The Pathogenic Role of Very Low Density Lipoprotein on Atrial Remodeling in the Metabolic Syndrome


{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Atrial Fibrillation (AF) Remains a Major Health Problem, Especially for Aging Populations
1.2. Atrial Remodeling and Atrial Cardiomyopathy Are Long-Term Changes Preceding the Occurrence of AF
1.3. MetS Is a Major Risk for Atrial Cardiomyopathy and AF
1.4. The Ambiguous Relationship between Dyslipidemia and AF in Clinical Studies
1.5. The Metabolism of VLDL
2. Cardiac Lipotoxicity
3. VLDL of MetS Exhibits Cytotoxicity to Atrial Myocytes
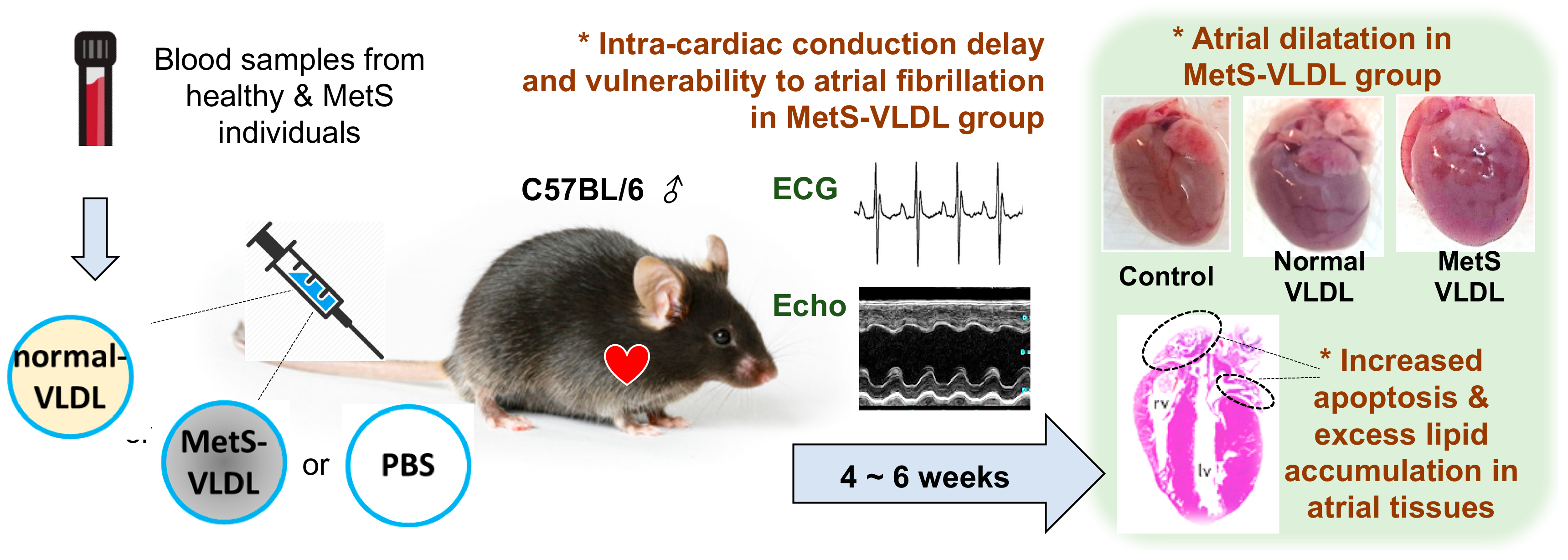
4. VLDL of MetS Induces Atrial Remodeling, Ventricular Hypertrophy, and Vulnerability to AF
5. VLDL of MetS Induces Intra-Cardiac Conduction Delay via Modulation of Cardiac Gap Junction Cx40/43
6. VLDL of MetS Suppresses the Store-Operated Calcium Entry (SOCE) and Downstream Calcineurin-Nuclear Factor of Activated T Cells (NFAT) Pathway
7. VLDL of MetS Disturbs Myofilament Regulation
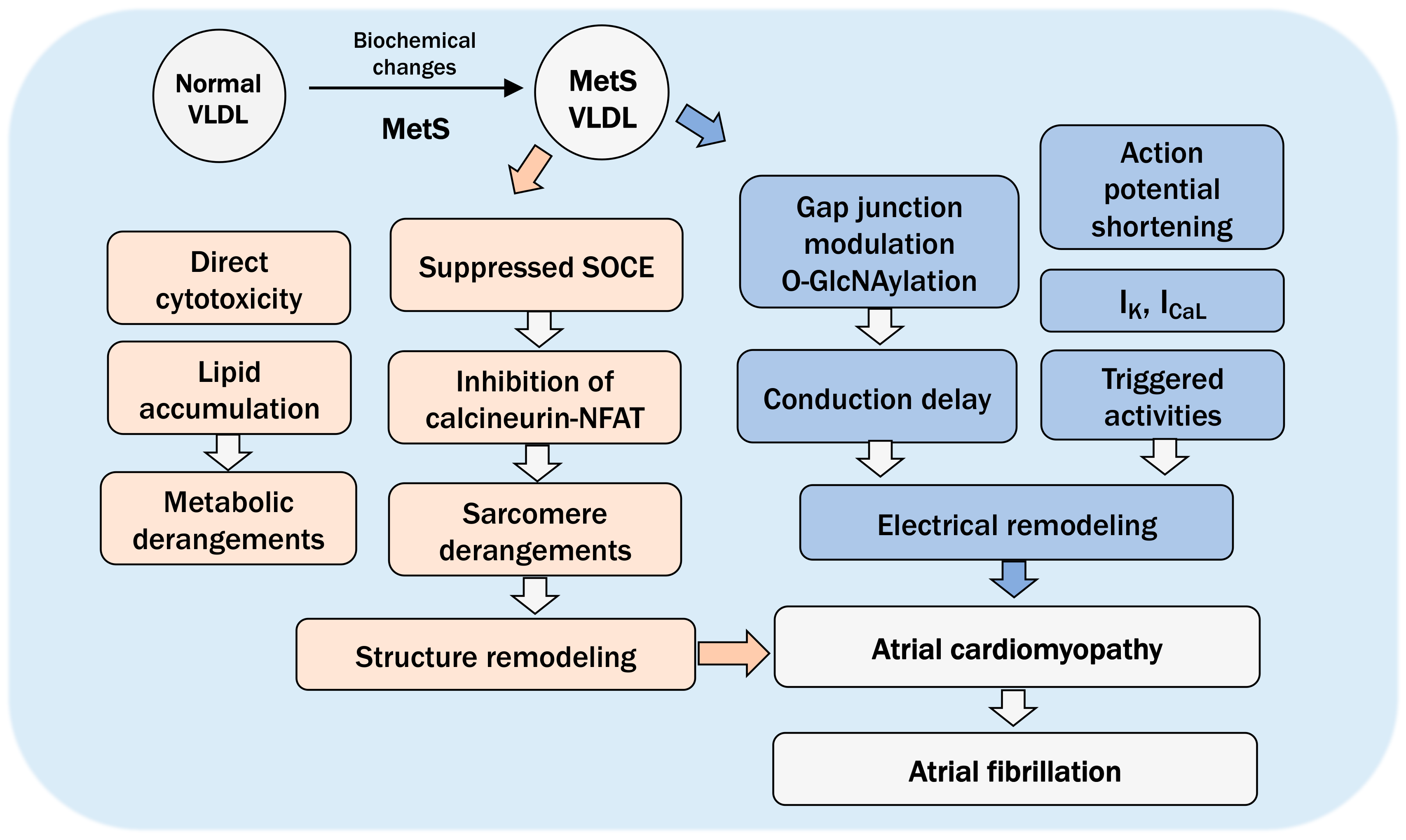
8. The Pathogenic Role of VLDL in Atrial Cardiomyopathy and Future Directions
Funding
Conflicts of Interest
References
- Wattigney, W.A.; Mensah, G.A.; Croft, J.B. Increasing Trends in Hospitalization for Atrial Fibrillation in the United States, 1985 Through 1999. Implic. Prim. Prev. 2003, 108, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Friberg, J.; Buch, P.; Scharling, H.; Gadsbphioll, N.; Jensen, G.B. Rising rates of hospital admissions for atrial fibrillation. Epidemiology 2003, 14, 666–672. [Google Scholar] [CrossRef]
- Hajhosseiny, R.; Matthews, G.K.; Lip, G.Y. Metabolic syndrome, atrial fibrillation, and stroke: Tackling an emerging epidemic. Heart Rhythm 2015, 12, 2332–2343. [Google Scholar] [CrossRef]
- Zakeri, R.; Van Wagoner, D.R.; Calkins, H.; Wong, T.; Ross, H.M.; Heist, E.K.; Meyer, T.E.; Kowey, P.R.; Mentz, R.J.; Cleland, J.G.; et al. The burden of proof: The current state of atrial fibrillation prevention and treatment trials. Heart Rhythm 2017, 14, 763–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, F.; Kwan, G.F.; Benjamin, E.J. Global epidemiology of atrial fibrillation. Nat. Rev. Cardiol. 2014, 11, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Van Wagoner, D.R.; Piccini, J.P.; Albert, C.M.; Anderson, M.E.; Benjamin, E.J.; Brundel, B.; Califf, R.M.; Calkins, H.; Chen, P.S.; Chiamvimonvat, N.; et al. Progress toward the prevention and treatment of atrial fibrillation: A summary of the Heart Rhythm Society Research Forum on the Treatment and Prevention of Atrial Fibrillation, Washington, DC, December 9-10, 2013. Heart Rhythm 2015, 12, e5–e29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, A.S.; Hylek, E.M.; Phillips, K.A.; Chang, Y.; Henault, L.E.; Selby, J.V.; Singer, D.E. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001, 285, 2370–2375. [Google Scholar] [CrossRef]
- Friberg, L.; Rosenqvist, M.; Lip, G.Y. Evaluation of risk stratification schemes for ischaemic stroke and bleeding in 182 678 patients with atrial fibrillation: the Swedish Atrial Fibrillation cohort study. Eur. Heart J. 2012, 33, 1500–1510. [Google Scholar] [CrossRef]
- Bai, D. Atrial fibrillation-linked GJA5/connexin40 mutants impaired gap junctions via different mechanisms. FEBS Lett. 2014, 588, 1238–1243. [Google Scholar] [CrossRef] [Green Version]
- Goette, A.; Kalman, J.M.; Aguinaga, L.; Akar, J.; Cabrera, J.A.; Chen, S.A.; Chugh, S.S.; Corradi, D.; D’Avila, A.; Dobrev, D.; et al. EHRA/HRS/APHRS/SOLAECE expert consensus on Atrial cardiomyopathies: definition, characterization, and clinical implication. Europace 2016. [Google Scholar] [CrossRef]
- Heijman, J.; Voigt, N.; Nattel, S.; Dobrev, D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ. Res. 2014, 114, 1483–1499. [Google Scholar] [CrossRef] [Green Version]
- Bombelli, M.; Facchetti, R.; Cuspidi, C.; Villa, P.; Dozio, D.; Brambilla, G.; Grassi, G.; Mancia, G. Prognostic Significance of Left Atrial Enlargement in a General Population: Results of the PAMELA Study. Hypertension 2014, 64, 1205–1211. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, M.K.; Dahl, J.S.; Henriksen, J.E.; Hey, T.M.; Høilund-Carlsen, P.F.; Beck-Nielsen, H.; Møller, J.E. Left atrial volume index: relation to long-term clinical outcome in type 2 diabetes. J. Am. Coll. Cardiol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Francia, P.; Ricotta, A.; Balla, C.; Adduci, C.; Semprini, L.; Frattari, A.; Modestino, A.; Mercanti, F.; Sensini, I.; Caprinozzi, M.; et al. P-wave duration in lead aVR and the risk of atrial fibrillation in hypertension. Ann. Noninvasive Electrocardiol. 2015, 20, 167–174. [Google Scholar] [CrossRef]
- Yin, X.; Zhao, Y.; Xi, Y.; Cheng, N.; Xia, Y.; Zhang, S.; Dong, Y.; Chang, D.; Cheng, J.; Yang, Y.; et al. The Early Stage of the Atrial Electroanatomic Remodeling as Substrates for Atrial Fibrillation in Hypertensive Patients. J. Am. Heart Assoc. 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J.; Haas, T.S.; Maron, M.S.; Lesser, J.R.; Browning, J.A.; Chan, R.H.; Olivotto, I.; Garberich, R.F.; Schwartz, R.S. Left atrial remodeling in hypertrophic cardiomyopathy and susceptibility markers for atrial fibrillation identified by cardiovascular magnetic resonance. Am. J. Cardiol. 2014, 113, 1394–1400. [Google Scholar] [CrossRef]
- Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001, 285, 2486–2497. [CrossRef]
- Chamberlain, A.M.; Agarwal, S.K.; Ambrose, M.; Folsom, A.R.; Soliman, E.Z.; Alonso, A. Metabolic syndrome and incidence of atrial fibrillation among blacks and whites in the Atherosclerosis Risk in Communities (ARIC) Study. Am. Heart J. 2010, 159, 850–856. [Google Scholar] [CrossRef] [Green Version]
- Nystrom, P.K.; Carlsson, A.C.; Leander, K.; de Faire, U.; Hellenius, M.L.; Gigante, B. Obesity, metabolic syndrome and risk of atrial fibrillation: A Swedish, prospective cohort study. PLoS ONE 2015, 10, e0127111. [Google Scholar] [CrossRef] [Green Version]
- Tanner, R.M.; Baber, U.; Carson, A.P.; Voeks, J.; Brown, T.M.; Soliman, E.Z.; Howard, V.J.; Muntner, P. Association of the metabolic syndrome with atrial fibrillation among United States adults (from the REasons for Geographic and Racial Differences in Stroke [REGARDS] Study). Am. J. Cardiol. 2011, 108, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Kwon, C.H.; Kim, H.; Kim, S.H.; Kim, B.S.; Kim, H.J.; Kim, D.K.; Han, S.W.; Ryu, K.H.; Dong Sung, J. The Impact of Metabolic Syndrome on the Incidence of Atrial Fibrillation: A Nationwide Longitudinal Cohort Study in South Korea. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.G.; Choi, K.J.; Han, S.; Hwang, K.W.; Kwon, C.H.; Park, G.M.; Won, K.B.; Ann, S.H.; Kim, J.; Kim, S.J.; et al. Metabolic Syndrome and the Risk of New-Onset Atrial Fibrillation in Middle-Aged East Asian Men. Circ. J. 2018, 82, 1763–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umetani, K.; Kodama, Y.; Nakamura, T.; Mende, A.; Kitta, Y.; Kawabata, K.; Obata, J.E.; Takano, H.; Kugiyama, K. High prevalence of paroxysmal atrial fibrillation and/or atrial flutter in metabolic syndrome. Circ. J. 2007, 71, 252–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polovina, M.; Hindricks, G.; Maggioni, A.; Piepoli, M.; Vardas, P.; Asanin, M.; Ethikic, D.; Ethuricic, N.; Milinkovic, I.; Seferovic, P.M. Association of metabolic syndrome with non-thromboembolic adverse cardiac outcomes in patients with atrial fibrillation. Eur. Heart J. 2018, 39, 4030–4039. [Google Scholar] [CrossRef]
- Lin, K.J.; Cho, S.I.; Tiwari, N.; Bergman, M.; Kizer, J.R.; Palma, E.C.; Taub, C.C. Impact of metabolic syndrome on the risk of atrial fibrillation recurrence after catheter ablation: systematic review and meta-analysis. J. Interv. Card. Electrophysiol. 2014, 39, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Wojcik, M.; Berkowitsch, A.; Kuniss, M.; Zaltsberg, S.; Pitschner, H.F.; Hamm, C.W.; Neumann, T. Outcomes of atrial fibrillation ablation in patients with metabolic syndrome. J. Am. Coll Cardiol. 2013, 61, 109–110. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.-T.; Chang, S.-H.; Chang, S.-N.; Hwang, J.-J.; Wu, C.-K.; Wang, Y.-C.; Tseng, C.-D.; Yeh, H.-M.; Lai, L.-P.; Chiang, F.-T.; et al. Additive effect of the metabolic syndrome score to the conventional CHADS2 score for the thromboembolic risk stratification of patients with atrial fibrillation. Heart Rhythm 2014, 11, 352–357. [Google Scholar] [CrossRef]
- Movahed, M.-R.; Hashemzadeh, M.; Mazen Jamal, M. Diabetes mellitus is a strong, independent risk for atrial fibrillation and flutter in addition to other cardiovascular disease. Int. J. Cardiol. 2005, 105, 315–318. [Google Scholar] [CrossRef]
- Huxley, R.R.; Filion, K.B.; Konety, S.; Alonso, A. Meta-Analysis of Cohort and Case–Control Studies of Type 2 Diabetes Mellitus and Risk of Atrial Fibrillation. Am. J. Cardiol. 2011, 108, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Verdecchia, P.; Angeli, F.; Reboldi, G. Hypertension and Atrial Fibrillation: Doubts and Certainties From Basic and Clinical Studies. Circ. Res. 2018, 122, 352–368. [Google Scholar] [CrossRef]
- Choisy, S.C.; Arberry, L.A.; Hancox, J.C.; James, A.F. Increased susceptibility to atrial tachyarrhythmia in spontaneously hypertensive rat hearts. Hypertension 2007, 49, 498–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okin, P.M.; Wachtell, K.; Devereux, R.B.; Harris, K.E.; Jern, S.; Kjeldsen, S.E.; Julius, S.; Lindholm, L.H.; Nieminen, M.S.; Edelman, J.M.; et al. Regression of electrocardiographic left ventricular hypertrophy and decreased incidence of new-onset atrial fibrillation in patients with hypertension. JAMA 2006, 296, 1242–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.N.; Wang, L.; Dong, J.Z.; Yu, R.H.; Long, D.Y.; Tang, R.B.; Sang, C.H.; Jiang, C.X.; Liu, N.; Bai, R.; et al. Electrocardiographic left ventricular hypertrophy predicts recurrence of atrial arrhythmias after catheter ablation of paroxysmal atrial fibrillation. Clin. Cardiol. 2018, 41, 797–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohne, L.J.; Johnson, D.; Rose, R.A.; Wilton, S.B.; Gillis, A.M. The Association Between Diabetes Mellitus and Atrial Fibrillation: Clinical and Mechanistic Insights. Front. Physiol. 2019, 10, 135. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Izquierdo, A.; Pereira, R.O.; Wende, A.R.; Punske, B.B.; Abel, E.D.; Tristani-Firouzi, M. The absence of insulin signaling in the heart induces changes in potassium channel expression and ventricular repolarization. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H747–H754. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.J.; Parise, H.; Levy, D.; D’Agostino, R.B., Sr.; Wolf, P.A.; Vasan, R.S.; Benjamin, E.J. Obesity and the risk of new-onset atrial fibrillation. JAMA 2004, 292, 2471–2477. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.G.; Han, K.D.; Choi, J.I.; Yung Boo, K.; Kim, D.Y.; Oh, S.K.; Lee, K.N.; Shim, J.; Kim, J.S.; Kim, Y.H. Impact of the Duration and Degree of Hypertension and Body Weight on New-Onset Atrial Fibrillation: A Nationwide Population-Based Study. Hypertension 2019. [Google Scholar] [CrossRef]
- Watanabe, H.; Tanabe, N.; Watanabe, T.; Darbar, D.; Roden, D.M.; Sasaki, S.; Aizawa, Y. Metabolic syndrome and risk of development of atrial fibrillation: The Niigata preventive medicine study. Circulation 2008, 117, 1255–1260. [Google Scholar] [CrossRef] [Green Version]
- Zanoli, L.; Di Pino, A.; Terranova, V.; Di Marca, S.; Pisano, M.; Di Quattro, R.; Ferrara, V.; Scicali, R.; Rabuazzo, A.M.; Fatuzzo, P.; et al. Inflammation and ventricular-vascular coupling in hypertensive patients with metabolic syndrome. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 1222–1229. [Google Scholar] [CrossRef] [Green Version]
- Mora, S.; Akinkuolie, A.O.; Sandhu, R.K.; Conen, D.; Albert, C.M. Paradoxical association of lipoprotein measures with incident atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2014, 7, 612–619. [Google Scholar] [CrossRef] [Green Version]
- Annoura, M.; Ogawa, M.; Kumagai, K.; Zhang, B.; Saku, K.; Arakawa, K. Cholesterol paradox in patients with paroxysmal atrial fibrillation. Cardiology 1999, 92, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Mourtzinis, G.; Kahan, T.; Bengtsson Bostrom, K.; Schioler, L.; Cedstrand Wallin, L.; Hjerpe, P.; Hasselstrom, J.; Manhem, K. Relation Between Lipid Profile and New-Onset Atrial Fibrillation in Patients With Systemic Hypertension (From the Swedish Primary Care Cardiovascular Database [SPCCD]). Am. J. Cardiol. 2018, 122, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Geng, J.; Ye, X.; Yuan, X.; Li, A.; Zhang, Z.; Xu, B.; Wang, Y. Change in lipid profile and risk of new-onset atrial fibrillation in patients with chronic heart failure: A 3-year follow-up observational study in a large Chinese hospital. Medicine 2018, 97, e12485. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gao, L.; Wang, Z.; Guan, B.; Guan, X.; Wang, B.; Han, X.; Xiao, X.; Waleed, K.B.; Chandran, C.; et al. Lipid profile and incidence of atrial fibrillation: A prospective cohort study in China. Clin. Cardiol. 2018, 41, 314–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Zhou, Q.; Shen, J.; Liu, G.; Zhou, W.; Wen, Y.; Luo, S. Lipid Profile and New-Onset Atrial Fibrillation in Patients With Acute ST-Segment Elevation Myocardial Infarction (An Observational Study in Southwest of China). Am. J. Cardiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Chen, N.; Wang, Q.; Zhuo, C.; Zhao, J.; Lv, N.; Huang, Y. Blood lipid levels and recurrence of atrial fibrillation after radiofrequency catheter ablation: a prospective study. J. Interv. Card. Electrophysiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Veronese, G.; Montomoli, J.; Schmidt, M.; Horvath-Puho, E.; Sorensen, H.T. Statin Use and Risk of Atrial Fibrillation or Flutter: A Population-based Case-Control Study. Am. J. Ther. 2015, 22, 186–194. [Google Scholar] [CrossRef]
- Zhou, X.; Du, J.L.; Yuan, J.; Chen, Y.Q. Statin therapy is beneficial for the prevention of atrial fibrillation in patients with coronary artery disease: a meta-analysis. Eur. J. Pharm. 2013, 707, 104–111. [Google Scholar] [CrossRef]
- Fauchier, L.; Clementy, N.; Babuty, D. Statin therapy and atrial fibrillation: systematic review and updated meta-analysis of published randomized controlled trials. Curr. Opin. Cardiol. 2013, 28, 7–18. [Google Scholar] [CrossRef]
- Alonso, A.; Yin, X.; Roetker, N.S.; Magnani, J.W.; Kronmal, R.A.; Ellinor, P.T.; Chen, L.Y.; Lubitz, S.A.; McClelland, R.L.; McManus, D.D.; et al. Blood lipids and the incidence of atrial fibrillation: the multi-ethnic study of atherosclerosis and the framingham heart study. J. Am. Heart Assoc. 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Kim, J.M.; Shin, D.G.; Kim, J.R.; Cho, K.H. Relation of atrial fibrillation (AF) and change of lipoproteins: male patients with AF exhibited severe pro-inflammatory and pro-atherogenic properties in lipoproteins. Clin. Biochem. 2014, 47, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Lee, J.H.; Kim, J.R.; Shin, D.G.; Lee, S.H.; Cho, K.H. Female patients with atrial fibrillation have increased oxidized and glycated lipoprotein properties and lower apolipoprotein A-I expression in HDL. Int. J. Mol. Med. 2011, 27, 841–849. [Google Scholar] [PubMed] [Green Version]
- Mittendorfer, B.; Yoshino, M.; Patterson, B.W.; Klein, S. VLDL Triglyceride Kinetics in Lean, Overweight, and Obese Men and Women. J. Clin. Endocrinol. Metab. 2016, 101, 4151–4160. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.V., 3rd; Patterson, B.W.; Okunade, A.; Klein, S. Fatty acid and very low density lipoprotein metabolism in obese African American and Caucasian women with type 2 diabetes. J. Lipid Res. 2012, 53, 2767–2772. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C.; Watts, G.F.; Nguyen, M.N.; Barrett, P.H. Apolipoproteins C-III and A-V as predictors of very-low-density lipoprotein triglyceride and apolipoprotein B-100 kinetics. Arter. Thromb. Vasc. Biol. 2006, 26, 590–596. [Google Scholar] [CrossRef] [Green Version]
- Castellani, L.W.; Nguyen, C.N.; Charugundla, S.; Weinstein, M.M.; Doan, C.X.; Blaner, W.S.; Wongsiriroj, N.; Lusis, A.J. Apolipoprotein AII is a regulator of very low density lipoprotein metabolism and insulin resistance. J. Biol. Chem. 2008, 283, 11633–11644. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, K.; Tokita, Y.; Tanaka, A.; Takahashi, S. The VLDL receptor plays a key role in the metabolism of postprandial remnant lipoproteins. Clin. Chim. Acta 2019, 495, 382–393. [Google Scholar] [CrossRef]
- Yakovlev, S.; Belkin, A.M.; Chen, L.; Cao, C.; Zhang, L.; Strickland, D.K.; Medved, L. Anti-VLDL receptor monoclonal antibodies inhibit fibrin-VLDL receptor interaction and reduce fibrin-dependent leukocyte transmigration. Thromb. Haemost. 2016, 116, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Galati, F.; Colonna, P.; Galati, A.; Ciardiello, C.; Bozzetti, M.P.; Massari, S. CETP TaqIB Polymorphism, Serum Lipid Levels And Risk Of Atrial Fibrillation: A Case-Control Study. J. Atr. Fibrillation 2014, 6, 964. [Google Scholar]
- Niu, Y.G.; Evans, R.D. Very-low-density lipoprotein: complex particles in cardiac energy metabolism. J. Lipids 2011, 2011, 189876. [Google Scholar] [CrossRef]
- Masuzaki, H.; Jingami, H.; Matsuoka, N.; Nakagawa, O.; Ogawa, Y.; Mizuno, M.; Yoshimasa, Y.; Yamamoto, T.; Nakao, K. Regulation of very-low-density lipoprotein receptor in hypertrophic rat heart. Circ. Res. 1996, 78, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, K.G.; Hiyama, Y.; Hu, Y.; Huggins, L.A.; Ramakrishnan, R.; Abumrad, N.A.; Shulman, G.I.; Blaner, W.S.; Goldberg, I.J. Chylomicron- and VLDL-derived lipids enter the heart through different pathways: in vivo evidence for receptor- and non-receptor-mediated fatty acid uptake. J. Biol. Chem. 2010, 285, 37976–37986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitman, S.C.; Daugherty, A.; Post, S.R. Macrophage colony-stimulating factor rapidly enhances beta-migrating very low density lipoprotein metabolism in macrophages through activation of a Gi/o protein signaling pathway. J. Biol. Chem. 2000, 275, 35807–35813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.; Kawarabayasi, Y.; Nakai, T.; Sakai, J.; Yamamoto, T. Rabbit very low density lipoprotein receptor: a low density lipoprotein receptor-like protein with distinct ligand specificity. Proc. Natl. Acad. Sci. USA 1992, 89, 9252–9256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, S.; Singh-Bist, A.; Natu, V.; Kraemer, F.B. Dietary regulation of the very low density lipoprotein receptor in mouse heart and fat. Horm. Metab. Res. 1997, 29, 524–529. [Google Scholar] [CrossRef]
- Argraves, K.M.; Battey, F.D.; MacCalman, C.D.; McCrae, K.R.; Gafvels, M.; Kozarsky, K.F.; Chappell, D.A.; Strauss, J.F., 3rd; Strickland, D.K. The very low density lipoprotein receptor mediates the cellular catabolism of lipoprotein lipase and urokinase-plasminogen activator inhibitor type I complexes. J. Biol. Chem. 1995, 270, 26550–26557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagyu, H.; Lutz, E.P.; Kako, Y.; Marks, S.; Hu, Y.; Choi, S.Y.; Bensadoun, A.; Goldberg, I.J. Very low density lipoprotein (VLDL) receptor-deficient mice have reduced lipoprotein lipase activity. Possible causes of hypertriglyceridemia and reduced body mass with VLDL receptor deficiency. J. Biol. Chem. 2002, 277, 10037–10043. [Google Scholar] [CrossRef] [Green Version]
- Wyne, K.L.; Pathak, K.; Seabra, M.C.; Hobbs, H.H. Expression of the VLDL receptor in endothelial cells. Arter. Thromb. Vasc. Biol. 1996, 16, 407–415. [Google Scholar] [CrossRef]
- Perman, J.C.; Bostrom, P.; Lindbom, M.; Lidberg, U.; StAhlman, M.; Hagg, D.; Lindskog, H.; Scharin Tang, M.; Omerovic, E.; Mattsson Hulten, L.; et al. The VLDL receptor promotes lipotoxicity and increases mortality in mice following an acute myocardial infarction. J. Clin. Invest. 2011, 121, 2625–2640. [Google Scholar] [CrossRef] [Green Version]
- Papageorgiou, I.; Viglino, C.; Brulhart-Meynet, M.C.; James, R.W.; Lerch, R.; Montessuit, C. Impaired stimulation of glucose transport in cardiac myocytes exposed to very low-density lipoproteins. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid Use and Misuse by the Heart. Circ. Res. 2016, 118, 1736–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elezaby, A.; Sverdlov, A.L.; Tu, V.H.; Soni, K.; Luptak, I.; Qin, F.; Liesa, M.; Shirihai, O.S.; Rimer, J.; Schaffer, J.E.; et al. Mitochondrial remodeling in mice with cardiomyocyte-specific lipid overload. J. Mol. Cell. Cardiol. 2015, 79, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drosatos, K.; Schulze, P.C. Cardiac lipotoxicity: molecular pathways and therapeutic implications. Curr. Heart Fail. Rep. 2013, 10, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Weijer, T.; Schrauwen-Hinderling, V.B.; Schrauwen, P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc Res. 2011, 92, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaffer, J.E. Lipotoxicity: when tissues overeat. Curr. Opin. Lipidol. 2003, 14, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Bukowska, A.; Schild, L.; Keilhoff, G.; Hirte, D.; Neumann, M.; Gardemann, A.; Neumann, K.H.; Röhl, F.W.; Huth, C.; Goette, A.; et al. Mitochondrial Dysfunction and Redox Signaling in Atrial Tachyarrhythmia. Exp. Biol. Med. 2008, 233, 558–574. [Google Scholar] [CrossRef]
- Opacic, D.; van Bragt, K.A.; Nasrallah, H.M.; Schotten, U.; Verheule, S. Atrial metabolism and tissue perfusion as determinants of electrical and structural remodelling in atrial fibrillation. Cardiovasc. Res. 2016, 109, 527–541. [Google Scholar] [CrossRef] [Green Version]
- Marfella, R.; Di Filippo, C.; Portoghese, M.; Barbieri, M.; Ferraraccio, F.; Siniscalchi, M.; Cacciapuoti, F.; Rossi, F.; D’Amico, M.; Paolisso, G. Myocardial lipid accumulation in patients with pressure-overloaded heart and metabolic syndrome. J. Lipid Res. 2009, 50, 2314–2323. [Google Scholar] [CrossRef] [Green Version]
- Nestel, P.J.; Poyser, A. Cholesterol content of the human atrium is related to plasma lipoprotein levels. Atherosclerosis 1978, 30, 177–183. [Google Scholar] [CrossRef]
- Chen, C.H.; Lu, J.; Chen, S.H.; Huang, R.Y.; Yilmaz, H.R.; Dong, J.; Elayda, M.A.; Dixon, R.A.; Yang, C.Y. Effects of electronegative VLDL on endothelium damage in metabolic syndrome. Diabetes Care 2012, 35, 648–653. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C.; Lin, H.T.; Ke, L.Y.; Wei, C.; Hsiao, Y.L.; Chu, C.S.; Lai, W.T.; Shin, S.J.; Chen, C.H.; Sheu, S.H.; et al. VLDL from Metabolic Syndrome Individuals Enhanced Lipid Accumulation in Atria with Association of Susceptibility to Atrial Fibrillation. Int J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.C.; Chen, C.C.; Tsai, W.C.; Lin, H.T.; Shiao, Y.L.; Sheu, S.H.; Wu, B.N.; Chen, C.H.; Lai, W.T. Very-Low-Density Lipoprotein of Metabolic Syndrome Modulates Gap Junctions and Slows Cardiac Conduction. Sci. Rep. 2017, 7, 12050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issad, T. O-GlcNAcylation of connexin 40: A sweet connection between diabetes and endothelial cell dysfunction? Focus on “O-GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice”. Am. J. Physiol. 2015, 309, C590–C592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Hojayev, B.; Jiang, N.; Wang, Z.V.; Tandan, S.; Rakalin, A.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. STIM1-dependent store-operated Ca2+ entry is required for pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2012, 52, 136–147. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, P. Socking It to cardiac hypertrophy: STIM1-mediated Ca2+ entry in the cardiomyocyte. Circulation 2011, 124, 766–768. [Google Scholar] [CrossRef] [Green Version]
- Shiou, Y.L.; Lin, H.T.; Ke, L.Y.; Wu, B.N.; Shin, S.J.; Chen, C.H.; Tsai, W.C.; Chu, C.S.; Lee, H.C. Very Low-Density Lipoproteins of Metabolic Syndrome Modulates STIM1, Suppresses Store-Operated Calcium Entry, and Deranges Myofilament Proteins in Atrial Myocytes. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Kurosaki, T.; Baba, Y. Ca2+ signaling and STIM1. Prog. Biophys. Mol. Biol. 2010, 103, 51–58. [Google Scholar] [CrossRef]
- Frazier, A.H.; Ramirez-Correa, G.A.; Murphy, A.M. Molecular mechanisms of sarcomere dysfunction in dilated and hypertrophic cardiomyopathy. Prog. Pediatr. Cardiol. 2011, 31, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Machackova, J.; Barta, J.; Dhalla, N.S. Myofibrillar remodelling in cardiac hypertrophy, heart failure and cardiomyopathies. Can. J. Cardiol. 2006, 22, 953–968. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C.; Wei, C.; Ke, L.Y.; Tsai, P.S.; Lin, H.T.; Shiao, Y.L.; Wu, B.N.; Chen, C.H.; Sheu, S.H. Atherogenic Very-Low-Density Lipoprotein Shortens Atrial Action Potential Duration by Increasing Potassium Currents and Calcium Transient. Biophys. J. 2015, 108, 586a. [Google Scholar]
- Nakajima, K.; Tokita, Y.; Tanaka, A. Hypothesis II: The majority of VLDL-apoB48 remnants in postprandial plasma are derived from the liver, not from the intestine. Clin. Chim. Acta 2019, 490, 12–16. [Google Scholar] [CrossRef]
- Frykman, P.K.; Brown, M.S.; Yamamoto, T.; Goldstein, J.L.; Herz, J. Normal plasma lipoproteins and fertility in gene-targeted mice homozygous for a disruption in the gene encoding very low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 1995, 92, 8453–8457. [Google Scholar] [CrossRef] [Green Version]
- Li, C.L.; Chu, C.H.; Lee, H.C.; Chou, M.C.; Liu, C.K.; Chen, C.H.; Ke, L.Y.; Chen, S.L. Immunoregulatory effects of very low density lipoprotein from healthy individuals and metabolic syndrome patients on glial cells. Immunobiology 2019, 224, 632–637. [Google Scholar] [CrossRef]
- Luchoomun, J.; Zhou, Z.; Bakillah, A.; Jamil, H.; Hussain, M.M. Assembly and secretion of VLDL in nondifferentiated Caco-2 cells stably transfected with human recombinant ApoB48 cDNA. Arter. Thromb. Vasc. Biol. 1997, 17, 2955–2963. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-C.; Lin, Y.-H. The Pathogenic Role of Very Low Density Lipoprotein on Atrial Remodeling in the Metabolic Syndrome. Int. J. Mol. Sci. 2020, 21, 891. https://doi.org/10.3390/ijms21030891
Lee H-C, Lin Y-H. The Pathogenic Role of Very Low Density Lipoprotein on Atrial Remodeling in the Metabolic Syndrome. International Journal of Molecular Sciences. 2020; 21(3):891. https://doi.org/10.3390/ijms21030891
Chicago/Turabian StyleLee, Hsiang-Chun, and Yi-Hsiung Lin. 2020. "The Pathogenic Role of Very Low Density Lipoprotein on Atrial Remodeling in the Metabolic Syndrome" International Journal of Molecular Sciences 21, no. 3: 891. https://doi.org/10.3390/ijms21030891
APA StyleLee, H.-C., & Lin, Y.-H. (2020). The Pathogenic Role of Very Low Density Lipoprotein on Atrial Remodeling in the Metabolic Syndrome. International Journal of Molecular Sciences, 21(3), 891. https://doi.org/10.3390/ijms21030891