Abstract
Alzheimer’s disease (AD), a main cause of dementia, is the most common neurodegenerative disease that is related to abnormal accumulation of the amyloid β (Aβ) protein. Despite decades of intensive research, the mechanisms underlying AD remain elusive, and the only available treatment remains symptomatic. Molecular understanding of the pathogenesis and progression of AD is necessary to develop disease-modifying treatment. Drosophila, as the most advanced genetic model, has been used to explore the molecular mechanisms of AD in the last few decades. Here, we introduce Drosophila AD models based on human Aβ and summarize the results of their genetic dissection. We also discuss the utility of functional genomics using the Drosophila system in the search for AD-associated molecular mechanisms in the post-genomic era.
1. Introduction
1.1. Genetics of Alzheimer’s Disease
Alzheimer’s disease (AD) is a progressive neurological disorder that results in irreversible loss of neurons, particularly in the cortex and hippocampus. As of 2019, over 50 million people worldwide have AD or a related dementia [1]; which leads to death within three to nine years after diagnosis [2].
In the brain of an AD patient, amyloid beta (Aβ)-containing senile plaques and neurofibrillary tangles (NFTs), the aggregates of hyperphosphorylated tau protein, are observed, which are the main hallmarks of AD [3]. Therefore, Aβ, the cleaved form of amyloid precursor protein (APP), and tau, a microtubule-binding protein, have been suggested to be important causative molecules in the pathology of AD [4,5]. Aβ can aggregate to form flexible soluble oligomers that are toxic to nerve cells [6]. F urthermore, the accumulation of Aβ protein causes AD-associated events such as formation of NFT, cell loss, vascular damage, and dementia [4]. Based on the increased prevalence of early onset AD (EOAD) in Down syndrome patients with three copies of the APP gene and in people with the APP gain of function mutation that increases Aβ levels, Aβ has been thought to be a major cause of AD [7,8,9]. Supporting this idea, gain-of-function mutations of presenilin 1/2, a gene encoding the components of γ-secretase that processes APP to Aβ, which increased accumulation of Aβ, have been also associated with EOAD [9]. On the other hand, hyperphosphorylated tau, a microtubule-associated protein that stabilizes the microtubules, aggregates to form the NFT, resulting in neuronal degeneration [10]. Pathological tau aggregation is found in the brains of most AD patients, and the association between Aβ accumulation and tau phosphorylation has been reported [10]. Therefore, abnormal aggregation of tau protein is considered an important event in the pathogenesis of AD.
However, in the last decade, all clinical trials targeting Aβ or tau have failed, and the only available treatment remains symptomatic [11]. Therefore, despite decades of intensive research, the cause of AD remains elusive. In fact, many studies have demonstrated that AD is a complicated multifactorial disorder and may be affected by the combination of various genetic and environmental factors [12,13]. A twin study suggested that, depending on the model applied, the heritability of AD is 58% to 79% even though nongenetic risk factors also play an essential role [14]. Therefore, it is important to identify various genetic risk factors associated with AD for early detection and intervention. A series of genome-wide association studies (GWAS) have identified several genetic risk loci for late onset AD (LOAD), which seem to cluster in patterns that suggest immunity, lipid processing, and endocytosis as important causal biological processes [13]. In addition, the functional relevance of many AD-related factors has been demonstrated through functional genomic studies using genetic models, such as nematodes, fruit flies, and zebra fish, as well as mice [15].
1.2. Drosophila Models of Alzheimer’s Disease
Owing to the advantages of the Drosophila system, including the ability to withstand various genetic manipulations that cannot be performed in mammals, it has been an important model in AD studies [16,17] (Figure 1). There are three main types of Drosophila AD models according to the transgenes used, and the first type is the γ-secretase-based model. γ-secretase complex components are functionally conserved in the fly, and its many targets, such as APP and Notch receptor, are also conserved [18,19]. Overexpression of wild-type or familial AD-mutant presenilin (psn), a gene coding a component of γ-secretase complex, induces intracellular calcium deficits, which are regarded as one of the earliest events of AD pathology [20], whereas deficiency of psn causes associative learning defects and synaptic abnormalities in Drosophila larvae [21]. Thus, it follows, studies using γ-secretase-based AD models have facilitated understanding of the role of Presenilin in both development and degeneration as well as verifying many modifiers and pathways.
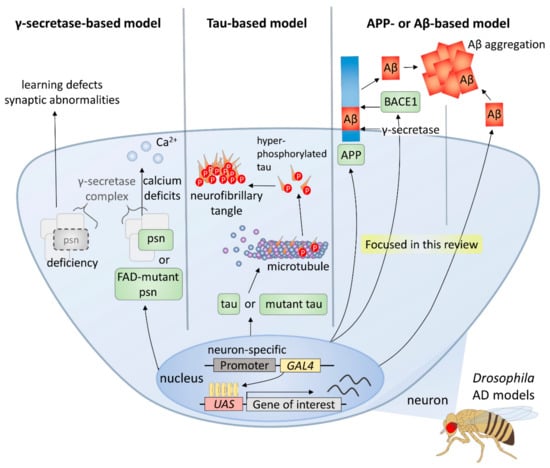
Figure 1.
Drosophila models for Alzheimer’s disease.
Furthermore, tau-based models have been established and used to study the role of tau in the formation of neurofibrillary tangles and neurotoxicity. For instance, several groups have shown that expression of human tau induces AD-like phenotypes in diverse Drosophila tissues [22,23]. A further study used a wild type or mutant human tau-expressing model to identify genetic modifiers of tau [24]. Moreover, the relationship between Aβ42 and tau has also been studied using Aβ42 and tau co-expression models [25].
Finally, most of the Drosophila AD models are based on APP or Aβ expression, since Aβ peptides, the major components of amyloid plaques, are considered to play the most important role in AD [26]. Because there is no conservation of both Aβ peptide sequence in APP and β-secretase in Drosophila, an essential condition for the generation of Aβ peptides, fly models expressing both human APP and BACE have been used [27,28,29]. In these models, AD-like phenotypes, such as age-dependent neuronal death, Aβ accumulation, and lethality are observed [27]. However, it has been found that APP-induced axonal defects are not caused by Aβ [30], and that the APP intracellular domain is involved in various processes such as axonal transport and synaptic plasticity [31]. Therefore, many groups have developed Drosophila AD models that directly express Aβ42 in the fly brain for a more direct study of the role of amyloid plaques in AD [32,33,34,35]. Each of the UAS-Aβ42 transgenes produced by these groups have differences in some part of the construct, such as the signal peptide, poly A tail, or the number of Aβ42 copies, which are directly related to the degree of Aβ peptide accumulation and intensity of AD-like phenotypes [36].
In this review, we will focus on the results obtained from models based on Aβ, the most commonly used AD models in Drosophila. The genetic modifiers found in studies using these Drosophila AD models to date suggest that several cellular pathways may be involved in the development of AD, and the results of these studies demonstrate the usefulness of the Drosophila model for finding related factors of multifactorial genetic diseases, such as AD.
2. AD-Related Mechanisms and Genetic Modifiers Identified Using the Drosophila Model
2.1. Amyloid Beta Accumulation
In the brain of Drosophila expressing Aβ42, age-dependent amyloid deposition was observed, as in human patient brains [37]. Moreover, ectopically expressed Aβ42 in Drosophila photoreceptors showed amyloidogenic and aggregating properties; the resistance to proteolytic cleavage, increased structural stability, and toxicity [32,35,38,39,40].
Recently, several studies showed the role of templated protein misfolding, referred to as seeding [41,42], which induces misfolding and aggregation of the normal soluble protein [43]. Consistently, Drosophila models have provided evidence for a link between the seeding mechanism and neurotoxicity in vivo on a short time scale [44].
2.1.1. Soluble Aβ Oligomer Toxicity and Aggregation
Soluble Aβ oligomer was observed in the CSF of human AD [45] and was more closely associated with disease severity than amyloid plaque, insoluble Aβ, or fibrillar species [46]. Moreover, in other studies using ELISA and Western blotting, the amount of soluble oligomer was found to be more decisive for cognitive deficits than the simple plaque counts [47], and these soluble peptides induced progressive neuronal loss [48]. Consistently, Aβ peptide generation in the Drosophila retina shows age-dependent neurodegeneration in retinal photoreceptor cells and precedes the formation of Aβ plaques, suggesting that the Aβ oligomer and protofibril mediate toxicity [27]. The structural importance of Aβ to generate oligomer is also proved in Drosophila. A flavonoid derivative that interferes and disorders the Aβ oligomer and inhibitors targeting the α-helix of Aβ prevented Aβ-induced neurotoxicity in a Drosophila transgenic AD model [49,50]. A study showed the genetic interaction of neuroserpin, a natural inhibitor of tissue-type plasminogen activator that forms a binary complex with Aβ and prevents mature fibril formation of Aβ, with Aβ ectopically expressed in vivo in the Drosophila AD model [51]. Moreover, recent studies have shown that the cytosolic and secreted forms of the heat shock protein 70 (HSP70) prevent Aβ42 self-aggregation by binding to Aβ42, in which this reduction in aggregation by HSP70 significantly improved the memory performance of flies expressing Aβ42 [52,53]. In contrast, the mammalian prion protein stabilized Aβ oligomers and enhanced Aβ neurotoxicity in Drosophila [54]. Meanwhile, a recent study suggested a new hypothesis of a Aβ aggregation mechanism that gangliosides are responsible for Aβ assembly, by showing that ectopic expression of ganglioside synthesis enzymes in Drosophila, such as β1,4-galactosyltransferases (B4GalT6) and α2,3-sialyltransferase (SAT1), accelerate Aβ assembly [55].
2.1.2. Aβ Degradation
Since the accumulation of Aβ is critical in AD pathology, the Aβ catabolic pathway-related factors should be very important in the control of AD. The in vivo function of neprilysin (NEP) and insulin degradation enzyme (IDE) that is involved in the Aβ catabolic pathway were tested in the Drosophila AD model. NEP belongs to the Aβ degrading enzymes, inhibition of which resulted in the pathologic deposition of Aβ in rats [56]. In the fly model, neuronal expression of human NEP led to significantly reduced intraneuronal deposits of Aβ42 in the brain and also suppressed Aβ42-induced neuron loss, suggesting that up-regulation of neuronal NEP activity is protective against intraneuronal Aβ42 accumulation and neuron loss [57]. IDE, a thiol metalloendopeptidase that cleaves small proteins, including insulin, has Aβ degrading activity [58], and IDE loss-of-function mutant mice showed elevated levels of neuronally secreted Aβ [58]. Consistent with mammals, the reduced lifespan of flies expressing APP and BACE in neurons was partially recovered by Drosophila Ide or human IDE expression, suggesting that IDE can inhibit the pathological processes associated with Aβ accumulation in vivo [59]. More recently, a study showed that partial knockout of neuronal Src homology 2B1 (SH2B1), an adaptor protein that is important for insulin receptor signaling, increased Aβ42 accumulation and had a detrimental effect on Aβ42-expressing flies, while overexpression of neuronal SH2B1 decreased Aβ42 accumulation and had beneficial effects [60]. These results suggested that the insulin signaling pathway plays important roles in Aβ metabolism.
The autophagic-lysosomal pathway is another important Aβ clearance pathway, the in vivo function of which in AD pathology is revealed in Drosophila. The hyperactivated PI3K/AKT/mTOR pathway, a negative-regulating pathway against autophagy, is linked to disrupted clearance of Aβ and tau [61] and alterations in this pathway are associated with autophagic dysfunction in the AD brain [62]. In Drosophila, genetic or pharmacologic inhibition of the PI3K/AKT/mTOR pathway improved Aβ-induced memory loss [63]. A recent study showed that ectopic expression of human Thioredoxin-80 (Trx80), a truncated form of Thioredoxin-1, prevents the toxic effects of Aβ and inhibits its aggregation [64]. In the same study, Gerenu and colleagues found that Trx80 exerts its protective activity through activation of autophagy. In addition, human phospholipase D3 (PLD3), a type-II transmembrane protein of the PLD family, exerts a neuroprotective effect against toxicity caused by Aβ when ectopically expressed in AD model flies, and the role of PLD3 in lysosome dynamics was considered to contribute to the beneficial effect of PLD3 [65]. Given the importance of autophagy in degenerative brain diseases, it is expected that more autophagy-related genes will be found as modifiers in AD pathology.
2.1.3. Intraneuronal Accumulation of Aβ
In addition to extracellular deposition, intraneuronal accumulation of Aβ has been revealed to be involved in pathological features of AD such as synaptic deficits, amyloid plaque formation, and cell death [66,67]. In the brains of AD patients, oligomeric Aβ is mainly localized in neurons, where it associates with lipid membranes [68]. As phosphoinositides, such as PI and PI4,5P, facilitate Aβ assembly in/on lipid membranes [69], their metabolizing enzymes affect AD pathogenesis by influencing neuronal accumulation of Aβ. Supporting this idea, a reduction in synaptojanin-1, which converts PI4,5P into PI4P, inhibited synaptic and behavioral impairments in APP transgenic mice [70,71]. In Drosophila, the functions of the PI4KIIIα complex, which controls the levels of plasmalemmal PI4P and PI4,5P, are well conserved. Genetic reduction of components, such as PI4KIIIα, rolling blackout (RBO), tetratricopeptide repeat domain 7 (TTC7), and Hyccin, of the PI4KIIIα complex suppressed the phenotypes of Aβ42-expressing flies by reduction of neuronal Aβ accumulation [72,73]. In addition, another genetic modifier screening study identified Drosophila orthologues of human 4-hydroxyphenylpyruvate dioxygenase (HPD) and proline rich mitotic checkpoint control factor (PRCC) as suppressors of intraneuronal accumulation of Aβ [74]. Although HPD functions as 4-hydroxyphenylpyruvate dioxygenase that catalyzes the conversion of 4-hydroxyphenylpyruvate to homogentisate and PRCC may have a role in pre-mRNA splicing, the molecular mechanisms underlying how these proteins affect the intraneuronal accumulation of Aβ remain to be elucidated.
2.2. Amyloid-Mediated Tauopathy
Since phosphorylation of tau plays an important role in AD, there is a lot of interest in the regulators of tau phosphorylation. Drosophila has a tau homolog protein containing conserved disease-related phosphorylation sites of human tau [75]. Several studies have reported that Aβ42 induces phosphorylation and pathology of tau in flies and further noted that tau plays an important role in the downstream processes of Aβ-induced toxicity [25,76,77,78]. Aβ42 enhances tau-induced toxicity such as axonal transport defects, neuronal dysfunction, and reduced survival in Aβ42/tau co-expressing flies [25]. These effects also have been consistently observed in several studies using cell and rodent models [79,80,81,82,83,84]. In double-transgenic mice, hyperphosphorylated tau aggregation was decreased by clearance of Aβ, while Aβ accumulation was not affected by increasing tau [82].
Interestingly, it is known that par-1, a Drosophila orthologue of microtubule/microtubule-associated protein affinity regulating kinase (MARK), and glycogen synthase kinase 3β (GSK3β), a component of the Wnt pathway, are critical for Aβ-induced tau phosphorylation in the fly model [25,78]. Several in vitro studies support the idea that Aβ promotes tau phosphorylation via GSK3β activity [85,86,87,88], and studies using mouse models indicate that MARK phosphorylates tau [89,90]. In human cell models, more than 40 tau phosphorylation sites are associated with AD [91,92,93,94]: Serine (Ser) 262 and Ser356 are phosphorylated by MARK [89,90], and Threonine (Thr) 231, Ser199, Ser202, Ser235, Ser396, and Ser404 by GSK3β [95,96,97,98]. In Drosophila, par-1 and GSK3β can also phosphorylate most of these phosphorylation sites of tau [99], suggesting that the catalytic function of the two kinases is conserved between insects and humans. Furthermore, it was first observed in flies that phosphorylation of tau by par-1 plays an important role in the subsequent phosphorylation of tau by GSK3β, suggesting that tau phosphorylation happens in a structurally arranged pattern [99,100]. When Ser262 and Ser356 in tau are substituted for non-phosphorylatable Ala, Aβ42-mediated tau phosphorylation at Thr231 by GSK3β is blocked [78].
In mice, Aβ42 toxicity occurs through Aβ-induced phosphorylation of tau, and reduction or clearance of tau alleviates phenotypes and toxicity of Aβ42; for instance, memory impairment, synaptic loss, neuron loss, and premature death [83,101]. Furthermore, a study showed that removal of endogenous Drosophila tau reduces Aβ42-induced locomotor dysfunction in flies [102]. However, another recent study revealed that deletion of endogenous Drosophila tau had no effect on Aβ42-induced premature death in the fly model [103]. Therefore, unlike in mammals, it is controversial whether endogenous Drosophila tau contributes to Aβ42-induced toxicity.
2.3. Modifiers Related to Stress-Responsive Pathways
2.3.1. Oxidative Stress
It has been known that Aβ42 causes oxidative stress, which is believed to be augmented in the brain of AD patients and animal models [104,105,106]. Based on the important role of oxidative stress in AD pathology, the interest in genes associated with oxidative stress has increased. Although several groups have shown that these genes affect AD pathology through screening studies in Drosophila [107,108,109,110,111], the effects of antioxidant genes, such as superoxide dismutase (SOD), on AD remain controversial.
Rival and colleagues carried out unbiased screening to identify genes whose expressions were changed by Aβ42 and found that antioxidative stress genes, such as Ferritin 1 heavy chain homolog (Fer1HCH), Ferritin 2 light chain homolog (Fer2LCH), catalase (CAT), and Sod2, reduced Aβ42-induced toxicity [107]. In other studies, Fer1HCH and Fer2LCH inhibited Aβ42-induced eye phenotype and premature death [108], while knockdown of Fer1HCH and Fer2LCH increased Aβ42-induced toxicity [109]. In addition, overexpression of sarah (sra), a Drosophila orthologue of protein-serine/threonine phosphatase regulator Down Syndrome Critical Region 1, increases the hydrogen peroxide susceptibility and enhances Aβ42-induced toxicity as a result of reducing the expression of Sod2, Sod3, and Glutathione S transferase D1 in Aβ42/sra-coexpressing flies [111].
In contrast, Favrin and colleagues identified 712 genes of whose expressions were changed in Aβ42-expressing flies and demonstrated that knockdown of Sod3, an extracellular superoxide dismutase gene whose expression increases in Aβ42-expressing flies, increases the locomotor defect and premature death in AD model flies [112]. Expression of wild type Sod1, another ROS-associated gene, decreases the lifespan of Aβ42-expressing flies, while a dominant negative form of Sod1 rescues premature death of the AD model flies [107]. These detrimental effects of Sods on Aβ42-expressing flies may be due to the toxic hydrogen peroxide overload, which occurs because of an imbalance between Sod, which produces hydrogen peroxide, and CAT, which catalyzes the decomposition of hydrogen peroxide to water and oxygen. Furthermore, the functional differences between these SODs may be also due to the distinct locations and prosthetic groups of the enzymes: Sod1 and Sod3 is Cu/ZnSOD in the cytoplasm and extracellular space, respectively, and Sod2 is MnSOD in the mitochondria [113,114]. Therefore, it is possible that there is a difference in ROS function between the mitochondria and cytoplasm in AD pathology.
It is believed that factors related with metals also play important roles in the cellular response to oxidative stress [115]. ROS generated by Aβ42 damages the cellular membrane, especially in the presence of metals such as copper, zinc, and iron [115]. Intake of copper or zinc enhances Aβ42 toxicity, while genetic inhibition of copper transporter 1B (Ctr1B) or Ctr1C, copper importers, or zinc/iron regulated transporter-related protein 1 (Zip1), a zinc importer, alleviates premature death and locomotor defects in AD model flies [116,117].
AD is strongly associated with oxidative stress, and many oxidative stress-related genes, including metals- and mitochondria-related genes, affect AD pathology. Although many antioxidant genes have been shown to have beneficial effects on AD, the protective effect of SOD remains controversial and further studies should be conducted.
2.3.2. Endoplasmic Reticulum Stress
Endoplasmic reticulum (ER) stress has also been implicated in AD [118] and can be induced by Aβ42 in cultured cells, mice, and flies [35,119,120,121,122]. During the course of ER stress, a fragment of activating transcription factor 6 (ATF6) moves into the nucleus and expresses ER stress response genes, including X-box binding protein 1 (XBP1) [123]. Expression of XBP1 is also promoted by Aβ42, and overexpression of spliced XBP1 (XBP1-S), the activated form of XBP1, reduces Aβ42 toxicity in Drosophila photoreceptors, whereas knockdown of endogenous XBP1 intensifies rough eye phenotype induced by Aβ42 [35]. Moreover, a recent study showed that overexpression of XBP1-S in the fly brain reduces Aβ42 levels and improves Aβ42-induced locomotor dysfunction, while reduction of endogenous XBP1 increases Aβ42 protein levels and enhances Aβ42-induced locomotor dysfunction [124]. Furthermore, chronic expression of Aβ42 activates protein kinase R-like endoplasmic reticulum kinase (PERK) and ATF6 pathways, both major branches of the unfolded protein response, as well as inositol-requiring enzyme 1 α-XBP1 pathway, and Aβ42-induced activation of PERK may have a beneficial effect on AD by Aβ42 clearance [124]. Therefore, Drosophila studies suggest that the ER stress response pathways may be implicated in the pathogenesis of AD.
2.4. Modifiers Involved in ERK Pathway or Cell Cycle
2.4.1. EGFR/ERK Signaling
Activation of the extracellular signal-regulated kinase (ERK) pathway, as well as other mitogen-activated protein kinases (MAPKs) that include c-jun N-terminal kinase (JNK) and p38 MAPK, has been observed in AD neurons and animal models [125,126,127,128]. The ERK pathway has been reported to play a crucial role in dead signaling in neurons [129,130,131], although it is commonly thought to be a survival signal [132,133]. In Drosophila, Aβ42 activates ERK, and pharmacological inhibition of ERK activity reduces neurotoxicity of Aβ42, suggesting that chronic activation of ERK is a crucial step in the progression of AD [131]. Furthermore, extracts of Chinese medical herbs, such as C. sativum and N. jatamansi that inhibit ERK activation, ameliorate AD phenotypes in cultured mammalian cells and flies [134,135]. Epidermal growth factor receptor (EGFR) signaling within a particular range is necessary to maintain homeostasis of mushroom bodies, which is required for neuronal plasticity, learning, and memory [136]. However, excessive EGFR aggravates short-term memory loss of Aβ42-expressing flies, while treatment with gefitinib or erlotinib, two EGFR inhibitors, suppresses Aβ42-induced memory loss [137].
2.4.2. Cell Cycle
Erroneous cell cycle re-entry (CCR) in neurons has been considered to be a crucial causative factor in neuronal death [138]. In AD brains, vulnerable neurons show activated cell cycle phenotypes, such as abnormally elevated cell cycle markers and re-expression of cell cycle regulators. Yet they are incapable of completing the cell cycle, resulting to the aberrant neuronal death [139,140]. The Aβ42 oligomer induces CCR in cultured primary neurons, and neuronal CCR takes place before accumulation of Aβ42 in APP-expressing rat brains [141,142]. Consistently, in the Drosophila brain, Aβ42 increases expression of Cyclin B, an important cell cycle protein, while genetic reduction of Cyclin B extends the lifespan and improves locomotor dysfunction of AD flies [143]. APP also induces erroneous CCR, and knockdown of polo, another key regulator of the cell cycle, partially rescues APP-induced locomotor dysfunction and retinal degeneration and prevents a shortened lifespan by repressing APP-induced CCR [144].
It is also known that Notch activation, which is essential for neuronal specification and development, is implicated in erroneous CCR and AD [145,146,147]. In the rodent model, kainic acid-induced activation of Notch results in neurodegeneration through erroneous CCR [146]. Moreover, genetic reduction of Delta, a ligand of Notch, and N-[N-(3,5-Difluorophen-acetyl)-Lalanyl]-S-phenylglycine t-butyl ester, a Notch inhibitor, rescued learning impairments and prevented the premature death of Aβ42-expressing flies [147].
2.5. Modifiers Related to Apoptosis
Apoptosis is a major pathway of neurodegenerative cell death in AD, and several apoptosis-related factors have been identified as genetic modifiers of AD pathology [23,148]. In Drosophila, apoptotic cell death induced by ectopic expression of Aβ42 was detected by several methods, including active caspase 3 antibody staining, TUNEL assay, and acridine orange staining in the fly brain [53,149]. Moreover, several studies have shown that anti-apoptotic proteins have protective effects on AD-like phenotypes in Aβ42- or tau-expressing flies [24,38,150]. For example, co-overexpression of baculovirus p35, a caspase inhibitor, partially inhibits Aβ42-induced cell death in fly photoreceptors [38], implying that Aβ42-mediated cell death occurs via both p35-sensitive caspase-dependent and -independent pathways. In contrast, another study showed that death-associated inhibitor of apoptosis 1 (DIAP1), a Drosophila homolog of the inhibitor of apoptosis proteins, almost completely suppressed the AD-like phenotype, including Aβ42-induced cell death in the fly brain [150]. Given that Dronc, an initiator of caspase, can be inhibited by DIAP1 but not by p35 [151,152,153], the difference in the degree of protective effects between p35 and DIAP1 suggests the role of Dronc in Aβ42-induced cell death. In addition, deficiency of the translocator protein 18 kDa (TSPO), an outer mitochondrial membrane protein that plays an important role in the regulation of apoptosis, rescues the reduced lifespan of Aβ42-expressing flies by decreasing apoptosis and caspase 3 and 7 activities [154].
JNK and the stress-activated protein kinase subfamily have been also implicated in AD pathology [155,156,157,158]. JNK signaling is up-regulated by Aβ42 and also contributes to Aβ42-induced cell death in Drosophila [38,150]. Furthermore, inhibition of JNK through genetic modification or pharmacological treatment alleviates Aβ42-induced neuronal cell death, reduced survival rate, and locomotor dysfunction in flies [150]. Additionally, night-time sleep loss of Aβ42-expressing flies is restored by JNK inhibition [159].
The detrimental effect of JNK in AD model flies has been also revealed through the modulation of JNK upstream and downstream factors [24,150,160,161]. Hemipterous (hep), a Drosophila homolog of the JNK kinase, enhances Tau-induced toxicity in fly eyes [24], while hep deficiency reduces neuronal cell death of Aβ42-expressing larvae [160]. Additionally, a deficiency of Drosophila forkhead box subgroup O (dFOXO), a downstream factor of JNK, suppressed Aβ42-induced neuronal cell death, the reduced survival rate, and locomotor dysfunction of Aβ42-expressing flies [150]. Expression of human amyloid precursor like protein-1 (APLP1), a component of the amyloid precursor protein family, in flies increases transcription of dFOXO target pro-apoptotic genes, hid and reaper, and APLP1-induced cell death is rescued by genetic reduction of dFOXO [161].
A recent study has highlighted another aspect of neuronal cell death in the brain of AD model flies [162]. Apoptosis promotes the elimination of impaired neurons from brain circuits, protecting the brain instead of attacking it. In the same study, Coelho and colleagues demonstrated that suppression of fitness-based removal of Aβ42-damaged neurons by knockdown of azot, the fitness checkpoint gene, and overexpression of DIAP1 exacerbated AD-like phenotypes of Aβ42-expressing flies, including degenerative vacuoles, a decreased lifespan, a locomotory defect, and a memory defect. This new finding shows that the role of neuronal cell death in the AD brain is more complex than previously thought.
2.6. Modifiers Related to Epigenetic Regulation
Based on its important function in neurons, epigenetic mechanisms have been suggested to play a pivotal role in AD pathophysiology [163]. Histone acetylation, which is regulated by the activities of histone deacetylases (HDAC) and histone acetyltransferases (HAT), has been primarily implicated in AD-like phenotypes in Drosophila. HDAC6 is a unique member of the HDAC family that acts mainly on cytoplasmic non-histone substrates [164] and increased in a postmortem study of human AD brain [165]. A study of mammals showed that Aβ-induced mitochondrial transport was rescued by inhibition of HDAC6 [166]. In Drosophila, microtubule defects in human tau-expressing flies were rescued by HDAC6 inhibition [167]. However, another study indicated that histone deacetylase inhibitor trichostatin A caused lethality and delayed development in Drosophila [168], as well as inducing neuronal death in mice [169].
γ-cleavage of APP releases an intracellular tail that forms a complex with Tip60, a member of the HAT family, which affects the pathophysiology of AD [170]. In APP-overexpressing transgenic mice, levels of Tip60 are increased [171]. In contrast, in Drosophila, Tip60 levels are decreased, while HDAC1 levels are increased, in the larval brain of APP-overexpressing flies, which resulted in epigenetic repression of neuroplasticity genes [172]. Moreover, specific loss of Tip60 activity enhanced APP-mediated lethality and neuronal apoptotic cell death in Drosophila, while overexpression of Tip60 diminished these defects and improved the learning and memory performance of APP-expressing larvae [172,173]. Another HAT family protein, the CREB-binding protein (CBP), has also been reported to play a neuroprotective role in the Drosophila AD model. The expression of CBP in Drosophila expressing Aβ42 in the retina rescued eye phenotype, apoptosis, neurodegeneration, and axonal targeting defects. This neuroprotective effect of CBP was found to be essential for the bromo, HAT, and poly glutamine stretch (BHQ) domain of CBP [174].
2.7. Modifiers Related With Synaptic Abnormalities
Failure of normal synaptic function might be one of the earliest measurable deficits in AD [175], and the decreases in synaptic density appear to occur early in the course of AD in mouse models and patients [176,177,178,179].
Aβ peptides have been suggested to have physiological roles in synaptic function [180], and Aβ oligomers specifically target molecular components that mediate synaptic plasticity [181]. In several studies, mutant APP transgenic mice showed synaptic dysfunction before plaque formation, suggesting that soluble Aβ levels in the cortex significantly correlate with the degree of synaptic loss [178,182,183,184]. Studies in Drosophila have shown that early memory defects and structural and/or functional synaptic defects are induced by expression of Aβ, and that Aβ peptides inhibit the formation and/or maturation of new synapses [37,63,185,186]. Thus, antibodies to neutralize soluble Aβ oligomers are suggested as treatment for early AD [187]. Consistent with these findings, various factors that act on normal synaptic function have been found to be AD-related factors, as follows.
2.7.1. Small GTPases
Small GTPases, such as Rho and Rac1, play a prominent role in the development of dendritic structure [188]. For example, the constitutive active (CA) form of RhoA reduces dendritic arbor growth in Drosophila [189], and CA Rac1 tends to form ruffle-like structure in rodent neurons [190].
Several studies implicated these small GTPases in AD pathology. It has been shown that RhoA expression was decreased in synapses and increased in dystrophic neurites in a mouse AD model, suggesting that RhoA might be associated with AD pathology [191]. In Drosophila, increased activation of Rho1, the Drosophila orthologue of vertebrate RhoA, by prenylation, a posttranslational modification facilitating the association of proteins with membranes, leads to age-dependent degeneration of the nervous system [192]. A recent study demonstrated that Rac1 activity was abnormally increased in the hippocampal tissues of AD patients and mouse AD models, and that inhibition of the elevated Rac1 activity rescued memory loss in both fly and mouse AD models [193].
2.7.2. Impaired Axonal Transport
Impaired transportation in neurons is regarded as an underlying cause of synaptic failure in AD [194], and overexpression of APP leads to axonal transport defects in fly and mouse models [195,196,197]. In the peripheral nerves of Drosophila larvae expressing APP, a “traffic jam” of vesicles was observed, and expression of scaffolding proteins, Fe65 and JIP1b, which interact with the APP intracellular domain, also induced the axonal transport defect [198].
2.7.3. Synaptic Proteins
It has been shown that the expression levels of genes involved in synaptic vesicle trafficking are decreased in the brain of AD patients [199]. In flies, expression of Bruchpilot, a homolog of ELKS/RAB6-interacting/CAST family member 2 (ERC2) that can affect the localization of Ca+2 channels in presynaptic release sites [200], was not significantly different in APP-and BACE-expressing larvae compared to the control group [201] and five-day-old Aβ-expressing flies, but showed reduced levels at 21 days of age [202]. Additionally, the levels of Discs-large, a homolog of DLG4 essential for the density of the synaptic glutamate receptor [203,204], were significantly reduced in fly larvae expressing APP and BACE [201]. A recent study proved the importance of the synaptic localization of a subset of synaptic proteins, including APP [205]. In this study, Furotani and colleagues demonstrated that knockdown of yata, a novel gene regulating the synaptic localization of β amyloid protein precursor-like and other synaptic proteins, rescued the phenotypes of APP-expressing flies, suggesting that the regulators of the synaptic localization of synaptic proteins are involved in AD pathology.
2.8. Mitochondrial Dysfunction or Mislocation
Mitochondrial dysfunction has been reported to contribute to the progression of AD pathology [206]. For example, Dynamin-related protein 1 (Drp1), a key regulator of mitochondrial fission, exerts a beneficial effect in Aβ42-expressing flies by protecting mitochondria [207]. In contrast, sra overexpression reduced the number of mitochondria and enhanced Aβ42 toxicity [111]. Also, the mislocation of mitochondria has been implicated in AD pathology. Mitochondria are dynamic organelles whose active movement is essential for the mobilization of the synaptic vesicle reserve pool [208]. In the brains of AD patients and the AD model mouse, disruption of mitochondrial transport resulted in the reduced distribution of mitochondria in the axon and dendrites [196]. Furthermore, mislocalization of mitochondria was observed in the brain of flies expressing Aβ42 [209]. Therefore, restoration of mitochondrial transportation could be a promising target mechanism that can be used to develop AD therapies.
2.9. Modifiers Related with Inflammation
The immune system has been known to be aberrantly activated in the brains of AD patients and contributes to AD pathogenesis [210,211]. As the immune response not only provides a protective role by promoting the clearance of toxic Aβ aggregates but also exerts a detrimental effect on AD pathology by up-regulating chronic inflammation, it has been regarded as a double-edged sword in neurodegenerative disease [212]. In particular, innate immunity and neuroinflammation are thought to be essential components of neurodegeneration in both fly and mammalian brains [213,214,215].
2.9.1. Phagocytic Receptor Draper
After Aβ is produced in the CNS, binding of soluble Aβ oligomer and fibrils to the microglial receptor produces a signal that results in the production of cytokines and chemokines [216]. In Drosophila, glial cells show functional and morphological similarity to those of mammals [217] and contribute to the protection of neurons, engulfing Aβ fibrils by phagocytosis in the fly AD model [218]. Glial phagocytosis may decrease with aging due to decreased levels of Draper, a Drosophila homolog of the mammalian Multiple EGF Like Domains 10, which is a conserved phagocytic receptor of glial cells [219], and up-regulation of Draper which can reverse the Aβ-related phenotype [218], suggesting that the phagocytic function of glial cells may exert a beneficial effect in neurodegenerative disease.
2.9.2. TREM2
Triggering Receptor Expressed on Myeloid cell 2 (TREM2) is another receptor that mediates phagocytosis on the microglial surface [220] and regulates inflammatory responses via Toll-like receptors (TLRs) in AD [221,222]. TREM2 suppresses inflammation through the inhibition of cytokine production [223] and has a protective effect in AD by reducing inflammation-induced neuronal damage [224]. TREM2 mutation correlates with a significantly increased risk of AD [225], and TREM2 deficiency promotes Aβ accumulation due to a dysfunctional microglial response [226]. There is no apparent Drosophila homolog of TREM2. However, a recent study using Drosophila AD models shows that glial expression of human TREM2 or human tyrosine kinase binding protein (TYROBP), the intracellular adaptor of TREM2, did not affect AD-like phenotypes of Aβ42-expressing flies, while glial expression of TREM2/TYROBP modifies molecular signatures induced by neuronal expression of Aβ42 [227]. Unlike Aβ42, TREM2/TYROBP expression in glial cells exacerbated tau-mediated AD pathology [227]. Therefore, further detailed research using various AD models, on the functions of TREM2 in AD pathology is needed.
2.9.3. Toll and IMD Pathways
Toll and immune deficiency (IMD) pathways are the major innate immune responsive pathways in Drosophila that regulate the production of antimicrobial peptides [228]. The Toll pathway that is mainly responsive to fungi, Gram-positive bacteria, and virulence factors, functions through the NF-kB family transcription factors, Dorsal and Dif, while the IMD pathway, responsive to Gram-negative bacteria, acts through another NF-kB family transcription factor, Relish.
In mammals, several TLRs are involved in Aβ uptake by microglial cells and activate innate immune responses to prevent Aβ accumulation in the CNS [221,229,230]. In AD brains, high expression of TLR was detected [231], and TLR-deficient mice showed increased Aβ deposits [221], suggesting that TLR is involved in Aβ clearance. However, loss-of-function mutations of Drosophila Toll (Tl) suppresses the pathological effects of human Aβ42 in the Drosophila AD model [232]. Moreover, the same study showed that deficiencies in the downstream components of the Toll pathway, including an adaptor protein Tube, the IRAK-like kinase Pelle, and Dorsal and Dif, ameliorated the rough eye phenotype of human Aβ42-expressing flies [232]. The difference between the mammalian and fruit fly results probably reflects the dual aspect of the Toll pathway, i.e., clearance and inflammation.
The IMD pathway has also been implicated in the toxicity of Aβ42 in Drosophila. The expression of peptidogylcan recognition protein SC1b (PGRP-SC1b), a suppressor of the IMD pathway, is up-regulated in Aβ42-expressing flies, and knockdown of PGRP-SC1b ameliorated the reduced lifespan and locomotory defect of the fly AD model [112], suggesting that the IMD pathway is protective against Aβ42 toxicity. In contrast, a recent study showed that the downregulation of Relish, a downstream transcription factor of the IMD pathway, in Drosophila astrocytes ameliorated the toxicity of Aβ42 as well as polyglutamine [233]. These confusing findings highlight the complexity of the role of the immune response in pathogenesis of AD.
3. Conclusions and Perspectives
In this study, we have reviewed the AD-associated genes found in the Drosophila models and their cellular pathways (Table 1 and Figure 2). Because AD is a multifactorial disease, various AD-related genetic factors and their functions need to be identified before developing accurate tests and treatments for this disease. Therefore, genetic studies focusing on the discovery of various AD-related genes are considered essential for identifying the causes of AD and developing therapies. With the recent application of GWAS and next-generation sequencing, 29 LOAD-related factors have been discovered, and research into the functions of these factors will broaden our understanding of AD etiology [234,235]. However, despite intensive research over the past decades, our understanding of the AD-related genes discovered to date is limited. In fact, a study showed that known AD-risk variants can explain only about 30% of AD variance, indicating that many genetic loci remain to be discovered [236]. Therefore, new approaches may be needed to boost the probability of identifying causal genes and pathways.

Table 1.
Genetic modifiers of amyloid precursor protein (APP) or Aβ-based Drosophila Alzheimer’s disease (AD) models.
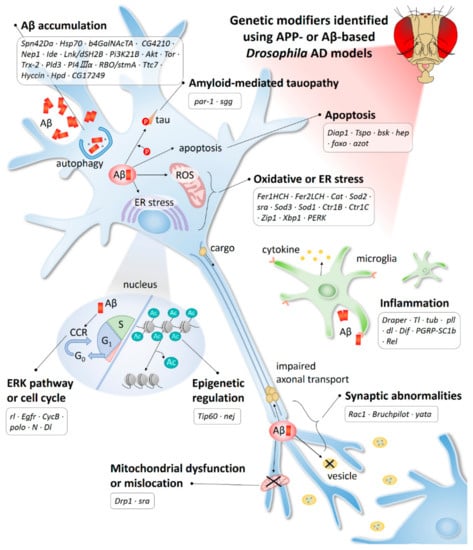
Figure 2.
AD-related mechanisms identified using the APP-or Aβ-based Drosophila model. Ac, acetylation; CCR, cell cycle re-entry; ER, endoplasmic reticulum; ROS, reactive oxygen species.
Functional genomic studies using Drosophila could be an alternative approach to finding new AD-related genes. The results of studies with Drosophila AD models described in this study are sufficient to show the feasibility of using Drosophila models to find new disease modifiers. While there are drawbacks—the Drosophila models are over simplified and have relatively low relevance to humans compared to mice—the advantages of using Drosophila models outweigh these drawbacks. Specifically, the Drosophila models have a wide variety of genetic tools, and it takes less than 10 days for the AD phenotype to appear, whereas it takes more than six months in the mouse AD model, although it varies from model to model [237], and allows for large-scale screening. In addition, the presence of RNAi for all genes annotated in its genome and simple crosses allow the screening of genetic modifiers for the toxicity of Aβ42 and tau. Therefore, more AD-related genes are expected to be discovered in the future thanks to Drosophila models. In particular, if there are limitations in the discovery of disease-related genes because of the small sample size in human GWAS studies, it will be possible to discover new genes by conducting a combined study of GWAS and Drosophila screening. Drosophila models will also be a good tool for in vivo functional studies of the genes discovered via human genetic studies. In the future, this combination of human genetics and Drosophila functional genomics is expected to be an important strategy for research into multifactorial diseases, such as AD.
In addition, Drosophila can also be used as a screening tool for drugs to modify AD symptoms or disease progression. Unfortunately, all recently developed AD treatment candidates have failed in the clinic and thus, the causative factors for the disease have come into question [238,239]. Despite such factors, AD progression, including accumulation of Aβ in the brain, begins long before the appearance of clinical symptoms such as memory loss, and most AD patients are diagnosed at a later stage of neurodegeneration. Recently, the FDA recognized this characteristic and issued an amendment to permit clinical trials very early in the disease [240]. As a result, it is anticipated that clinical trials of new drugs for these early diagnosis and early stage patients will increase rapidly. Therefore, finding more drug candidates will be another important task in this field.
Drosophila is a well-characterized insect with various phenotypes and is a model system that can be used to rapidly screen many drug candidates in vivo. Indeed, many studies to date have used the Drosophila AD model to screen for small molecules that can modify AD symptoms or validate the in vivo efficacy of developed drug candidates [29,241,242,243]. In addition, Drosophila has been used to screen natural products and traditional medicines that are expected to have therapeutic and prophylactic properties in AD [244,245]. In the future, the superiority of the Drosophila model as an in vivo drug screening system is expected to increase its utility.
Author Contributions
Conceptualization, K.S.C.; formal analysis, B.C. and S.-Y.W.; data curation, B.C. and Y.J.; writing—original draft preparation, K.S.C., Y.J. and J.H.L.; writing—review and editing, K.S.C., S.-Y.W., B.C., Y.J. and J.H.L.; visualization, B.C.; supervision, K.S.C. and S.-Y.W.; project administration, K.S.C. and S.-Y.W.; funding acquisition, K.S.C. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI18C0041).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | Amyloid β |
| APLP1 | Amyloid precursor like protein 1 |
| APP | Amyloid precursor protein |
| ATF6 | Activating transcription factor 6 |
| B4GALT6 | β1,4-galactosyltransferases |
| BHQ | Bromo, HAT and poly glutamine stretch |
| CA | Constitutive active |
| CAT | Catalase |
| CCR | Cell cycle reentry |
| CBP | CREB-binding protein |
| Ctr1B | Copper transporter 1B |
| dFOXO | Drosophila forkhead box subgroup O |
| DIAP1 | Death-associated inhibitor of apoptosis 1 |
| Drp1 | Dynamin-related protein 1 |
| EGFR | Epidermal growth factor receptor |
| EOAD | Early onset AD |
| ER | Endoplasmic reticulum |
| ERC2 | ELKS/RAB6-interacting/CAST family member 2 |
| ERK | Extracellular signal-regulated kinase |
| Fer1HCH | Ferritin 1 heavy chain homologue |
| Fer2LCH | Ferritin 2 light chain homologue |
| GSK3β | Glycogen synthase kinase 3β |
| GWAS | Genome-wide association studies |
| HAT | Histone acetyltransferases |
| HDAC | Histone deacetylases |
| hep | hemipterous |
| HPD | 4-hydroxyphenylpyruvate dioxygenase |
| Hsp70 | Heat shock protein 70 |
| IDE | Insulin degradation enzyme |
| IMD | Immune deficiency |
| JNK | c-jun N-terminal kinase |
| LOAD | Late onset AD |
| MARK | Microtubule affinity regulating kinase |
| MAPKs | Mitogen-activated protein kinases |
| NEP | Neprilysin |
| NFTs | Neurofibrillary tangles |
| PERK | Protein kinase R-like endoplasmic reticulum kinase |
| PGRP-SC1b | Peptidogylcan recognition protein SC1b |
| PLD3 | Phospholipase D3 |
| PRCC | Proline rich mitotic checkpoint control factor |
| psn | presenilin |
| RBO | Rolling blackout |
| SAT1 | α2,3-sialyltransferase |
| Ser | Serine |
| SH2B1 | Src homology 2B1 |
| SOD | Superoxide dismutase |
| sra | sarah |
| Thr | Threonine |
| TLRs | Toll-like receptors |
| TREM2 | Triggering receptor expressed on myeloid cell 2 |
| Trx80 | Thioredoxin-80 |
| TSPO | Translocator protein 18 kDa |
| TTC7 | Tetratricopeptide repeat domain 7 |
| XBP1 | X-box binding protein 1 |
| XBP1-S | spliced XBP1 |
| Zip1 | zinc/iron regulated transporter-related protein 1 |
References
- Alzheimer’s Disease International. Executive summary. In World Alzheimer Report 2019: Attitudes to Dementia; Alzheimer’s Disease International (ADI): London, UK, 2019; pp. 1–160. [Google Scholar]
- Cummings, J.L. Alzheimer’s disease. N. Engl. J. Med. 2004, 351, 56–67. [Google Scholar] [CrossRef]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Mudher, A.; Lovestone, S. Alzheimer’s disease–do tauists and baptists finally shake hands? Trends Neurosci. 2002, 25, 22–26. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, K.; Wisniewski, H.; Wen, G. Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann. Neurol. 1985, 17, 278–282. [Google Scholar] [CrossRef]
- Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N–terminus of β–amyloid. Nat. Genet. 1992, 1, 345. [Google Scholar] [CrossRef]
- Hardy, J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997, 20, 154–159. [Google Scholar] [CrossRef]
- Ballatore, C.; Lee, V.M.-Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663. [Google Scholar] [CrossRef]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef]
- Bettens, K.; Sleegers, K.; Van Broeckhoven, C. Genetic insights in Alzheimer’s disease. Lancet Neurol. 2013, 12, 92–104. [Google Scholar] [CrossRef]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, D.; De Deyn, P.P. Animal models in the drug discovery pipeline for Alzheimer’s disease. Br. J. Pharmacol. 2011, 164, 1285–1300. [Google Scholar] [CrossRef]
- Tan, F.H.P.; Azzam, G. Drosophila melanogaster: Deciphering Alzheimer’s Disease. Malays. J. Med. Sci. 2017, 24, 6–20. [Google Scholar] [CrossRef]
- Yeates, C.J.; Sarkar, A.; Kango-Singh, M.; Singh, A. Unraveling Alzheimer’s Disease Using Drosophila. In Insights into Human Neurodegeneration: Lessons Learnt from Drosophila; Mutsuddi, M., Mukherjee, A., Eds.; Springer: Singapore, 2019; pp. 251–277. [Google Scholar]
- Guo, Y.; Livne-Bar, I.; Zhou, L.; Boulianne, G.L. Drosophila presenilin is required for neuronal differentiation and affects notch subcellular localization and signaling. J. Neurosci. 1999, 19, 8435–8442. [Google Scholar] [CrossRef]
- Ye, Y.; Fortini, M.E. Apoptotic activities of wild-type and Alzheimer’s disease-related mutant presenilins in Drosophila melanogaster. J. Cell Biol. 1999, 146, 1351–1364. [Google Scholar] [CrossRef]
- Michno, K.; Knight, D.; Campusano, J.M.; van de Hoef, D.; Boulianne, G.L. Intracellular calcium deficits in Drosophila cholinergic neurons expressing wild type or FAD-mutant presenilin. PLoS ONE 2009, 4, e6904. [Google Scholar] [CrossRef]
- Knight, D.; Iliadi, K.; Charlton, M.P.; Atwood, H.L.; Boulianne, G.L. Presynaptic plasticity and associative learning are impaired in a Drosophila presenilin null mutant. Dev. Neurobiol. 2007, 67, 1598–1613. [Google Scholar] [CrossRef]
- Wittmann, C.W.; Wszolek, M.F.; Shulman, J.M.; Salvaterra, P.M.; Lewis, J.; Hutton, M.; Feany, M.B. Tauopathy in Drosophila: Neurodegeneration without neurofibrillary tangles. Science 2001, 293, 711–714. [Google Scholar] [CrossRef]
- Jackson, G.R.; Wiedau-Pazos, M.; Sang, T.K.; Wagle, N.; Brown, C.A.; Massachi, S.; Geschwind, D.H. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 2002, 34, 509–519. [Google Scholar] [CrossRef]
- Shulman, J.M.; Feany, M.B. Genetic modifiers of tauopathy in Drosophila. Genetics 2003, 165, 1233–1242. [Google Scholar] [PubMed]
- Folwell, J.; Cowan, C.M.; Ubhi, K.K.; Shiabh, H.; Newman, T.A.; Shepherd, D.; Mudher, A. Aβ exacerbates the neuronal dysfunction caused by human tau expression in a Drosophila model of Alzheimer’s disease. Exp. Neurol. 2010, 223, 401–409. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Greeve, I.; Kretzschmar, D.; Tschape, J.A.; Beyn, A.; Brellinger, C.; Schweizer, M.; Nitsch, R.M.; Reifegerste, R. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J. Neurosci. 2004, 24, 3899–3906. [Google Scholar] [CrossRef]
- Carmine-Simmen, K.; Proctor, T.; Tschape, J.; Poeck, B.; Triphan, T.; Strauss, R.; Kretzschmar, D. Neurotoxic effects induced by the Drosophila amyloid-beta peptide suggest a conserved toxic function. Neurobiol. Dis. 2009, 33, 274–281. [Google Scholar] [CrossRef]
- Chakraborty, R.; Vepuri, V.; Mhatre, S.D.; Paddock, B.E.; Miller, S.; Michelson, S.J.; Delvadia, R.; Desai, A.; Vinokur, M.; Melicharek, D.J. Characterization of a Drosophila Alzheimer’s disease model: Pharmacological rescue of cognitive defects. PLoS ONE 2011, 6, e20799. [Google Scholar] [CrossRef]
- Stokin, G.B.; Almenar-Queralt, A.; Gunawardena, S.; Rodrigues, E.M.; Falzone, T.; Kim, J.; Lillo, C.; Mount, S.L.; Roberts, E.A.; McGowan, E.; et al. Amyloid precursor protein-induced axonopathies are independent of amyloid-beta peptides. Hum. Mol. Genet. 2008, 17, 3474–3486. [Google Scholar] [CrossRef]
- Muller, T.; Meyer, H.E.; Egensperger, R.; Marcus, K. The amyloid precursor protein intracellular domain (AICD) as modulator of gene expression, apoptosis, and cytoskeletal dynamics-relevance for Alzheimer’s disease. Prog. Neurobiol. 2008, 85, 393–406. [Google Scholar] [CrossRef]
- Finelli, A.; Kelkar, A.; Song, H.J.; Yang, H.; Konsolaki, M. A model for studying Alzheimer’s Abeta42-induced toxicity in Drosophila melanogaster. Mol. Cell. Neurosci. 2004, 26, 365–375. [Google Scholar] [CrossRef]
- Crowther, D.C.; Kinghorn, K.J.; Miranda, E.; Page, R.; Curry, J.A.; Duthie, F.A.; Gubb, D.C.; Lomas, D.A. Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience 2005, 132, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Iijima, K.; Iijima-Ando, K. Drosophila models of Alzheimer’s amyloidosis: The challenge of dissecting the complex mechanisms of toxicity of amyloid-β 42. J. Alzheimers Dis. 2008, 15, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Casas-Tinto, S.; Zhang, Y.; Sanchez-Garcia, J.; Gomez-Velazquez, M.; Rincon-Limas, D.E.; Fernandez-Funez, P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum. Mol. Genet. 2011, 20, 2144–2160. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.; Lee, S.; Shin, M.; Lee, J.H.; Suh, Y.S.; Hwang, S.; Yun, H.S.; Cho, K.S. Phenotypic differences between Drosophila Alzheimer’s disease models expressing human Aβ42 in the developing eye and brain. Anim. Cells Syst. 2017, 21, 160–168. [Google Scholar] [CrossRef]
- Iijima, K.; Chiang, H.C.; Hearn, S.A.; Hakker, I.; Gatt, A.; Shenton, C.; Granger, L.; Leung, A.; Iijima-Ando, K.; Zhong, Y. Abeta42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLoS ONE 2008, 3, e1703. [Google Scholar] [CrossRef]
- Tare, M.; Modi, R.M.; Nainaparampil, J.J.; Puli, O.R.; Bedi, S.; Fernandez-Funez, P.; Kango-Singh, M.; Singh, A. Activation of JNK signaling mediates amyloid-ss-dependent cell death. PLoS ONE 2011, 6, e24361. [Google Scholar] [CrossRef]
- Moran, M.T.; Tare, M.; Kango-Singh, M.; Singh, A. Homeotic Gene teashirt (tsh) has a neuroprotective function in amyloid-beta 42 mediated neurodegeneration. PLoS ONE 2013, 8, e80829. [Google Scholar] [CrossRef]
- Steffensmeier, A.M.; Tare, M.; Puli, O.R.; Modi, R.; Nainaparampil, J.; Kango-Singh, M.; Singh, A. Novel neuroprotective function of apical-basal polarity gene crumbs in amyloid beta 42 (aβ42) mediated neurodegeneration. PLoS ONE 2013, 8, e78717. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247. [Google Scholar] [CrossRef]
- Eisele, Y.S. From Soluble A β to Progressive A β Aggregation: Could prion-like templated misfolding play a role? Brain Pathol. 2013, 23, 333–341. [Google Scholar] [CrossRef]
- Sowade, R.F.; Jahn, T.R. Seed-induced acceleration of amyloid-β mediated neurotoxicity in vivo. Nat. Commun. 2017, 8, 512. [Google Scholar] [CrossRef]
- Pitschke, M.; Prior, R.; Haupt, M.; Riesner, D. Detection of single amyloid β-protein aggregates in the cerebrospinal fluid of Alzheimer’s patients by fluorescence correlation spectroscopy. Nat. Med. 1998, 4, 832. [Google Scholar] [CrossRef]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Vbeyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- Näslund, J.; Haroutunian, V.; Mohs, R.; Davis, K.L.; Davies, P.; Greengard, P.; Buxbaum, J.D. Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA 2000, 283, 1571–1577. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Kumar, A.; Srivastava, S.; Tripathi, S.; Singh, S.K.; Srikrishna, S.; Sharma, A. Molecular insight into amyloid oligomer destabilizing mechanism of flavonoid derivative 2-(4‘-benzyloxyphenyl)-3-hydroxy-chromen-4-one through docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2016, 34, 1252–1263. [Google Scholar] [CrossRef]
- Nerelius, C.; Sandegren, A.; Sargsyan, H.; Raunak, R.; Leijonmarck, H.; Chatterjee, U.; Fisahn, A.; Imarisio, S.; Lomas, D.; Crowther, D. α-Helix targeting reduces amyloid-β peptide toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 9191–9196. [Google Scholar] [CrossRef]
- Kinghorn, K.J.; Crowther, D.C.; Sharp, L.K.; Nerelius, C.; Davis, R.L.; Chang, H.T.; Green, C.; Gubb, D.C.; Johansson, J.; Lomas, D.A. Neuroserpin binds Aβ and is a neuroprotective component of amyloid plaques in Alzheimer disease. J. Biol. Chem. 2006, 281, 29268–29277. [Google Scholar] [CrossRef]
- Martín-Peña, A.; Rincón-Limas, D.E.; Fernandez-Fúnez, P. Engineered Hsp70 chaperones prevent Aβ42-induced memory impairments in a Drosophila model of Alzheimer’s disease. Sci. Rep. 2018, 8, 9915. [Google Scholar] [CrossRef]
- Fernandez-Funez, P.; Sanchez-Garcia, J.; de Mena, L.; Zhang, Y.; Levites, Y.; Khare, S.; Golde, T.E.; Rincon-Limas, D.E. Holdase activity of secreted Hsp70 masks amyloid-beta42 neurotoxicity in Drosophila. Proc. Natl. Acad. Sci. USA 2016, 113, E5212–E5221. [Google Scholar] [CrossRef]
- Younan, N.D.; Chen, K.-F.; Rose, R.-S.; Crowther, D.C.; Viles, J.H. Prion protein stabilizes amyloid-β (Aβ) oligomers and enhances Aβ neurotoxicity in a Drosophila model of Alzheimer’s disease. J. Biol. Chem. 2018, 293, 13090–13099. [Google Scholar] [CrossRef]
- Yamasaki, Y.; Tsuda, L.; Suzuki, A.; Yanagisawa, K. Induction of ganglioside synthesis in Drosophila brain accelerates assembly of amyloid β protein. Sci. Rep. 2018, 8, 8345. [Google Scholar] [CrossRef]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.-J.; Hama, E.; Sekine-Aizawa, Y. Identification of the major Aβ 1–42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143. [Google Scholar] [CrossRef]
- Iijima-Ando, K.; Hearn, S.A.; Granger, L.; Shenton, C.; Gatt, A.; Chiang, H.-C.; Hakker, I.; Zhong, Y.; Iijima, K. Overexpression of neprilysin reduces alzheimer amyloid-β42 (Aβ42)-induced neuron loss and intraneuronal Aβ42 deposits but causes a reduction in cAMP-responsive element-binding protein-mediated transcription, age-dependent axon pathology, and premature death in Drosophila. J. Biol. Chem. 2008, 283, 19066–19076. [Google Scholar]
- Qiu, W.Q.; Walsh, D.M.; Ye, Z.; Vekrellis, K.; Zhang, J.; Podlisny, M.B.; Rosner, M.R.; Safavi, A.; Hersh, L.B.; Selkoe, D.J. Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J. Biol. Chem. 1998, 273, 32730–32738. [Google Scholar] [CrossRef]
- Tsuda, M.; Kobayashi, T.; Matsuo, T.; Aigaki, T. Insulin-degrading enzyme antagonizes insulin-dependent tissue growth and Aβ-induced neurotoxicity in Drosophila. FEBS Lett. 2010, 584, 2916–2920. [Google Scholar] [CrossRef]
- Shen, Y.; Xia, Y.; Meng, S.; Lim, N.K.; Wang, W.; Huang, F. SH2B1 is involved in the accumulation of amyloid-β 42 in Alzheimer’s disease. J. Alzheimers Dis. 2017, 55, 835–847. [Google Scholar] [CrossRef]
- O’Neill, C. PI3-kinase/Akt/mTOR signaling: Impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp. Gerontol. 2013, 48, 647–653. [Google Scholar] [CrossRef]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef]
- Chiang, H.-C.; Wang, L.; Xie, Z.; Yau, A.; Zhong, Y. PI3 kinase signaling is involved in Aβ-induced memory loss in Drosophila. Proc. Natl. Acad. Sci. USA 2010, 107, 7060–7065. [Google Scholar] [CrossRef]
- Gerenu, G.; Persson, T.; Goikolea, J.; Calvo-Garrido, J.; Loera-Valencia, R.; Pottmeier, P.; Santiago, C.; Poska, H.; Presto, J.; Cedazo-Minguez, A. Thioredoxin-80 protects against amyloid-beta pathology through autophagic-lysosomal pathway regulation. Mol. Psychiatry 2019, 1–14. [Google Scholar] [CrossRef]
- Demirev, A.V.; Song, H.-L.; Cho, M.-H.; Cho, K.; Peak, J.-J.; Yoo, H.J.; Kim, D.-H.; Yoon, S.-Y. V232M substitution restricts a distinct O-glycosylation of PLD3 and its neuroprotective function. Neurobiol. Dis. 2019, 129, 182–194. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499. [Google Scholar] [CrossRef]
- Gouras, G.K.; Tampellini, D.; Takahashi, R.H.; Capetillo-Zarate, E. Intraneuronal β-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol. 2010, 119, 523–541. [Google Scholar] [CrossRef]
- Hong, S.; Ostaszewski, B.L.; Yang, T.; O’Malley, T.T.; Jin, M.; Yanagisawa, K.; Li, S.; Bartels, T.; Selkoe, D.J. Soluble Aβ oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 2014, 82, 308–319. [Google Scholar] [CrossRef]
- McLaurin, J.; Chakrabartty, A. Characterization of the interactions of Alzheimer β-amyloid peptides with phospholipid membranes. Eur. J. Biochem. 1997, 245, 355–363. [Google Scholar] [CrossRef]
- McIntire, L.B.J.; Berman, D.E.; Myaeng, J.; Staniszewski, A.; Arancio, O.; Di Paolo, G.; Kim, T.-W. Reduction of synaptojanin 1 ameliorates synaptic and behavioral impairments in a mouse model of Alzheimer’s disease. J. Neurosci. 2012, 32, 15271–15276. [Google Scholar] [CrossRef]
- Zhu, L.; Zhong, M.; Zhao, J.; Rhee, H.; Caesar, I.; Knight, E.M.; Volpicelli-Daley, L.; Bustos, V.; Netzer, W.; Liu, L. Reduction of synaptojanin 1 accelerates Aβ clearance and attenuates cognitive deterioration in an Alzheimer mouse model. J. Biol. Chem. 2013, 288, 32050–32063. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.-A.; Jiang, L.-X.; Liu, H.-Y.; Zhang, B.-Z.; Lim, N.; Li, Q.-Y.; Huang, F.-D. Downregulation of RBO-PI4KIIIα facilitates Aβ42 secretion and ameliorates neural deficits in Aβ42-expressing Drosophila. J. Neurosci. 2017, 37, 4928–4941. [Google Scholar] [CrossRef]
- Sun, M.; Zhao, Y.; Han, M.; Zhang, B.; Zhang, X.; Zhang, Q.; Lim, N.K.-H.; Wang, W.-A.; Huang, F.-D. TTC7 and Hyccin regulate neuronal Aβ 42 accumulation and its associated neural deficits in Aβ 42-expressing Drosophila. J. Alzheimers Dis. 2018, 65, 1–10. [Google Scholar] [CrossRef]
- Belfiori-Carrasco, L.F.; Marcora, M.S.; Bocai, N.I.; Ceriani, M.F.; Morelli, L.; Castaño, E.M. A novel genetic screen identifies modifiers of age-dependent amyloid β toxicity in the Drosophila brain. Front. Aging Neurosci. 2017, 9, 61. [Google Scholar] [CrossRef][Green Version]
- Heidary, G.; Fortini, M.E. Identification and characterization of the Drosophila tau homolog. Mech. Dev. 2001, 108, 171–178. [Google Scholar] [CrossRef]
- Fulga, T.A.; Elson-Schwab, I.; Khurana, V.; Steinhilb, M.L.; Spires, T.L.; Hyman, B.T.; Feany, M.B. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat. Cell Biol. 2007, 9, 139. [Google Scholar] [CrossRef]
- Iijima, K.; Gatt, A.; Iijima-Ando, K. Tau Ser262 phosphorylation is critical for Aβ42-induced tau toxicity in a transgenic Drosophila model of Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, 2947–2957. [Google Scholar] [CrossRef]
- Ando, K.; Maruko-Otake, A.; Ohtake, Y.; Hayashishita, M.; Sekiya, M.; Iijima, K.M. Stabilization of microtubule-unbound tau via tau phosphorylation at Ser262/356 by Par-1/MARK contributes to augmentation of AD-related phosphorylation and Aβ42-induced tau toxicity. PLoS Genet. 2016, 12, e1005917. [Google Scholar] [CrossRef]
- Götz, J.; Chen, F.; Van Dorpe, J.; Nitsch, R. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef]
- Lewis, J.; Dickson, D.W.; Lin, W.-L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.-H.; Sahara, N.; Skipper, L.; Yager, D. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef]
- Pennanen, L.; Götz, J. Different tau epitopes define Aβ 42-mediated tau insolubility. Biochem. Biophys. Res. Commun. 2005, 337, 1097–1101. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Cheng, D.; Jouleh, B.; Torp, R.; LaFerla, F.M. Genetically augmenting tau levels does not modulate the onset or progression of Aβ pathology in transgenic mice. J. Neurochem. 2007, 102, 1053–1063. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef]
- Hu, S.; Begum, A.N.; Jones, M.R.; Oh, M.S.; Beech, W.K.; Beech, B.H.; Yang, F.; Chen, P.; Ubeda, O.J.; Kim, P.C.; et al. GSK3 inhibitors show benefits in an Alzheimer’s disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol. Dis. 2009, 33, 193–206. [Google Scholar] [CrossRef]
- Takashima, A.; Murayama, M.; Murayama, O.; Kohno, T.; Honda, T.; Yasutake, K.; Nihonmatsu, N.; Mercken, M.; Yamaguchi, H.; Sugihara, S.; et al. Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc. Natl. Acad. Sci. USA 1998, 95, 9637–9641. [Google Scholar] [CrossRef]
- Song, M.S.; Rauw, G.; Baker, G.B.; Kar, S. Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur. J. Neurosci. 2008, 28, 1989–2002. [Google Scholar] [CrossRef]
- Yang, T.; Knowles, J.K.; Lu, Q.; Zhang, H.; Arancio, O.; Moore, L.A.; Chang, T.; Wang, Q.; Andreasson, K.; Rajadas, J.; et al. Small molecule, non-peptide p75 ligands inhibit Abeta-induced neurodegeneration and synaptic impairment. PLoS ONE 2008, 3, e3604. [Google Scholar] [CrossRef]
- Noh, M.Y.; Koh, S.H.; Kim, Y.; Kim, H.Y.; Cho, G.W.; Kim, S.H. Neuroprotective effects of donepezil through inhibition of GSK-3 activity in amyloid-beta-induced neuronal cell death. J. Neurochem. 2009, 108, 1116–1125. [Google Scholar] [CrossRef]
- Drewes, G.; Trinczek, B.; Illenberger, S.; Biernat, J.; Schmitt-Ulms, G.; Meyer, H.E.; Mandelkow, E.M.; Mandelkow, E. Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark). A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J. Biol. Chem. 1995, 270, 7679–7688. [Google Scholar] [CrossRef]
- Drewes, G.; Ebneth, A.; Preuss, U.; Mandelkow, E.M.; Mandelkow, E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 1997, 89, 297–308. [Google Scholar] [CrossRef]
- Hasegawa, M.; Morishima-Kawashima, M.; Takio, K.; Suzuki, M.; Titani, K.; Ihara, Y. Protein sequence and mass spectrometric analyses of tau in the Alzheimer’s disease brain. J. Biol. Chem. 1992, 267, 17047–17054. [Google Scholar]
- Hanger, D.P.; Brion, J.P.; Gallo, J.M.; Cairns, N.J.; Luthert, P.J.; Anderton, B.H. Tau in Alzheimer’s disease and Down’s syndrome is insoluble and abnormally phosphorylated. Biochem. J. 1991, 275 Pt 1, 99–104. [Google Scholar] [CrossRef]
- Hanger, D.P.; Betts, J.C.; Loviny, T.L.; Blackstock, W.P.; Anderton, B.H. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer’s disease brain using nanoelectrospray mass spectrometry. J. Neurochem. 1998, 71, 2465–2476. [Google Scholar] [CrossRef]
- Morishima-Kawashima, M.; Hasegawa, M.; Takio, K.; Suzuki, M.; Yoshida, H.; Titani, K.; Ihara, Y. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J. Biol. Chem. 1995, 270, 823–829. [Google Scholar] [CrossRef]
- Sperber, B.R.; Leight, S.; Goedert, M.; Lee, V.M. Glycogen synthase kinase-3 beta phosphorylates tau protein at multiple sites in intact cells. Neurosci. Lett. 1995, 197, 149–153. [Google Scholar] [CrossRef]
- Singh, T.J.; Haque, N.; Grundke-Iqbal, I.; Iqbal, K. Rapid Alzheimer-like phosphorylation of tau by the synergistic actions of non-proline-dependent protein kinases and GSK-3. FEBS Lett. 1995, 358, 267–272. [Google Scholar] [CrossRef]
- Singh, T.J.; Wang, J.Z.; Novak, M.; Kontzekova, E.; Grundke-Iqbal, I.; Iqbal, K. Calcium/calmodulin-dependent protein kinase II phosphorylates tau at Ser-262 but only partially inhibits its binding to microtubules. FEBS Lett. 1996, 387, 145–148. [Google Scholar] [CrossRef]
- Sengupta, A.; Kabat, J.; Novak, M.; Wu, Q.; Grundke-Iqbal, I.; Iqbal, K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch. Biochem. Biophys. 1998, 357, 299–309. [Google Scholar] [CrossRef]
- Nishimura, I.; Yang, Y.; Lu, B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell 2004, 116, 671–682. [Google Scholar] [CrossRef]
- Kosmidis, S.; Grammenoudi, S.; Papanikolopoulou, K.; Skoulakis, E.M. Differential effects of Tau on the integrity and function of neurons essential for learning in Drosophila. J. Neurosci. 2010, 30, 464–477. [Google Scholar] [CrossRef]
- Leroy, K.; Ando, K.; Laporte, V.; Dedecker, R.; Suain, V.; Authelet, M.; Heraud, C.; Pierrot, N.; Yilmaz, Z.; Octave, J.N.; et al. Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am. J. Pathol. 2012, 181, 1928–1940. [Google Scholar] [CrossRef]
- Sofola, O.; Kerr, F.; Rogers, I.; Killick, R.; Augustin, H.; Gandy, C.; Allen, M.J.; Hardy, J.; Lovestone, S.; Partridge, L. Inhibition of GSK-3 ameliorates Abeta pathology in an adult-onset Drosophila model of Alzheimer’s disease. PLoS Genet. 2010, 6, e1001087. [Google Scholar] [CrossRef]
- Burnouf, S.; Gronke, S.; Augustin, H.; Dols, J.; Gorsky, M.K.; Werner, J.; Kerr, F.; Alic, N.; Martinez, P.; Partridge, L. Deletion of endogenous Tau proteins is not detrimental in Drosophila. Sci. Rep. 2016, 6, 23102. [Google Scholar] [CrossRef]
- Klein, J.A.; Ackerman, S.L. Oxidative stress, cell cycle, and neurodegeneration. J. Clin. Investig. 2003, 111, 785–793. [Google Scholar] [CrossRef]
- Moreira, P.I.; Santos, M.S.; Oliveira, C.R.; Shenk, J.C.; Nunomura, A.; Smith, M.A.; Zhu, X.; Perry, G. Alzheimer disease and the role of free radicals in the pathogenesis of the disease. CNS Neurol. Disord. Drug Targets 2008, 7, 3–10. [Google Scholar]
- Eckert, A.; Schmitt, K.; Gotz, J. Mitochondrial dysfunction—The beginning of the end in Alzheimer’s disease? Separate and synergistic modes of tau and amyloid-beta toxicity. Alzheimers Res. Ther. 2011, 3, 15. [Google Scholar] [CrossRef]
- Rival, T.; Page, R.M.; Chandraratna, D.S.; Sendall, T.J.; Ryder, E.; Liu, B.; Lewis, H.; Rosahl, T.; Hider, R.; Camargo, L.M.; et al. Fenton chemistry and oxidative stress mediate the toxicity of the beta-amyloid peptide in a Drosophila model of Alzheimer’s disease. Eur. J. Neurosci. 2009, 29, 1335–1347. [Google Scholar] [CrossRef]
- Ott, S.; Dziadulewicz, N.; Crowther, D.C. Iron is a specific cofactor for distinct oxidation- and aggregation-dependent Abeta toxicity mechanisms in a Drosophila model. Dis. Model. Mech. 2015, 8, 657–667. [Google Scholar] [CrossRef]
- Liu, B.; Moloney, A.; Meehan, S.; Morris, K.; Thomas, S.E.; Serpell, L.C.; Hider, R.; Marciniak, S.J.; Lomas, D.A.; Crowther, D.C. Iron promotes the toxicity of amyloid beta peptide by impeding its ordered aggregation. J. Biol. Chem. 2011, 286, 4248–4256. [Google Scholar] [CrossRef]
- Dias-Santagata, D.; Fulga, T.A.; Duttaroy, A.; Feany, M.B. Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J. Clin. Investig. 2007, 117, 236–245. [Google Scholar] [CrossRef]
- Lee, S.; Bang, S.M.; Hong, Y.K.; Lee, J.H.; Jeong, H.; Park, S.H.; Liu, Q.F.; Lee, I.S.; Cho, K.S. The calcineurin inhibitor Sarah (Nebula) exacerbates Abeta42 phenotypes in a Drosophila model of Alzheimer’s disease. Dis. Model. Mech. 2016, 9, 295–306. [Google Scholar] [CrossRef]
- Favrin, G.; Bean, D.M.; Bilsland, E.; Boyer, H.; Fischer, B.E.; Russell, S.; Crowther, D.C.; Baylis, H.A.; Oliver, S.G.; Giannakou, M.E. Identification of novel modifiers of Abeta toxicity by transcriptomic analysis in the fruitfly. Sci. Rep. 2013, 3, 3512. [Google Scholar] [CrossRef]
- Muller, F.L.; Lustgarten, M.S.; Jang, Y.; Richardson, A.; Van Remmen, H. Trends in oxidative aging theories. Free Radic. Biol. Med. 2007, 43, 477–503. [Google Scholar] [CrossRef]
- Jung, I.; Kim, T.Y.; Kim-Ha, J. Identification of Drosophila SOD3 and its protective role against phototoxic damage to cells. FEBS Lett. 2011, 585, 1973–1978. [Google Scholar] [CrossRef]
- Bush, A.I. The metallobiology of Alzheimer’s disease. Trends Neurosci. 2003, 26, 207–214. [Google Scholar] [CrossRef]
- Lang, M.; Wang, L.; Fan, Q.; Xiao, G.; Wang, X.; Zhong, Y.; Zhou, B. Genetic inhibition of solute-linked carrier 39 family transporter 1 ameliorates abeta pathology in a Drosophila model of Alzheimer’s disease. PLoS Genet. 2012, 8, e1002683. [Google Scholar] [CrossRef]
- Lang, M.; Fan, Q.; Wang, L.; Zheng, Y.; Xiao, G.; Wang, X.; Wang, W.; Zhong, Y.; Zhou, B. Inhibition of human high-affinity copper importer Ctr1 orthologous in the nervous system of Drosophila ameliorates Abeta42-induced Alzheimer’s disease-like symptoms. Neurobiol. Aging 2013, 34, 2604–2612. [Google Scholar] [CrossRef]
- Lindholm, D.; Wootz, H.; Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006, 13, 385. [Google Scholar] [CrossRef]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J. Cell Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef]
- Costa, R.O.; Ferreiro, E.; Cardoso, S.M.; Oliveira, C.R.; Pereira, C.M. ER stress-mediated apoptotic pathway induced by Abeta peptide requires the presence of functional mitochondria. J. Alzheimers Dis. 2010, 20, 625–636. [Google Scholar] [CrossRef]
- Costa, R.O.; Ferreiro, E.; Oliveira, C.R.; Pereira, C.M. Inhibition of mitochondrial cytochrome c oxidase potentiates Abeta-induced ER stress and cell death in cortical neurons. Mol. Cell. Neurosci. 2013, 52, 1–8. [Google Scholar] [CrossRef]
- Kang, E.B.; Kwon, I.S.; Koo, J.H.; Kim, E.J.; Kim, C.H.; Lee, J.; Yang, C.H.; Lee, Y.I.; Cho, I.H.; Cho, J.Y. Treadmill exercise represses neuronal cell death and inflammation during Abeta-induced ER stress by regulating unfolded protein response in aged presenilin 2 mutant mice. Apoptosis 2013, 18, 1332–1347. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Marcora, M.S.; Belfiori-Carrasco, L.F.; Bocai, N.I.; Morelli, L.; Castano, E.M. Amyloid-beta42 clearance and neuroprotection mediated by X-box binding protein 1 signaling decline with aging in the Drosophila brain. Neurobiol. Aging 2017, 60, 57–70. [Google Scholar] [CrossRef]
- Perry, G.; Roder, H.; Nunomura, A.; Takeda, A.; Friedlich, A.L.; Zhu, X.; Raina, A.K.; Holbrook, N.; Siedlak, S.L.; Harris, P.L.; et al. Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. Neuroreport 1999, 10, 2411–2415. [Google Scholar] [CrossRef]
- Zhu, X.; Castellani, R.J.; Takeda, A.; Nunomura, A.; Atwood, C.S.; Perry, G.; Smith, M.A. Differential activation of neuronal ERK, JNK/SAPK and p38 in Alzheimer disease: The ‘two hit’ hypothesis. Mech. Ageing Dev. 2001, 123, 39–46. [Google Scholar] [CrossRef]
- Dineley, K.T.; Westerman, M.; Bui, D.; Bell, K.; Ashe, K.H.; Sweatt, J.D. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease. J. Neurosci. 2001, 21, 4125–4133. [Google Scholar] [CrossRef]
- Ma, Q.L.; Harris-White, M.E.; Ubeda, O.J.; Simmons, M.; Beech, W.; Lim, G.P.; Teter, B.; Frautschy, S.A.; Cole, G.M. Evidence of Abeta- and transgene-dependent defects in ERK-CREB signaling in Alzheimer’s models. J. Neurochem. 2007, 103, 1594–1607. [Google Scholar] [CrossRef]
- Zhu, X.; Lee, H.-G.; Raina, A.K.; Perry, G.; Smith, M.A. The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals 2002, 11, 270–281. [Google Scholar] [CrossRef]
- Cheung, E.C.; Slack, R.S. Emerging role for ERK as a key regulator of neuronal apoptosis. Sci. STKE. 2004, 2004, pe45. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, S.; Hong, Y.K.; Hwang, S.; Lee, J.H.; Bang, S.M.; Kim, Y.K.; Koo, B.S.; Lee, I.S.; Cho, K.S. Suppressive effects of SuHeXiang Wan on amyloid-beta42-induced extracellular signal-regulated kinase hyperactivation and glial cell proliferation in a transgenic Drosophila model of Alzheimer’s disease. Biol. Pharm. Bull. 2013, 36, 390–398. [Google Scholar] [CrossRef]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef]
- Cobb, M.H. MAP kinase pathways. Prog. Biophys. Mol. Biol. 1999, 71, 479–500. [Google Scholar] [CrossRef]
- Liu, Q.F.; Jeong, H.; Lee, J.H.; Hong, Y.K.; Oh, Y.; Kim, Y.M.; Suh, Y.S.; Bang, S.; Yun, H.S.; Lee, K.; et al. Coriandrum sativum suppresses Abeta42-Induced ROS increases, glial cell proliferation, and ERK activation. Am. J. Chin. Med. 2016, 44, 1325–1347. [Google Scholar] [CrossRef]
- Liu, Q.F.; Jeon, Y.; Sung, Y.W.; Lee, J.H.; Jeong, H.; Kim, Y.M.; Yun, H.S.; Chin, Y.W.; Jeon, S.; Cho, K.S.; et al. Nardostachys jatamansi ethanol extract ameliorates Abeta42 cytotoxicity. Biol. Pharm. Bull. 2018, 41, 470–477. [Google Scholar] [CrossRef]
- Rahn, T.; Leippe, M.; Roeder, T.; Fedders, H. EGFR signaling in the brain is necessary for olfactory learning in Drosophila larvae. Learn. Mem. 2013, 20, 194–200. [Google Scholar] [CrossRef][Green Version]
- Wang, L.; Chiang, H.C.; Wu, W.; Liang, B.; Xie, Z.; Yao, X.; Ma, W.; Du, S.; Zhong, Y. Epidermal growth factor receptor is a preferred target for treating amyloid-beta-induced memory loss. Proc. Natl. Acad. Sci. USA 2012, 109, 16743–16748. [Google Scholar] [CrossRef]
- Herrup, K. Post-mitotic role of the cell cycle machinery. Curr. Opin. Cell Biol. 2013, 25, 711–716. [Google Scholar] [CrossRef]
- Lee, H.G.; Casadesus, G.; Zhu, X.; Castellani, R.J.; McShea, A.; Perry, G.; Petersen, R.B.; Bajic, V.; Smith, M.A. Cell cycle re-entry mediated neurodegeneration and its treatment role in the pathogenesis of Alzheimer’s disease. Neurochem. Int. 2009, 54, 84–88. [Google Scholar] [CrossRef]
- Crews, L.; Rockenstein, E.; Masliah, E. APP transgenic modeling of Alzheimer’s disease: Mechanisms of neurodegeneration and aberrant neurogenesis. Brain Struct. Funct. 2010, 214, 111–126. [Google Scholar] [CrossRef]
- Varvel, N.H.; Bhaskar, K.; Patil, A.R.; Pimplikar, S.W.; Herrup, K.; Lamb, B.T. Abeta oligomers induce neuronal cell cycle events in Alzheimer’s disease. J. Neurosci. 2008, 28, 10786–10793. [Google Scholar] [CrossRef]
- Seward, M.E.; Swanson, E.; Norambuena, A.; Reimann, A.; Cochran, J.N.; Li, R.; Roberson, E.D.; Bloom, G.S. Amyloid-beta signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J. Cell Sci. 2013, 126, 1278–1286. [Google Scholar] [CrossRef]
- Kong, Y.; Li, K.; Fu, T.; Wan, C.; Zhang, D.; Song, H.; Zhang, Y.; Liu, N.; Gan, Z.; Yuan, L. Quercetin ameliorates Abeta toxicity in Drosophila AD model by modulating cell cycle-related protein expression. Oncotarget 2016, 7, 67716–67731. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Zhao, Y.; Huang, X.; Chen, C.; Sun, L.; Zhuang, L.; Xue, L. Loss of Polo ameliorates APP-induced Alzheimer’s disease-like symptoms in Drosophila. Sci. Rep. 2015, 5, 16816. [Google Scholar] [CrossRef] [PubMed]
- Alberi, L.; Hoey, S.E.; Brai, E.; Scotti, A.L.; Marathe, S. Notch signaling in the brain: In good and bad times. Ageing Res. Rev. 2013, 12, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Marathe, S.; Liu, S.; Brai, E.; Kaczarowski, M.; Alberi, L. Notch signaling in response to excitotoxicity induces neurodegeneration via erroneous cell cycle reentry. Cell Death Differ. 2015, 22, 1775–1784. [Google Scholar] [CrossRef]
- Kong, Y.; Wu, J.; Zhang, D.; Wan, C.; Yuan, L. The Role of miR-124 in Drosophila Alzheimer’s Disease Model by Targeting Delta in Notch Signaling Pathway. Curr. Mol. Med. 2015, 15, 980–989. [Google Scholar] [CrossRef]
- Roth, K.A. Caspases, apoptosis, and Alzheimer disease: Causation, correlation, and confusion. J. Neuropathol. Exp. Neurol. 2001, 60, 829–838. [Google Scholar] [CrossRef]
- Wu, S.C.; Cao, Z.S.; Chang, K.M.; Juang, J.L. Intestinal microbial dysbiosis aggravates the progression of Alzheimer’s disease in Drosophila. Nat. Commun. 2017, 8, 24. [Google Scholar] [CrossRef]
- Hong, Y.K.; Lee, S.; Park, S.H.; Lee, J.H.; Han, S.Y.; Kim, S.T.; Kim, Y.K.; Jeon, S.; Koo, B.S.; Cho, K.S. Inhibition of JNK/dFOXO pathway and caspases rescues neurological impairments in Drosophila Alzheimer’s disease model. Biochem. Biophys. Res. Commun. 2012, 419, 49–53. [Google Scholar] [CrossRef]
- Hawkins, C.J.; Yoo, S.J.; Peterson, E.P.; Wang, S.L.; Vernooy, S.Y.; Hay, B.A. The Drosophila caspase DRONC cleaves following glutamate or aspartate and is regulated by DIAP1, HID, and GRIM. J. Biol. Chem. 2000, 275, 27084–27093. [Google Scholar]
- Meier, P.; Silke, J.; Leevers, S.J.; Evan, G.I. The Drosophila caspase DRONC is regulated by DIAP1. EMBO J. 2000, 19, 598–611. [Google Scholar] [CrossRef]
- Yu, S.Y.; Yoo, S.J.; Yang, L.; Zapata, C.; Srinivasan, A.; Hay, B.A.; Baker, N.E. A pathway of signals regulating effector and initiator caspases in the developing Drosophila eye. Development 2002, 129, 3269–3278. [Google Scholar] [PubMed]
- Lin, R.; Angelin, A.; Da Settimo, F.; Martini, C.; Taliani, S.; Zhu, S.; Wallace, D.C. Genetic analysis of dTSPO, an outer mitochondrial membrane protein, reveals its functions in apoptosis, longevity, and Ab42-induced neurodegeneration. Aging Cell 2014, 13, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Hasegawa, M.; Jakes, R.; Lawler, S.; Cuenda, A.; Cohen, P. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett. 1997, 409, 57–62. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Betts, J.C.; Blackstock, W.P.; Nebreda, A.R.; Anderton, B.H. Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: Differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase-3beta. J. Neurochem. 2000, 74, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Rottkamp, C.A.; Boux, H.; Takeda, A.; Perry, G.; Smith, M.A. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2000, 59, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar] [CrossRef]
- Song, Q.; Feng, G.; Huang, Z.; Chen, X.; Chen, Z.; Ping, Y. Aberrant axonal arborization of PDF neurons induced by Abeta42-mediated JNK activation underlies sleep disturbance in an Alzheimer’s model. Mol. Neurobiol. 2017, 54, 6317–6328. [Google Scholar] [CrossRef]
- Hong, Y.K.; Park, S.H.; Lee, S.; Hwang, S.; Lee, M.J.; Kim, D.; Lee, J.H.; Han, S.Y.; Kim, S.T.; Kim, Y.K.; et al. Neuroprotective effect of SuHeXiang Wan in Drosophila models of Alzheimer’s disease. J. Ethnopharmacol. 2011, 134, 1028–1032. [Google Scholar] [CrossRef]
- Wang, X.; Ma, Y.; Zhao, Y.; Chen, Y.; Hu, Y.; Chen, C.; Shao, Y.; Xue, L. APLP1 promotes dFoxO-dependent cell death in Drosophila. Apoptosis 2015, 20, 778–786. [Google Scholar] [CrossRef]
- Coelho, D.S.; Schwartz, S.; Merino, M.M.; Hauert, B.; Topfel, B.; Tieche, C.; Rhiner, C.; Moreno, E. Culling less fit neurons protects against amyloid-β-induced brain damage and cognitive and motor decline. Cell Rep. 2018, 25, 3661–3673. [Google Scholar] [CrossRef]
- Lord, J.; Cruchaga, C. The epigenetic landscape of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, N.; Rao, P.; Burkhardt, S.; Sananbenesi, F.; Schlüter, O.M.; Bradke, F.; Lu, J.; Fischer, A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Dolan, P.J.; Johnson, G.V. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 2008, 106, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Choi, H.; Jung, E.S.; Lee, W.; Oh, S.; Jeon, N.L.; Mook-Jung, I. HDAC6 inhibitor blocks amyloid beta-induced impairment of mitochondrial transport in hippocampal neurons. PLoS ONE 2012, 7, e42983. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhao, K.; Wu, J.; Xu, Z.; Jin, S.; Zhang, Y.Q. HDAC6 mutations rescue human tau-induced microtubule defects in Drosophila. Proc. Natl. Acad. Sci. USA 2013, 110, 4604–4609. [Google Scholar] [CrossRef]
- Pile, L.; Lee, F.-H.; Wassarman, D. The histone deacetylase inhibitor trichostatin A influences the development of Drosophila melanogaster. Cell. Mol. Life. Sci. 2001, 58, 1715–1718. [Google Scholar] [CrossRef]
- Jeong, M.R.; Hashimoto, R.; Senatorov, V.V.; Fujimaki, K.; Ren, M.; Lee, M.S.; Chuang, D.-M. Valproic acid, a mood stabilizer and anticonvulsant, protects rat cerebral cortical neurons from spontaneous cell death: A role of histone deacetylase inhibition. FEBS Lett. 2003, 542, 74–78. [Google Scholar] [CrossRef]
- Cao, X.; Südhof, T.C. A transcriptively active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 2001, 293, 115–120. [Google Scholar] [CrossRef]
- Baek, S.H.; Ohgi, K.A.; Rose, D.W.; Koo, E.H.; Glass, C.K.; Rosenfeld, M.G. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-κB and β-amyloid precursor protein. Cell 2002, 110, 55–67. [Google Scholar] [CrossRef]
- Panikker, P.; Xu, S.-J.; Zhang, H.; Sarthi, J.; Beaver, M.; Sheth, A.; Akhter, S.; Elefant, F. Restoring Tip60 HAT/HDAC2 balance in the neurodegenerative brain relieves epigenetic transcriptional repression and reinstates cognition. J. Neurosci. 2018, 38, 4569–4583. [Google Scholar] [CrossRef]
- Pirooznia, S.K.; Sarthi, J.; Johnson, A.A.; Toth, M.S.; Chiu, K.; Koduri, S.; Elefant, F. Tip60 HAT activity mediates APP induced lethality and apoptotic cell death in the CNS of a Drosophila Alzheimer’s disease model. PLoS ONE 2012, 7, e41776. [Google Scholar] [CrossRef] [PubMed]
- Cutler, T.; Sarkar, A.; Moran, M.; Steffensmeier, A.; Puli, O.R.; Mancini, G.; Tare, M.; Gogia, N.; Singh, A. Drosophila eye model to study neuroprotective role of CREB binding protein (CBP) in Alzheimer’s disease. PLoS ONE 2015, 10, e0137691. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Hsia, A.Y.; Masliah, E.; McConlogue, L.; Yu, G.-Q.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Malenka, R.C.; Nicoll, R.A.; Mucke, L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 1999, 96, 3228–3233. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.-F.; Kuo, Y.-M.; Roher, A.E.; Brachova, L.; Shen, Y.; Sue, L.; Beach, T.; Kurth, J.H.; Rydel, R.E.; Rogers, J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 1999, 155, 853–862. [Google Scholar] [CrossRef]
- Koistinaho, M.; Ort, M.; Cimadevilla, J.M.; Vondrous, R.; Cordell, B.; Koistinaho, J.; Bures, J.; Higgins, L.S. Specific spatial learning deficits become severe with age in β-amyloid precursor protein transgenic mice that harbor diffuse β-amyloid deposits but do not form plaques. Proc. Natl. Acad. Sci. USA 2001, 98, 14675–14680. [Google Scholar] [CrossRef]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP processing and synaptic function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef]
- Townsend, M.; Shankar, G.M.; Mehta, T.; Walsh, D.M.; Selkoe, D.J. Effects of secreted oligomers of amyloid β-protein on hippocampal synaptic plasticity: A potent role for trimers. J. Physiol. 2006, 572, 477–492. [Google Scholar] [CrossRef]
- Moechars, D.; Dewachter, I.; Lorent, K.; Reversé, D.; Baekelandt, V.; Naidu, A.; Tesseur, I.; Spittaels, K.; Van Den Haute, C.; Checler, F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 1999, 274, 6483–6492. [Google Scholar] [CrossRef]
- Chapman, P.F.; White, G.L.; Jones, M.W.; Cooper-Blacketer, D.; Marshall, V.J.; Irizarry, M.; Younkin, L.; Good, M.A.; Bliss, T.; Hyman, B.T. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat. Neurosci. 1999, 2, 271. [Google Scholar] [CrossRef] [PubMed]
- Fitzjohn, S.M.; Morton, R.A.; Kuenzi, F.; Rosahl, T.W.; Shearman, M.; Lewis, H.; Smith, D.; Reynolds, D.S.; Davies, C.H.; Collingridge, G.L. Age-related impairment of synaptic transmission but normal long-term potentiation in transgenic mice that overexpress the human APP695SWE mutant form of amyloid precursor protein. J. Neurosci. 2001, 21, 4691–4698. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Duan, J.; Ran, D.; Fan, Z.; Yan, Y.; Huang, N.; Gu, H.; Zhu, Y. Amyloid-β depresses excitatory cholinergic synaptic transmission in Drosophila. Neurosci. Bull. 2012, 28, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Iijima, K.; Liu, H.-P.; Chiang, A.-S.; Hearn, S.A.; Konsolaki, M.; Zhong, Y. Dissecting the pathological effects of human Aβ40 and Aβ42 in Drosophila: A potential model for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 6623–6628. [Google Scholar] [CrossRef]
- Klyubin, I.; Walsh, D.M.; Lemere, C.A.; Cullen, W.K.; Shankar, G.M.; Betts, V.; Spooner, E.T.; Jiang, L.; Anwyl, R.; Selkoe, D.J.; et al. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat. Med. 2005, 11, 556–561. [Google Scholar] [CrossRef]
- Newey, S.E.; Velamoor, V.; Govek, E.E.; Van Aelst, L. Rho GTPases, dendritic structure, and mental retardation. J. Neurobiol. 2005, 64, 58–74. [Google Scholar] [CrossRef]
- Lee, T.; Winter, C.; Marticke, S.S.; Lee, A.; Luo, L. Essential roles of Drosophila RhoA in the regulation of neuroblast proliferation and dendritic but not axonal morphogenesis. Neuron 2000, 25, 307–316. [Google Scholar] [CrossRef]
- Nakayama, A.Y.; Harms, M.B.; Luo, L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J. Neurosci. 2000, 20, 5329–5338. [Google Scholar] [CrossRef]
- Huesa, G.; Baltrons, M.A.; Gómez-Ramos, P.; Morán, A.; García, A.; Hidalgo, J.; Francés, S.; Santpere, G.; Ferrer, I.; Galea, E. Altered distribution of RhoA in Alzheimer’s disease and AβPP overexpressing mice. J. Alzheimers Dis. 2010, 19, 37–56. [Google Scholar] [CrossRef]
- Cook, M.; Mani, P.; Wentzell, J.S.; Kretzschmar, D. Increased RhoA prenylation in the loechrig (loe) mutant leads to progressive neurodegeneration. PLoS ONE 2012, 7, e44440. [Google Scholar] [CrossRef]
- Wu, W.; Du, S.; Shi, W.; Liu, Y.; Hu, Y.; Xie, Z.; Yao, X.; Liu, Z.; Ma, W.; Xu, L. Inhibition of Rac1-dependent forgetting alleviates memory deficits in animal models of Alzheimer’s disease. Protein Cell. 2019, 10, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.A.; Burns, M.; Binder, L.I.; Kanaan, N.M.; LaPointe, N.; Bosco, D.A.; Brown, R.H.; Brown, H.; Tiwari, A.; Hayward, L. Axonal transport defects in neurodegenerative diseases. J. Neurosci. 2009, 29, 12776–12786. [Google Scholar] [CrossRef] [PubMed]
- Gunawardena, S.; Goldstein, L.S. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron 2001, 32, 389–401. [Google Scholar] [CrossRef]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef]
- Shaw, J.L.; Chang, K.T. Nebula/DSCR1 upregulation delays neurodegeneration and protects against APP-induced axonal transport defects by restoring calcineurin and GSK-3β signaling. PLoS Genet. 2013, 9, e1003792. [Google Scholar] [CrossRef]
- Rusu, P.; Jansen, A.; Soba, P.; Kirsch, J.; Löwer, A.; Merdes, G.; Kuan, Y.H.; Jung, A.; Beyreuther, K.; Kjaerulff, O. Axonal accumulation of synaptic markers in APP transgenic Drosophila depends on the NPTY motif and is paralleled by defects in synaptic plasticity. Eur. J. Neurosci. 2007, 25, 1079–1086. [Google Scholar] [CrossRef]
- Yao, P.J.; Zhu, M.; Pyun, E.I.; Brooks, A.I.; Therianos, S.; Meyers, V.E.; Coleman, P.D. Defects in expression of genes related to synaptic vesicle traffickingin frontal cortex of Alzheimer’s disease. Neurobiol. Dis. 2003, 12, 97–109. [Google Scholar] [CrossRef]
- Kittel, R.J.; Wichmann, C.; Rasse, T.M.; Fouquet, W.; Schmidt, M.; Schmid, A.; Wagh, D.A.; Pawlu, C.; Kellner, R.R.; Willig, K.I. Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science 2006, 312, 1051–1054. [Google Scholar] [CrossRef]
- Mhatre, S.D.; Satyasi, V.; Killen, M.; Paddock, B.E.; Moir, R.D.; Saunders, A.J.; Marenda, D.R. Synaptic abnormalities in a Drosophila model of Alzheimer’s disease. Dis. Model. Mech. 2014, 7, 373–385. [Google Scholar] [CrossRef]
- Huang, J.-K.; Ma, P.-L.; Ji, S.-Y.; Zhao, X.-L.; Tan, J.-X.; Sun, X.-J.; Huang, F.-D. Age-dependent alterations in the presynaptic active zone in a Drosophila model of Alzheimer’s disease. Neurobiol. Dis. 2013, 51, 161–167. [Google Scholar] [CrossRef]
- Thomas, U.; Ebitsch, S.; Gorczyca, M.; Koh, Y.; Hough, C.; Woods, D.; Gundelfinger, E.; Budnik, V. Synaptic targeting and localization of discs-large is a stepwise process controlled by different domains of the protein. Curr. Biol. 2000, 10, 1108–1117. [Google Scholar] [CrossRef]
- Chen, K.; Featherstone, D.E. Discs-large (DLG) is clustered by presynaptic innervation and regulates postsynaptic glutamate receptor subunit composition in Drosophila. BMC Biol. 2005, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Furotani, K.; Kamimura, K.; Yajima, T.; Nakayama, M.; Enomoto, R.; Tamura, T.; Okazawa, H.; Sone, M. Suppression of the synaptic localization of a subset of proteins including APP partially ameliorates phenotypes of the Drosophila Alzheimer’s disease model. PLoS ONE 2018, 13, e0204048. [Google Scholar] [CrossRef] [PubMed]
- Verri, M.; Pastoris, O.; Dossena, M.; Aquilani, R.; Guerriero, F.; Cuzzoni, G.; Venturini, L.; Ricevuti, G.; Bongiorno, A.I. Mitochondrial alterations, oxidative stress and neuroinflammation in Alzheimer’s disease. Int. J. Immunopathol. Pharmacol. 2012, 25, 345–353. [Google Scholar] [CrossRef]
- Lv, F.; Yang, X.; Cui, C.; Su, C. Exogenous expression of Drp1 plays neuroprotective roles in the Alzheimer’s disease in the Abeta42 transgenic Drosophila model. PLoS ONE 2017, 12, e0176183. [Google Scholar] [CrossRef] [PubMed]
- Verstreken, P.; Ly, C.V.; Venken, K.J.; Koh, T.-W.; Zhou, Y.; Bellen, H.J. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef]
- Iijima-Ando, K.; Hearn, S.A.; Shenton, C.; Gatt, A.; Zhao, L.; Iijima, K. Mitochondrial mislocalization underlies Aβ42-induced neuronal dysfunction in a Drosophila model of Alzheimer’s disease. PLoS ONE 2009, 4, e8310. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef]
- Petersen, A.J.; Rimkus, S.A.; Wassarman, D.A. ATM kinase inhibition in glial cells activates the innate immune response and causes neurodegeneration in Drosophila. Proc. Natl. Acad. Sci. USA 2012, 109, E656–E664. [Google Scholar] [CrossRef]
- Cao, Y.; Chtarbanova, S.; Petersen, A.J.; Ganetzky, B. Dnr1 mutations cause neurodegeneration in Drosophila by activating the innate immune response in the brain. Proc. Natl. Acad. Sci. USA 2013, 110, E1752–E1760. [Google Scholar] [CrossRef]
- Lee, C.D.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef]
- Coutinho-Budd, J.; Freeman, M.R. Probing the enigma: Unraveling glial cell biology in invertebrates. Curr. Opin. Neurobiol. 2013, 23, 1073–1079. [Google Scholar] [CrossRef]
- Ray, A.; Speese, S.D.; Logan, M.A. Glial draper rescues Aβ toxicity in a Drosophila model of Alzheimer’s disease. J. Neurosci. 2017, 37, 11881–11893. [Google Scholar] [CrossRef]
- Purice, M.D.; Speese, S.D.; Logan, M.A. Delayed glial clearance of degenerating axons in aged Drosophila is due to reduced PI3K/Draper activity. Nat. Commun. 2016, 7, 12871. [Google Scholar] [CrossRef]
- Ulland, T.K.; Colonna, M. TREM2—A key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef]
- Tahara, K.; Kim, H.-D.; Jin, J.-J.; Maxwell, J.A.; Li, L.; Fukuchi, K.-I. Role of toll-like receptor signalling in Aβ uptake and clearance. Brain 2006, 129, 3006–3019. [Google Scholar] [CrossRef]
- Cameron, B.; Landreth, G.E. Inflammation, microglia, and Alzheimer’s disease. Neurobiol. Dis. 2010, 37, 503–509. [Google Scholar] [CrossRef]
- Sessa, G.; Podini, P.; Mariani, M.; Meroni, A.; Spreafico, R.; Sinigaglia, F.; Colonna, M.; Panina, P.; Meldolesi, J. Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur. J. Neurosci. 2004, 20, 2617–2628. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, J.-T.; Zhu, X.-C.; Tan, L. TREM2 in Alzheimer’s disease. Mol. Neurobiol. 2013, 48, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, M.; Wang, M.; Fujisaki, N.; Sakakibara, Y.; Quan, X.; Ehrlich, M.E.; De Jager, P.L.; Bennett, D.A.; Schadt, E.E.; Gandy, S. Integrated biology approach reveals molecular and pathological interactions among Alzheimer’s Aβ42, Tau, TREM2, and TYROBP in Drosophila models. Genome Med. 2018, 10, 26. [Google Scholar] [CrossRef]
- Khush, R.S.; Leulier, F.; Lemaitre, B. Drosophila immunity: Two paths to NF-kappaB. Trends Immunol. 2001, 22, 260–264. [Google Scholar] [CrossRef]
- Richard, K.L.; Filali, M.; Préfontaine, P.; Rivest, S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid β1–42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. J. Neurosci. 2008, 28, 5784–5793. [Google Scholar] [CrossRef]
- Scholtzova, H.; Kascsak, R.J.; Bates, K.A.; Boutajangout, A.; Kerr, D.J.; Meeker, H.C.; Mehta, P.D.; Spinner, D.S.; Wisniewski, T. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. J. Neurosci. 2009, 29, 1846–1854. [Google Scholar] [CrossRef]
- Walker, D.G.; Tang, T.M.; Lue, L.-F. Increased expression of toll-like receptor 3, an anti-viral signaling molecule, and related genes in Alzheimer’s disease brains. Exp. Neurol. 2018, 309, 91–106. [Google Scholar] [CrossRef]
- Tan, L.; Schedl, P.; Song, H.-J.; Garza, D.; Konsolaki, M. The Toll→ NFκB signaling pathway mediates the neuropathological effects of the human Alzheimer’s Aβ42 polypeptide in Drosophila. PLoS ONE 2008, 3, e3966. [Google Scholar] [CrossRef]
- Li, Y.; Sibon, O.; Dijkers, P. Inhibition of NF-κB in astrocytes is sufficient to delay neurodegeneration induced by proteotoxicity in neurons. J. Neuroinflammation 2018, 15, 261. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Tanzi, R.E. Alzheimer disease risk genes: 29 and counting. Nat. Rev. Neurol. 2019, 15, 191. [Google Scholar] [CrossRef] [PubMed]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Ridge, P.G.; Hoyt, K.B.; Boehme, K.; Mukherjee, S.; Crane, P.K.; Haines, J.L.; Mayeux, R.; Farrer, L.A.; Pericak-Vance, M.A.; Schellenberg, G.D. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol. Aging 2016, 41, e13–e200. [Google Scholar] [CrossRef]
- Hall, A.M.; Roberson, E.D. Mouse models of Alzheimer’s disease. Brain Res. Bull. 2012, 88, 3–12. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A.; Dolgin, E. Failed Alzheimer’s trial does not kill leading theory of disease. Nature 2016, 540, 15. [Google Scholar] [CrossRef]
- Sabbagh, M.N.; Hendrix, S.; Harrison, J.E. FDA position statement “Early Alzheimer’s disease: Developing drugs for treatment, Guidance for Industry”. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 13–19. [Google Scholar]
- Wang, X.; Perumalsamy, H.; Kwon, H.W.; Na, Y.-E.; Ahn, Y.-J. Effects and possible mechanisms of action of acacetin on the behavior and eye morphology of Drosophila models of Alzheimer’s disease. Sci. Rep. 2015, 5, 16127. [Google Scholar] [CrossRef]
- Lee, B.I.; Lee, S.; Suh, Y.S.; Lee, J.S.; Kim, A.k.; Kwon, O.Y.; Yu, K.; Park, C.B. Photoexcited porphyrins as a strong suppressor of β-Amyloid aggregation and synaptic toxicity. Angew. Chem. Int. Ed. 2015, 54, 11472–11476. [Google Scholar] [CrossRef]
- Lee, B.I.; Suh, Y.S.; Chung, Y.J.; Yu, K.; Park, C.B. Shedding light on Alzheimer’s β-amyloidosis: Photosensitized methylene blue inhibits self-assembly of β-amyloid peptides and disintegrates their aggregates. Sci. Rep. 2017, 7, 7523. [Google Scholar] [CrossRef]
- Deshpande, P.; Gogia, N.; Singh, A. Exploring the efficacy of natural products in alleviating Alzheimer’s disease. Neural. Regen. Res. 2019, 14, 1321. [Google Scholar] [PubMed]
- Lee, S.; Bang, S.M.; Lee, J.W.; Cho, K.S. Evaluation of traditional medicines for neurodegenerative diseases using Drosophila models. Evid. Based Complement. Altern. Med. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).