Untargeted Metabolomics for Metabolic Diagnostic Screening with Automated Data Interpretation Using a Knowledge-Based Algorithm

 , ,
, , _Verhoeven-Duif.png)

Abstract
:1. Introduction
2. Results
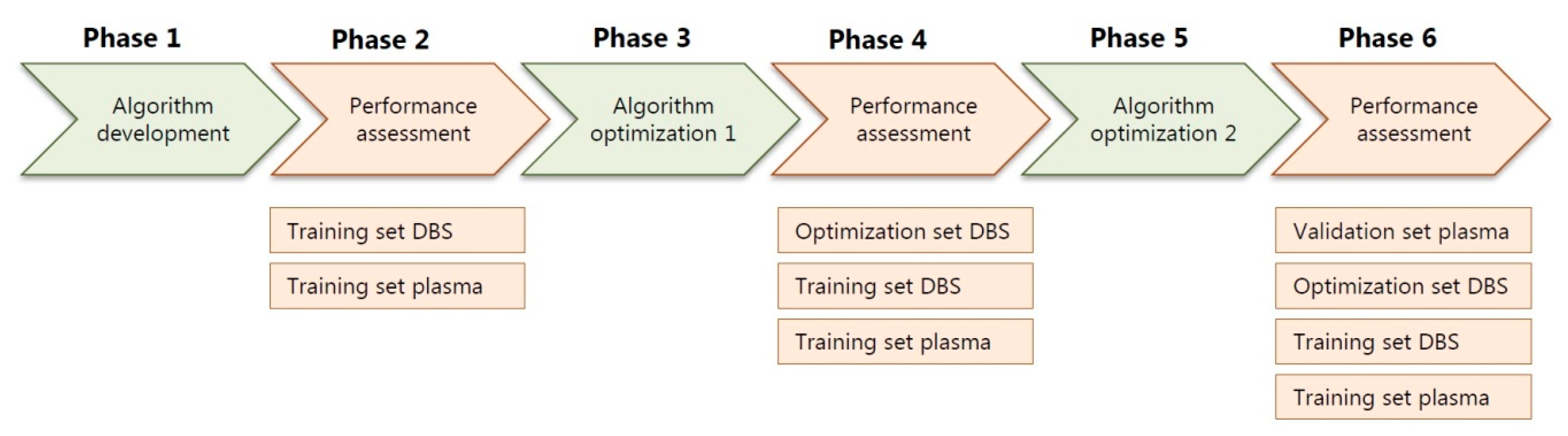
2.1. Training Sets, Optimization Sets and Patient Samples
2.2. Validation Set, Patient Samples
2.3. Performance Assessment of All Patient Samples
2.4. Control Samples
2.5. R Shiny App to Aid Insight in Automated Data Interpretation
3. Discussion
4. Materials and Methods
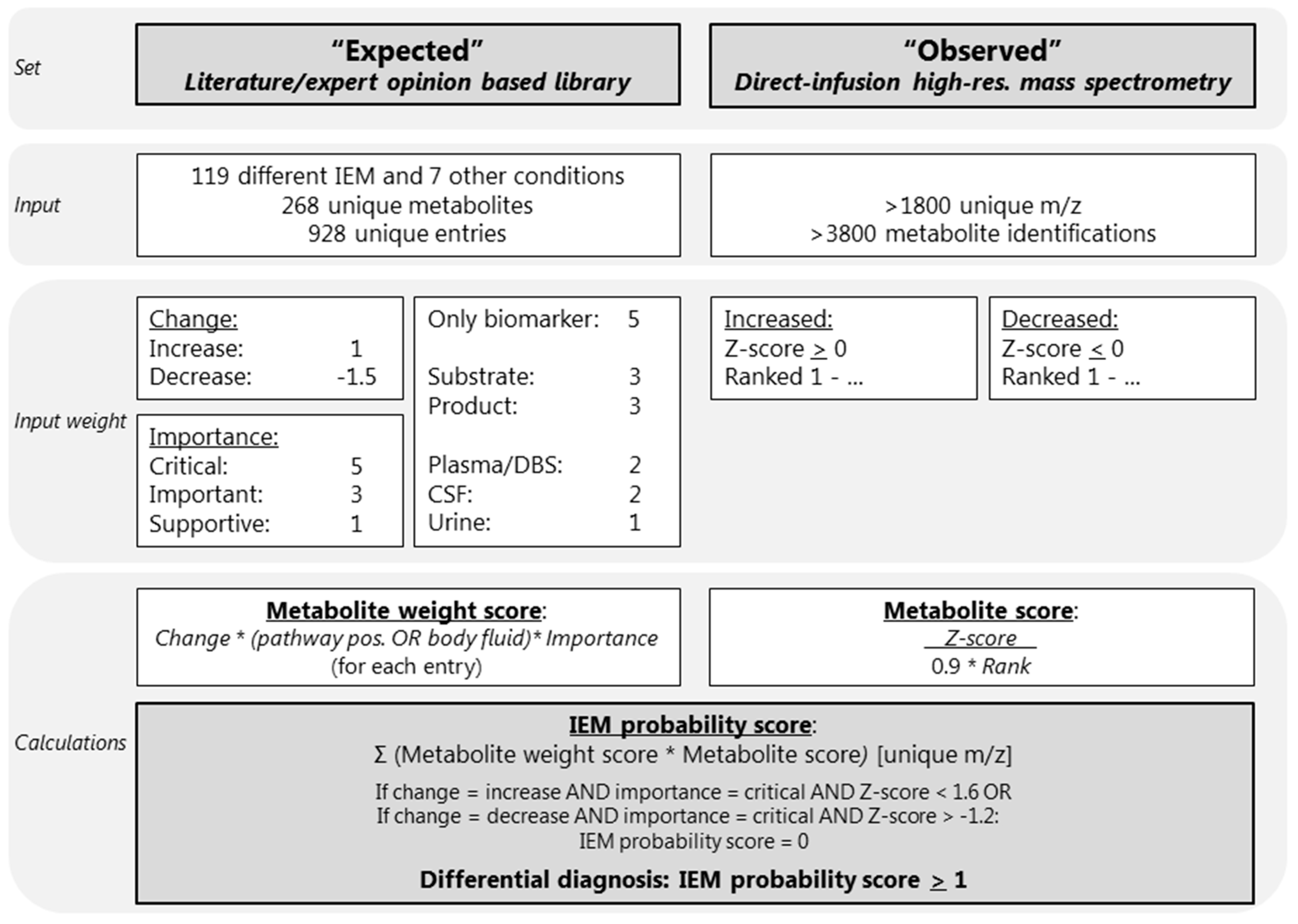
4.1. Development of an IEM-Panel and Automated Data Interpretation
4.2. Patient Inclusion
4.3. Sample Inclusion
4.4. Input Parameter: Expected Library
4.5. Input Parameter: Observed Metabolite Alterations Using Untargeted Metabolomics
4.6. Automated Data Interpretation
4.7. R Shiny App to Aid Automated Data Interpretation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CSF | cerebrospinal fluid |
| DBS | dried blood spots |
| DD | differential diagnosis |
| HMDB | Human Metabolome Database |
| IEM | inborn error of metabolism |
| m/z | mass to charge ratio |
| NGMS | next generation metabolic screening |
| OMIM | Online Mendelian Inheritance in Man |
References
- Miller, M.J.; Kennedy, A.D.; Eckhart, A.D.; Burrage, L.C.; Wulff, J.E.; Miller, L.A.; Milburn, M.V.; Ryals, J.A.; Beaudet, A.L.; Sun, Q.; et al. Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. J. Inherit. Metab. Dis. 2015, 38, 1029–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coene, K.L.M.; Kluijtmans, L.A.J.; van der Heeft, E. Next-generation metabolic screening: Targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients. J. Inherit. Metab. Dis. 2018, 41, 337–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haijes, H.A.; Willemsen, M.; van der Ham, M.; Gerrits, J.; Pras-Raves, M.L.; Prinsen, H.C.M.T.; van Hasselt, P.M.; de Sain-van der Velden, M.G.M.; Verhoeven-Duif, N.M.; Jans, J.J.M. Direct infusion based metabolomics identifies metabolic disease in patients’ dried blood spots and plasma. Metabolites 2019, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haijes, H.A.; van der Ham, M.; Gerrits, J.; van Hasselt, P.M.; Prinsen, H.C.M.T.; de Sain-van der Velden, M.G.M.; Verhoeven-Duif, N.M.; Jans, J.J.M. Direct-infusion based metabolomics unveils biochemical profiles of inborn errors of metabolism in cerebrospinal fluid. Mol. Genet. Metab. 2019, 127, 51–57. [Google Scholar] [CrossRef] [PubMed]
- European Research Network for Evaluation and Improvement of Screening, Diagnosis and Treatment of Inherited Disorders of Metabolism. Annual Report 2017. Available online: https://www.erndim.org/store/docs/DOC4322ERNDIMAnnualRepor-HETAEBUV245881-19-10-2018.pdf (accessed on 31 January 2020).
- Smedley, D.; Robinson, P.N. Phenotype-driven strategies for exome prioritization of human Mendelian disease genes. Genome Med. 2015, 7, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aitken, S.; Firth, H.V.; McRae, J.; Halachev, M.; Kini, U.; Parker, M.J.; Lees, M.M.; Lachlan, K.; Sarkar, A.; Joss, S.; et al. Finding diagnostically useful patterns in quantitative phenotypic data. Am. J. Hum. Genet. 2019, 105, 933–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haijes, H.A.; de Sain-van der Velden, M.G.M.; Prinsen, H.C.M.T.; Willems, A.P.; van der Ham, M.; Gerrits, J.; Couse, M.H.; Friedman, J.M.; van Karnebeek, C.D.M.; Selby, K.A.; et al. Aspartylglycosamine is a biomarker for NGLY1-CDDG, a congenital disorder of deglycosylation. Mol. Genet Metab. 2019, 127, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Abela, L.; Simmons, L.; Steindl, K.; Schmitt, B.; Mastrangelo, M.; Joset, P.; Papuc, M.; Sticht, H.; Baumer, A.; Crowther, L.M.; et al. N(8)-acetylspermidine as a potential biomarker for Snyder-Robinson syndrome identified by clinical metabolomics. J. Inherit Metab. Dis. 2016, 39, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.D.; Pappan, K.L.; Donti, T.; Delgado, M.R.; Shinawi, M.; Pearson, T.S.; Lalani, S.R.; Craigen, W.E.; Sutton, V.R.; Evans, A.M.; et al. 2-Pyrrolidinone and succinimide as clinical screening biomarkers for GABA-transaminase deficiency: Anti-seizure medications impact accurate diagnosis. Front Neurosci. 2019, 13, 394. [Google Scholar] [CrossRef] [PubMed]
- Burrage, L.C.; Thistlethwaite, L.; Stroup, B.M.; Sun, Q.; Miller, M.J.; Nagamani, S.C.S.; Craigen, W.; Scaglia, F.; Sutton, V.R.; Graham, B.; et al. Untargeted metabolomics profiling reveals multiple pathway perturbations and new clinical biomarkers in urea cycle disorders. Genet. Med. 2019, 21, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; van Karnebeek, C.D.M.; Vockley, J.; Blau, N. A proposed nosology of inborn errors of metabolism. Genet Med. 2019, 21, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Domínguez, R.; Castilla-Quintero, R.; García-Barrera, T.; Gómez-Ariza, J.L. Development of a metabolomics approach based on urine samples and direct infusion mass spectrometry. Anal. Biochem. 2014, 465, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, 801–807. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Training Sets | Optimization Set | Validation Set | ||
|---|---|---|---|---|
| Matrix | DBS | Plasma | DBS | Plasma |
| Samples | 110 | 86 | 96 | 115 |
| Patients | 42 | 38 | 96 | 115 |
| IEM | 23 | 21 | 53 | 58 |
| Correct IEM in DD (n; %) | 86/110; 78% | 68/86; 79% | 68/96; 71% | 83/115; 72% |
| Correct IEM in top 3 of DD (n; %) | 74/110; 67% | 36/86; 42% | 60/96; 63% | 65/115; 57% |
| Correct IEM ranked first (n; %) | 46/110; 42% | 28/86; 33% | 38/96; 40% | 43/115; 37% |
| Length DD (median; (5th–95th)) | 8; [2–14] | 12; [3–25] | 8; [1–23] | 10; [3–22] |
| Training Sets | Optimization Set | Validation Set | ||
|---|---|---|---|---|
| Matrix | DBS | Plasma | DBS | Plasma |
| Samples | 105 | 84 | 66 | 83 |
| Individuals | 30 | 28 | 48 | 28 |
| Length DD (median; (5th–95th)) | 2; (0–12) | 3; (0–11) | 2; (0–8) | 3; (0–10) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haijes, H.A.; van der Ham, M.; Prinsen, H.C.M.T.; Broeks, M.H.; van Hasselt, P.M.; de Sain-van der Velden, M.G.M.; Verhoeven-Duif, N.M.; Jans, J.J.M. Untargeted Metabolomics for Metabolic Diagnostic Screening with Automated Data Interpretation Using a Knowledge-Based Algorithm. Int. J. Mol. Sci. 2020, 21, 979. https://doi.org/10.3390/ijms21030979
Haijes HA, van der Ham M, Prinsen HCMT, Broeks MH, van Hasselt PM, de Sain-van der Velden MGM, Verhoeven-Duif NM, Jans JJM. Untargeted Metabolomics for Metabolic Diagnostic Screening with Automated Data Interpretation Using a Knowledge-Based Algorithm. International Journal of Molecular Sciences. 2020; 21(3):979. https://doi.org/10.3390/ijms21030979
Chicago/Turabian StyleHaijes, Hanneke A., Maria van der Ham, Hubertus C.M.T. Prinsen, Melissa H. Broeks, Peter M. van Hasselt, Monique G.M. de Sain-van der Velden, Nanda M. Verhoeven-Duif, and Judith J.M. Jans. 2020. "Untargeted Metabolomics for Metabolic Diagnostic Screening with Automated Data Interpretation Using a Knowledge-Based Algorithm" International Journal of Molecular Sciences 21, no. 3: 979. https://doi.org/10.3390/ijms21030979
APA StyleHaijes, H. A., van der Ham, M., Prinsen, H. C. M. T., Broeks, M. H., van Hasselt, P. M., de Sain-van der Velden, M. G. M., Verhoeven-Duif, N. M., & Jans, J. J. M. (2020). Untargeted Metabolomics for Metabolic Diagnostic Screening with Automated Data Interpretation Using a Knowledge-Based Algorithm. International Journal of Molecular Sciences, 21(3), 979. https://doi.org/10.3390/ijms21030979