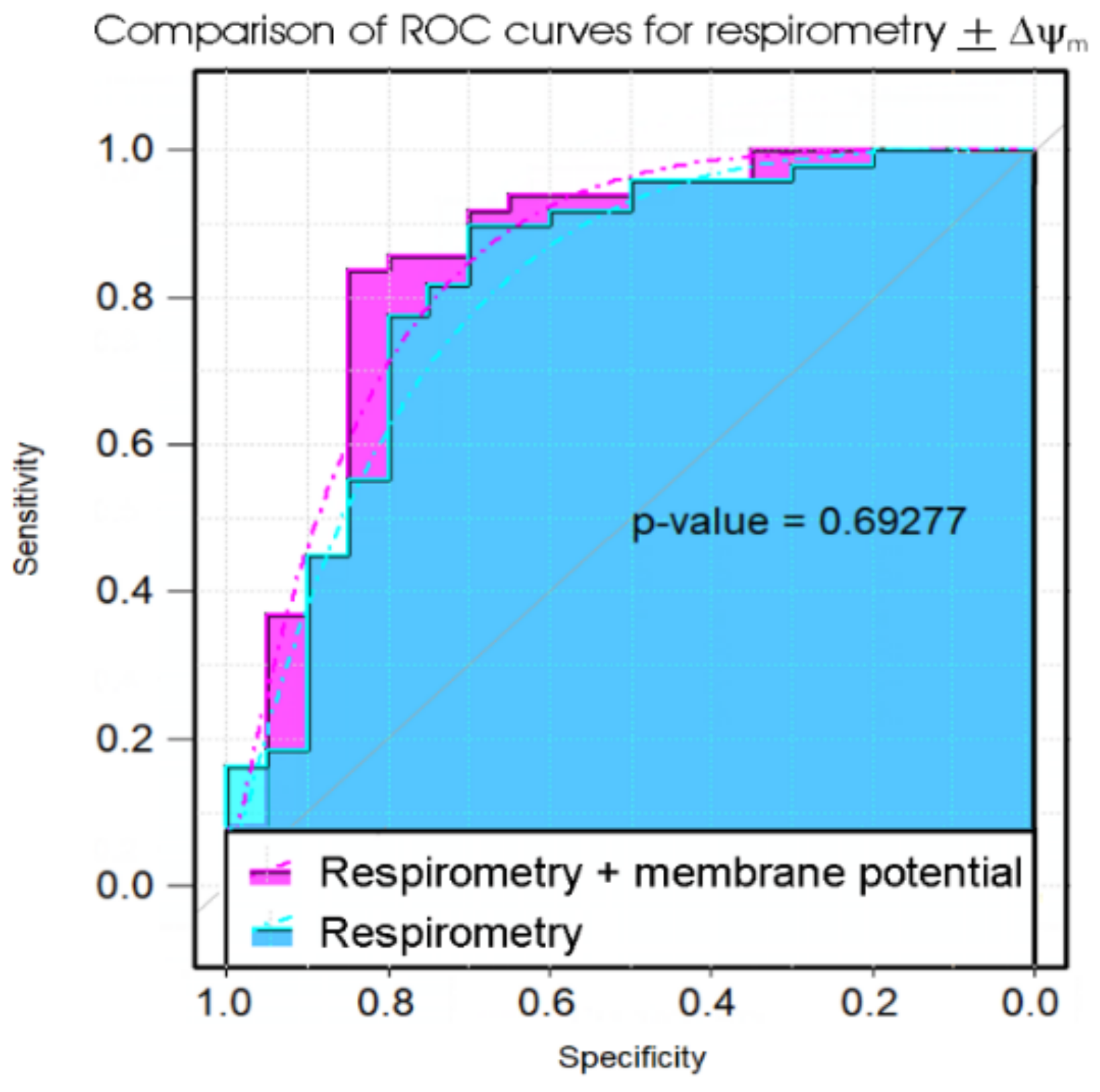
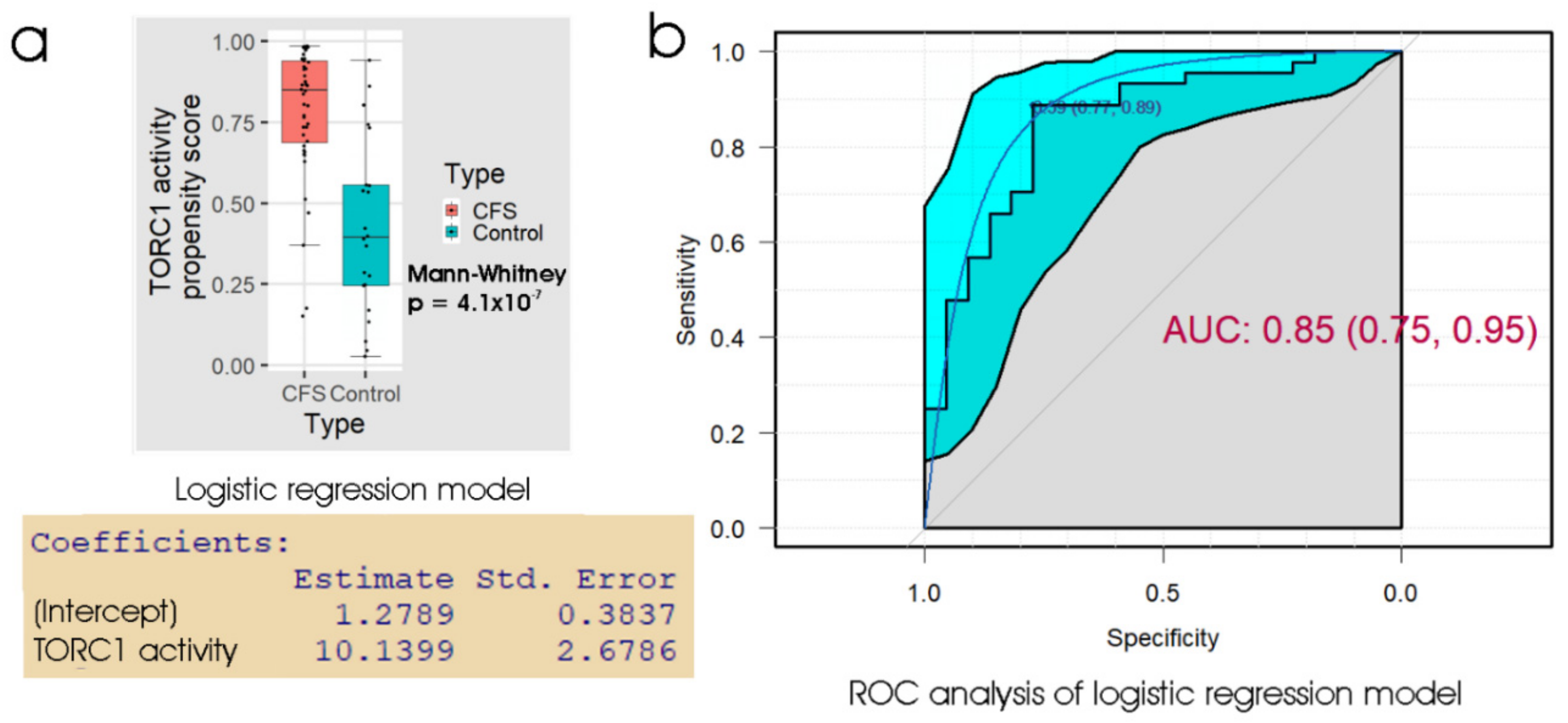
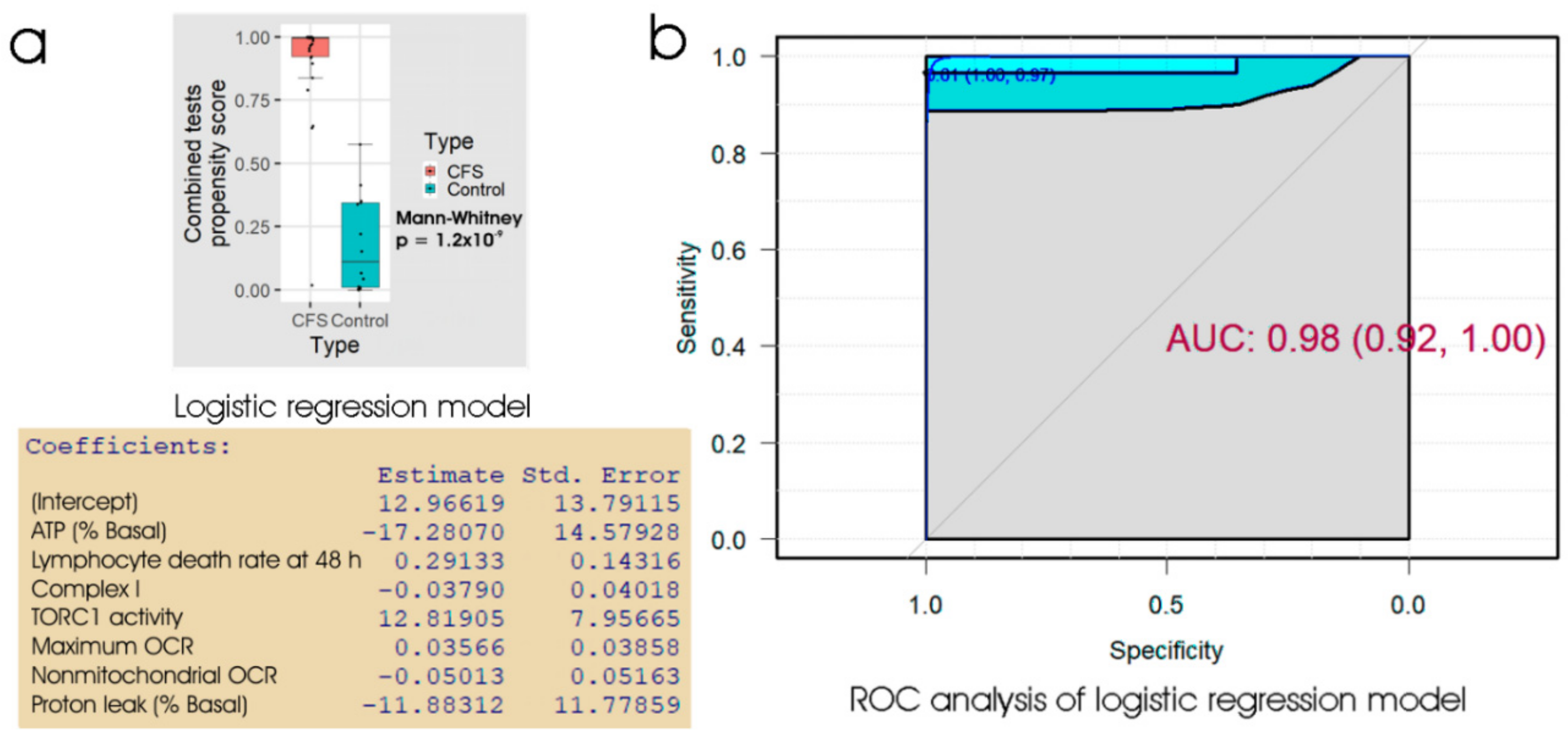
Of the biomarker tests we examined, the lymphocyte death rates are the simplest and cheapest, and would provide the quickest result in a clinical setting. We found that the fraction of dead lymphocytes after frozen storage and culture for 48 h could distinguish between ME/CFS and control samples with a high sensitivity but only modest specificity. Although ex vivo lymphocyte death rates were elevated compared to controls on all three days of culture after recovery from frozen storage, assessing the proportion of dead cells in the culture at more than one time point did not improve the discriminatory value of the test. Of the remaining three types of test, the measurement of mitochondrial membrane potential produced no significant improvement over the respirometric assay of mitochondrial function, so we discarded its use in the remaining analysis. Both the respiratory function assay and the TORC1 activity assay produced similar results to the lymphocyte death assay—high sensitivity combined with low specificity. However, when all three tests were combined using multiple logistic regression, we found very high sensitivity and specificity, with only two from a total of 43 samples (one patient and one control) being misclassified. Except for the recently reported lymphocyte impedance response to salt stress [
31], this combination of three tests (lymphocyte death rate, lymphoblast mitochondrial respiratory function and lymphoblast TORC1 activity) thus achieves better accuracy and reliability (sensitivity, specificity and AUC) for discriminating ME/CFS than has been previously reported using other blood-based molecular tests [
20,
21,
22,
23].
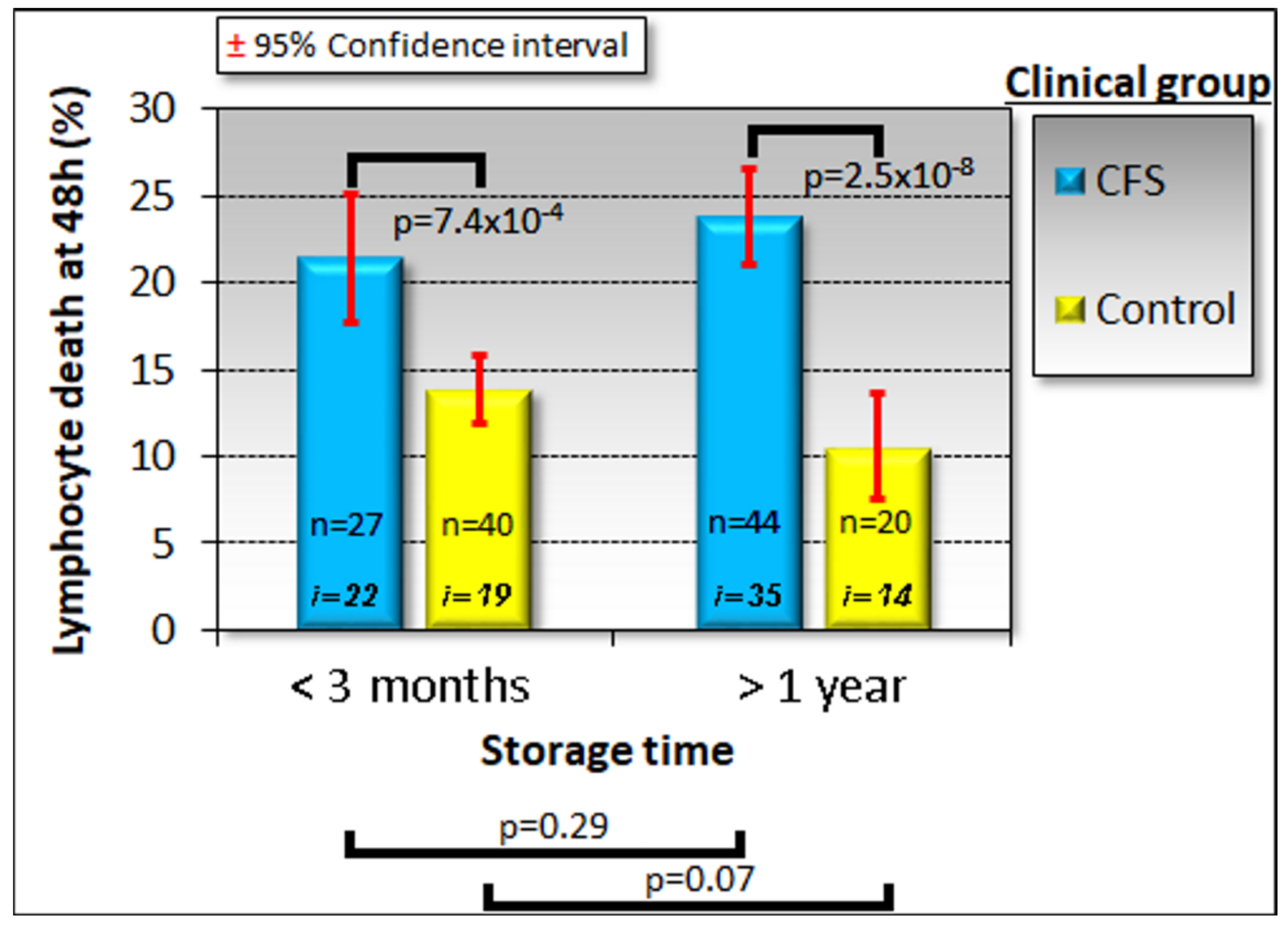
Because of the time, expertise and expense associated with lymphoblast isolation and testing, we explored whether a two-stage test would be suitable. Our results suggested a test protocol which can discriminate between ME/CFS patients and healthy individuals with near-perfect accuracy. In this protocol, the frozen lymphocyte viability after 48 h in culture would be used as an initial screening test. With its high sensitivity, low cost and speed, it requires only a small blood volume allowing most of the sample to be utilised for subsequent confirmatory tests. We have also demonstrated that the test result is stable over long periods of frozen storage. These practical advantages lend great value to the protocol in a broader clinical context and set it apart from methods which would proceed directly to highly specialised testing. Applied to the subset of 43 samples for which we have results from all three tests, this initial screening would have failed to detect only one patient who would have tested positive if all three tests had been combined. One patient and one control sample were misclassified, regardless of using the staged protocol or the combined tests, so that the staged protocol would have misclassified only one additional patient. We therefore propose that a staged protocol combining the clinical suitability of the lymphocyte screening step and the discriminatory power of the entire protocol provides a promising diagnostic biomarker for ME/CFS. The patient and their clinician could choose on the basis of the results from the lymphocyte death rate test, whether or not to proceed with the slower, more expensive confirmatory tests.
One limitation of the viability assay we used in this work, Trypan Blue staining, is that although it is very fast and simple in principle, it requires microscopy and cell counting by a person skilled in the art. It would be valuable in future work to determine if some of the many other cell viability assays that are commercially available might lend themselves more readily to use in a reproducible way by less skilled personnel. This would facilitate higher throughput and greater accuracy in the results in a clinical setting.
A further limitation of the testing protocol suggested here is that it has so far been fully tested on a relatively small sample of 29 patients and 14 controls for whom we have a complete data set from all tests. The testing protocol here will need to be refined using much larger samples in order to both validate it and to determine more accurately the weighting coefficients that should be applied in the logit function (from the logistic regression). At this stage, we can nonetheless conclude that the cell-based blood biomarkers used here are amongst the most promising candidates so far identified for potential use in diagnosing ME/CFS.
A limitation of all biomarkers so far proposed for ME/CFS is that it is not yet known how specific they are vís a vís other illnesses which cause chronic fatigue and/or post exertional malaise and with which ME/CFS may potentially be confused. It would be useful to examine the viability levels of frozen lymphocytes in similar diseases to assess how specific the elevated death rate of frozen lymphocytes is to ME/CFS patients. It has been documented previously, that frozen PBMC viability is also reduced in paediatric Dengue fever [
38]. However, the strength of the frozen lymphocyte viability test is its high sensitivity as a screening step to successfully detect ME/CFS individuals and correctly triage true-positive ME/CFS samples towards the subsequent confirmatory tests. It seems possible that the number and specific biological nature of these subsequent measurements means that the resulting molecular read-out is likely to be unique to ME/CFS, particularly in combination with the patient’s clinical history. To be confused with ME/CFS in the confirmatory tests of mitochondrial respiratory function and TORC1 activity, other illnesses would not only need to cause reduced viability of frozen lymphocytes, they would also need to confer the same pattern of molecular abnormalities upon the derived lymphoblasts—decreased Complex V efficiency, elevated proton leak as a proportion of basal metabolic rate, as well as increases in maximum respiratory capacity, Complex I activity, nonmitochondrial oxygen consumption and TORC1 activity. It is worth noting that lymphoblasts from patients with Parkinson’s disease, another complex chronic disorder, exhibit a quite different pattern of abnormalities related to mitochondrial function [
39]. Of course, Parkinson’s disease patients are unlikely to be confused clinically with ME/CFS patients in the first place. Thus, although the combination of cellular and molecular phenotypes reported here may well be unique to ME/CFS, it will be essential in future work to determine its specificity in relation to other, similar diseases.
Despite the clarity of the differences we have observed in frozen lymphocyte viability, the underlying reason for the elevated death of ME/CFS lymphocytes after storage remains undetermined. Impaired mitochondrial respiratory function, including Complex V impairment, has long been known to result in apoptotic cell death in ex vivo lymphoid cells [
40]. However, we have not demonstrated whether the lymphocyte death we observe is apoptotic or whether it is one of the other known forms of eukaryotic cell death. Whatever the specific cell death pathway involved, it may reflect an inability of ME/CFS patient cells to adequately respond to cellular damage or stress. In this case, such an insult could be introduced by freezing, which is well understood to damage biological systems by the formation of ice crystals, but has also been documented to specifically affect PBMC viability, function, and expression of stress response genes [
41,
42]. While the elevated death rate of the ME/CFS lymphocytes could reflect a greater mechanical susceptibility to immediate structural damage by freezing, there remains another possibility. Compared with controls, the number of dead ME/CFS lymphocytes continued to increase at a faster rate than the controls over multiple days in culture. This suggests that underlying and ongoing cytopathological processes could be contributing towards cell death in culture of previously frozen lymphocytes.
4. Materials and Methods
4.1. Participant Cohort
Participants were assessed and samples collected by trained staff from La Trobe University and the CFS Discovery Clinic, Melbourne, Australia. Testing was carried out at participant homes when the severity of illness precluded patients from travelling. Participants were selected using the Canadian Consensus Criteria [
4] assessed for postorthostatic tachycardia syndrome comorbidity, and asked to complete the Depression, Anxiety and Stress Scale questionnaire and the Epworth Sleepiness Scale questionnaire. ME/CFS-specific severity assessments were also conducted using Richardson and Lidbury’s weighted standing time [
7]. For PBMC isolation, 15 mL of blood was taken per participant in heparin-treated vacutainer tubes (BD). Patients with other known reasons for fatigue were excluded.
For most of the work reported here, we used the same age- and gender-matched participant groups as reported in the accompanying paper [
32]. This cohort included 51 ME/CFS patients (86% female, median age 51, age range 26–71) and 22 healthy controls (68% female, median age 43, age range 21–67). However, because of limitations on the supply of lymphocytes, we were able to assay lymphocyte death rates in culture only in a subset of these participants—35 patients (89% female, median age 52, age range 26–71) and 14 controls (71% female, median age 42, age range 21–58). Furthermore, these lymphocyte samples had been stored for significant time periods ( > one year). Therefore, we took the opportunity afforded by a recent influx of new participant samples to include more recently obtained samples prepared in the three month period prior to submission of this manuscript. This additional cohort contained 13 new ME/CFS participants (85% female, median age 38, age range 22–70) and 19 new control individuals (42% female, median age 29, age range 19–55). While the age distribution in the new cohort was not significantly different between the patient and control groups (Fisher exact test,
p > 0.1), the gender proportions were significantly different (
p = 0.02). As a consequence, the gender proportions in the total cohort also differed significantly (
p = 0.002) between the patient and control groups used for the lymphocyte death rates. Nevertheless, this additional cohort allowed us to verify our previous finding [
32], that the lymphocyte death rates in culture were not age- or gender-dependent, and to confirm published reports that lymphocyte viability is stable for long periods in frozen storage [
33,
43].
Participants were recruited and samples obtained with approvals by the Australian National University Human Research Ethics Committee (Reference 2015/193, accepted by the La Trobe University Human Ethics Committee on 26 February 2016) and the La Trobe University Human Ethics Committee (Reference HEC19316, approved on 26 August 2019).
4.2. PBMC Isolation from Blood Sample
This method has been described in an accompanying paper [
32]. Briefly, lymphocytes were isolated by Ficoll-Paque density centrifugation and counted. For immortalization, 5 × 10
6 cells were set aside and resuspended in 5 mL Roswell Park Memorial Institute (RPMI) 1640 without L-glutamine (Life Technologies) supplemented with 1X Glutamax (Life Technologies, Carlsbad, California, United States), 10% fetal bovine serum (FBS) and 1% Penicillin/Streptomycin. Excess lymphocytes were separated into aliquots of 5 × 10
6 cells, harvested and resuspended in 250 µL of Recovery™ Cell Culture Freezing Medium (Life Technologies, Carlsbad, California, United States) and stored at 0 °C.
4.3. Immortalisation of Lymphocytes
One mL of culture supernatant from B95.8 cells expressing Epstein-Barr virus (EBV) was added to 5 × 106 cells in 5 mL RPMI 1640. Per well, 150 µL of the mix was seeded in a 96-well U-bottom plate, then incubated for one hour within a humidified 5% CO2 incubator at 37 °C. A final concentration of 500 ng/mL Cyclosporin A (Sigma, St. Louis, MO, USA) was then added to each well. Cultures were fed weekly by replacing half of the medium with the same formulation, without disturbing the cells. This process was repeated over a period of approximately three weeks until the cells were confluent and growing rapidly, after which the lymphoblast cultures were processed as described in the following section.
4.4. Lymphoblast Cultures
Confluent lymphoblasts were transferred to T25 flasks in growth medium (Minimum Essential Medium α (Life Technologies, Carlsbad, California, United States), supplemented with 10% FBS and 1% Penicillin/Streptomycin), where they were cultured within a humidified 5% CO
2 incubator at 37 °C. Lymphoblast storage in Recovery™ Cell Culture Freezing Medium at −80 °C has been previously described in detail [
32].
Prior to commencing experiments, lymphoblast lines were cultured over as short a time and as few passages (2–5) as possible. For a set of triplicate, independent experiments, harvest, assay and conduct of experiments occurred over approximately one week. Two immortalised lymphoblast cell lines created from healthy donor blood were utilised as internal controls to normalise for variation between experiments where appropriate.
4.5. Viable Cell Counts
Lymphoblast or lymphocyte (PBMC) viable counts for all applications were determined by staining with Trypan blue (Thermo-Fisher Scientific, Waltham, Massachusetts, United States) prior to haemocytometer cell counting. Trypan blue-stained cells were counted as dead and unstained, intact cells as viable. For the unimmortalised lymphocyte viability measurements over time, frozen aliquots were thawed in a 37 °C water bath, pelleted at 1000 × g for 2 min and resuspended in 1 mL RPMI 1640 without L-glutamine supplemented with 1X Glutamax, 10% FBS and 1% Penicillin/Streptomycin. The cells were then washed at 1000 × g for 2 min and resuspended in fresh medium of the same formulation. They were then seeded in 96-well U-bottom plate at a density of 1 × 106 cells/mL and kept in a humidified 5% CO2 incubator at 37 °C over the course of the experiment. Each well was mixed gently by pipette before sampling to ensure counting of a homogeneous cell suspension.
4.6. Mitochondrial Stress Test (Seahorse Respirometry)
Oxygen consumption rates (OCR) of 8 × 10
5 viable PBMCs or lymphoblasts per well were measured using the Seahorse XFe24 Extracellular Flux Analyser with Seahorse XF24 FluxPaks (Agilent Technologies, Chicopee, Massachusetts, USA). Immortalised lymphoblasts were cultured in 3 mL growth medium per well in 6-well Costar plates prior to Seahorse experiments. Seahorse assays were carried out as previously described in detail [
39]. Oxygen consumption rates (OCR in pmol/min) were measured (basal OCR) prior to and after successive injection of 1 µM oligomycin (ATP synthase inhibitor), 1 µM CCCP (carbonyl cyanide m-chlorophenyl hydrazone, an uncoupling protonophore), 1 µM rotenone (Complex I inhibitor) and 5 µM antimycin A (Complex III inhibitor). From the resulting data, we determined the OCR associated with respiratory ATP synthesis (oligomycin-sensitive), the maximum OCR in CCCP-uncoupled mitochondria and the rotenone-sensitive OCR attributable to uncoupled Complex I activity, the antimycin-sensitive Complex II/III activity, the OCR by mitochondrial functions (e.g., protein import) other than ATP synthesis that are Δψm-driven (so-called ‘proton leak’), non-respiratory oxygen consumption (e.g., by cellular and mitochondrial oxygenases and oxidases), and the respiratory ‘spare-capacity’ (excess capacity of the respiratory electron transport chain that is not being used in basal respiration).
4.7. 4E-BP1 Phosphorylation Levels (TORC1 Activity)
TORC1 activity in ME/CFS lymphoblast lysates was measured as previously described [
32] using a time-resolved FRET-based multiwell plate assay of the phosphorylation state of 4E-BP1, a major TORC1 substrate (Cisbio Bioassays, Codolet, France).
Lymphoblasts were harvested, resuspended in growth medium at 2.75 × 105 cells/mL and plated in four replicates at 5 × 104 cells/well in a 96-well plate. Cells were incubated at 5% CO2 / 37 °C for 2 h, with two of the replicates subjected to TOR inhibition by 0.5 µM TORIN2. Lysis buffer was added to each well as per manufacturer instructions and the plate mixed on an orbital shaker for 40 min before plating each sample into a 384 well white plate (Corning, New York, USA)—incorporating various controls and antibody mix (anti- 4E-BP1 antibody labelled with d2 acceptor, and anti-phospho-4E-BP1 antibody labelled with Eu3+-cryptate donor) according to manufacturer instructions. After a 2 h incubation at room temperature the plate was scanned by the Clariostar plate reader (BMG, Ortenberg, Germany) and the ratio of the FRET signal from anti-phospho-4E-BP1 antibody to the donor fluorescence signal from anti-4E-BP1 antibody was measured. Internal normalisation control lymphoblasts were included within each assay in case of between-experiment variation.
4.8. Quantification and Statistical Analysis of Biochemical Assays
Data was analysed using Microsoft Excel with the WinStat add-in (Fitch, R.K.,
http://www.winstat.com) and R [
44] using the packages R Commander [
45], REzy [
46], Rattle [
47], pROC [
36] and stats. The linear discriminant analysis (WinStat) used as prior probabilities for the proportions of the ME/CFS and control samples in the total cohort. In the logistic regression (REzy, R Commander), the propensity score was calculated from the fitted logit function. The propensity score represents a probability that the sample in question is from an ME/CFS patient. For both methods, we used either a single independent variable or a set of five key respirometric parameters that are significantly altered in ME/CFS lymphoblasts. For lymphoblast TORC1 activity, the independent variable was the logarithm of the normalised phosphorylation level of 4E-BP1, a specific cellular substrate of TORC1. For lymphocyte death rate it was the percentage of dead lymphocytes after 48 h in culture medium. For respirometry, the five key parameters used were the fraction of the basal O
2 consumption rate (OCR) attributable to ATP synthesis by Complex V and the use of the proton gradient in other mitochondrial membrane transport processes (the proton leak), maximum CCCP-uncoupled OCR, the maximum uncoupled Complex I activity and the nonmitochondrial OCR. The outcomes of both the linear discriminant analysis and the logistic regression were expressed as a “confusion matrix”, showing a cross tabulation of the actual source of the sample (ME/CFS or control) and the classification produced by the method in question. From this, the error rates (false positives and false negatives) were calculated.
Linear discriminant analysis allocates an individual sample to the group whose average it is closest to, on the test criteria being used, while logistic regression makes the assignment on the basis of which group the sample has the higher likelihood of belonging to (propensity score > 0.5). Although arbitrary, these criteria for determining test thresholds make intuitive sense. However, in neither case are they designed to minimise the overall error rate, so that they can yield large differences between the false positive and false negative rates. We therefore also performed Receiver Operating Characteristic (ROC) analysis on the outcomes of the logistic regression, during which the sensitivity (fraction of positives that are correct) is plotted against specificity (fraction of negatives that are correct). Since ROC analysis depends only on the rank order of observations in the data set, when there is only a single independent measure being tested, it produces identical results for the raw and for rescaled or transformed data from the corresponding linear discriminant or logistic regression analysis. Only the scale changes on which the threshold is measured. For consistency, and to facilitate comparisons between different assays, we have conducted ROC analysis on the propensity scores from logistic regression throughout this paper. The AUC (area under the ROC curve) with 95% confidence limits was calculated as an indicator of the usefulness of the biomarker in question in distinguishing ME/CFS from control samples. The “best” threshold value for the biomarker in question was defined in the ROC analysis as the point on the ROC curve which maximised the sum of the sensitivity and specificity (i.e., minimised the sum of the errors). ROC curve comparisons by the bootstrapping method were done as described by Robin et al. (2011) using 2000 replicates [
36]. Confidence limits for the sensitivities (on the vertical axis of ROC curves) were used to plot 95% confidence limits for the ROC curve itself.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






