Using Transcriptomic Analysis to Assess Double-Strand Break Repair Activity: Towards Precise in Vivo Genome Editing

 ,
, 
 ,
,  , and
, and
Abstract
:1. Introduction
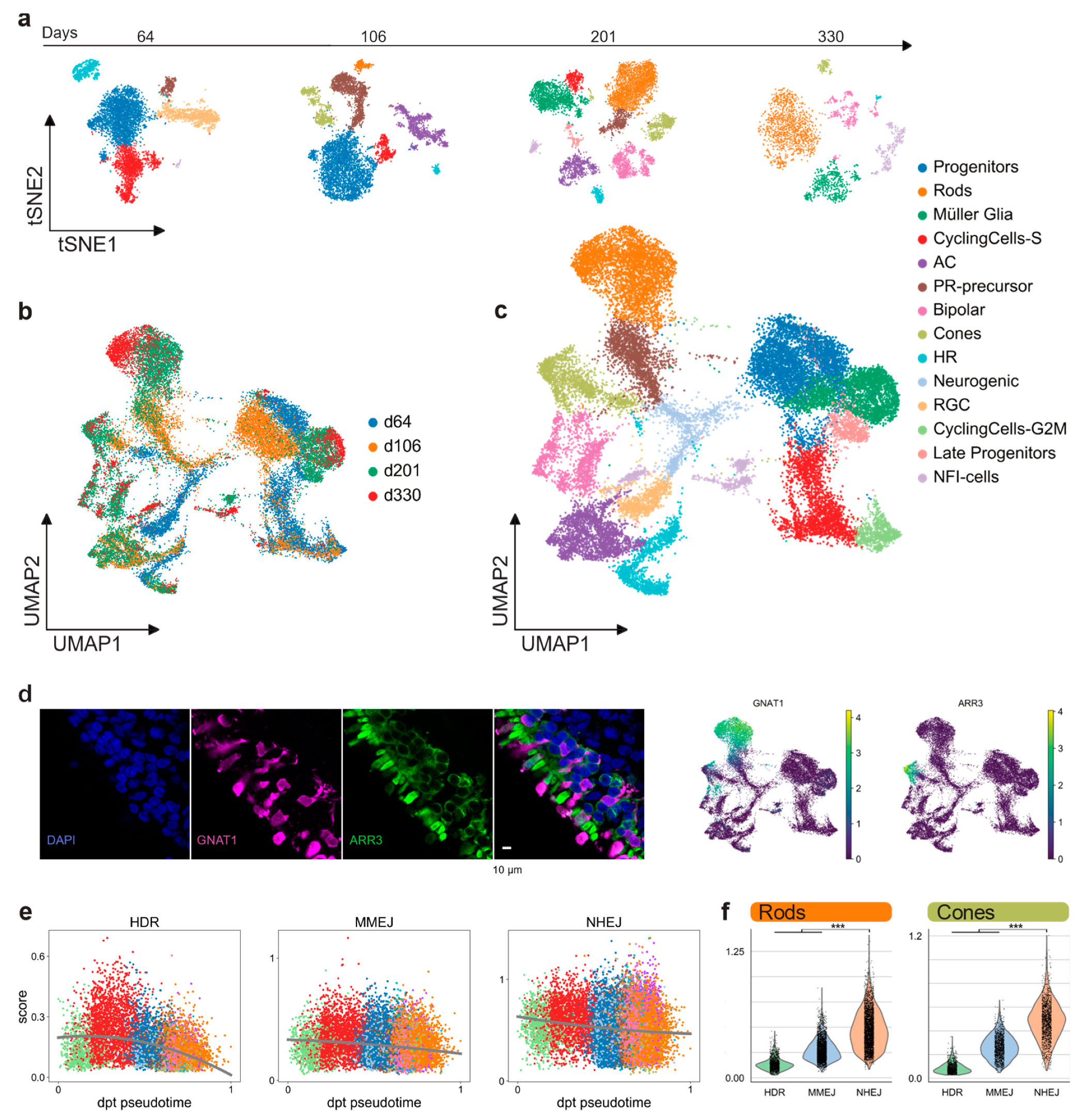
2. Results
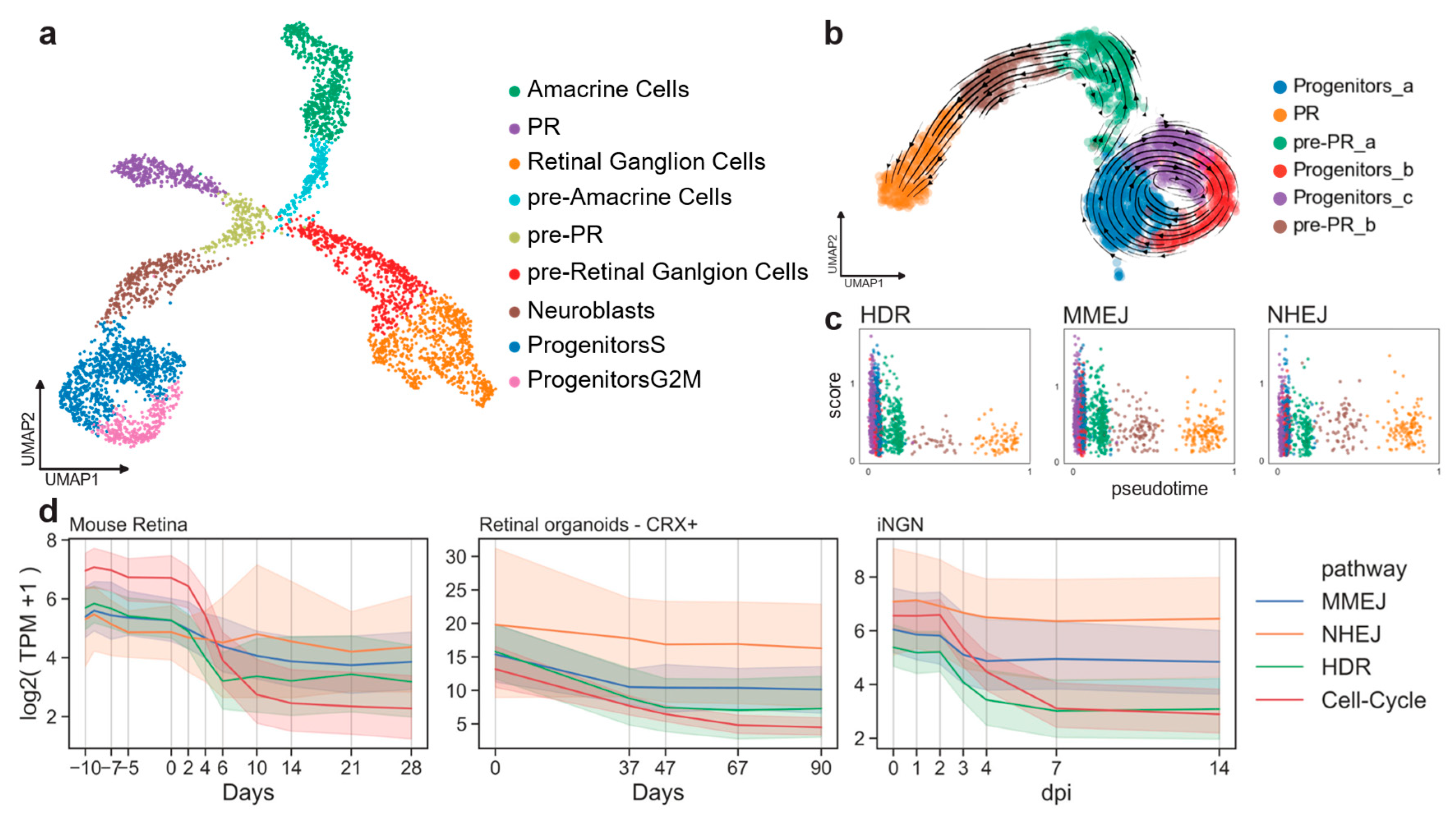
2.1. Homology Repair Correlates with Cell-Cycle Activity
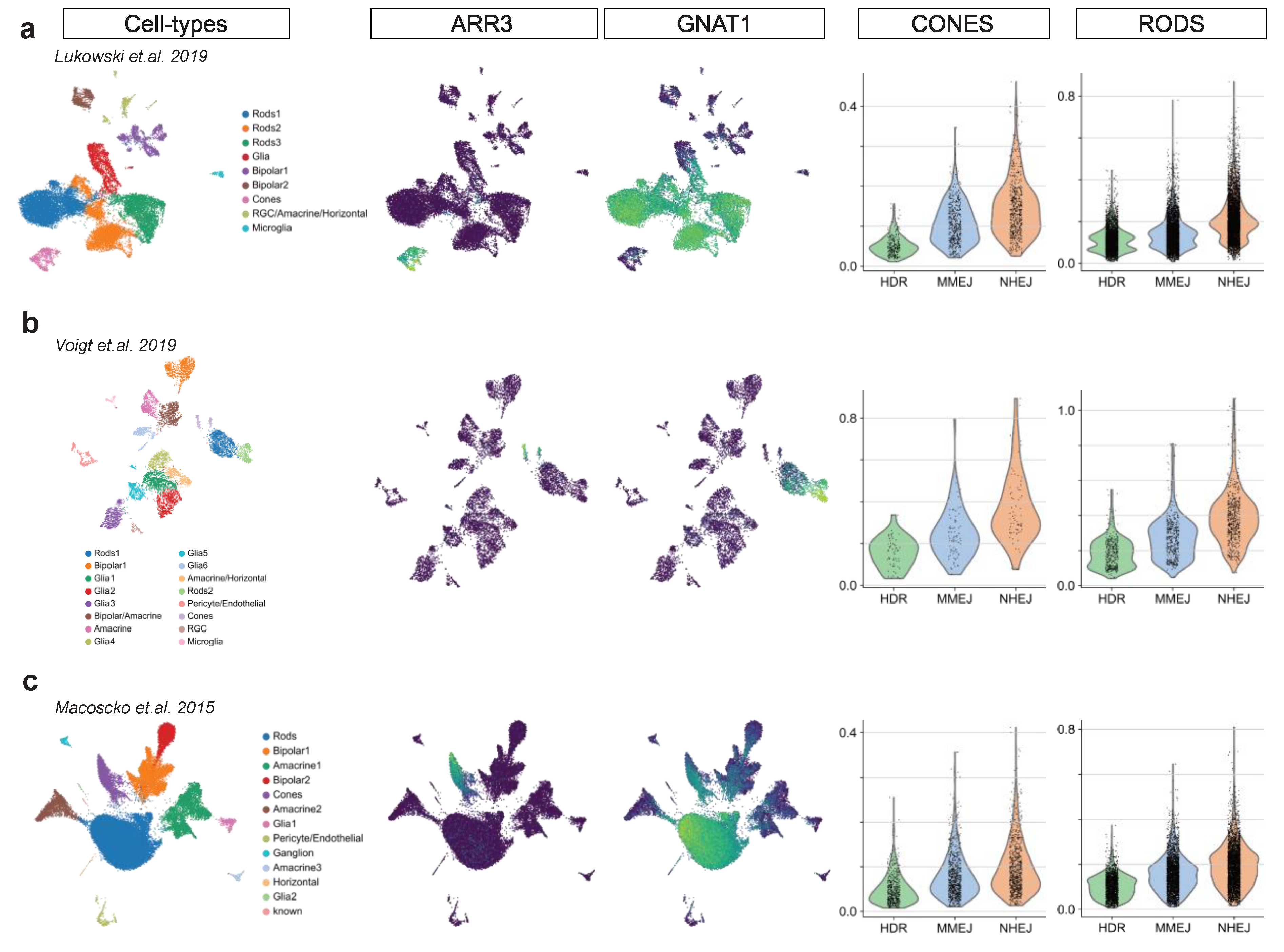
2.2. Photoreceptor DSB Pathway Activity
2.3. Comparison of Different Mammal Species and in Vitro Testbeds
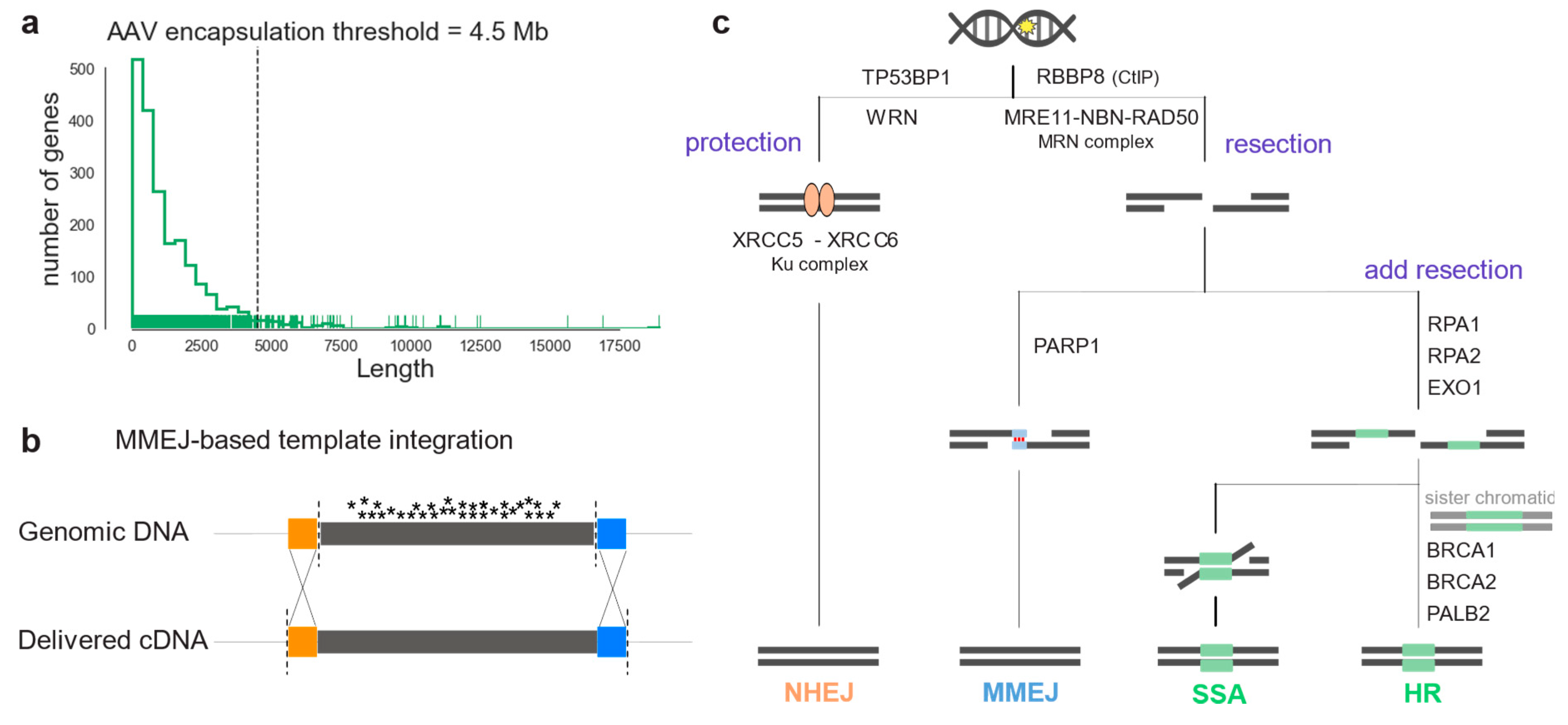
3. Discussion
4. Materials and Methods
4.1. Curated Gene List for DSB Pathways
4.2. Datasets
4.3. Computational Analysis of RNA-Seq Data
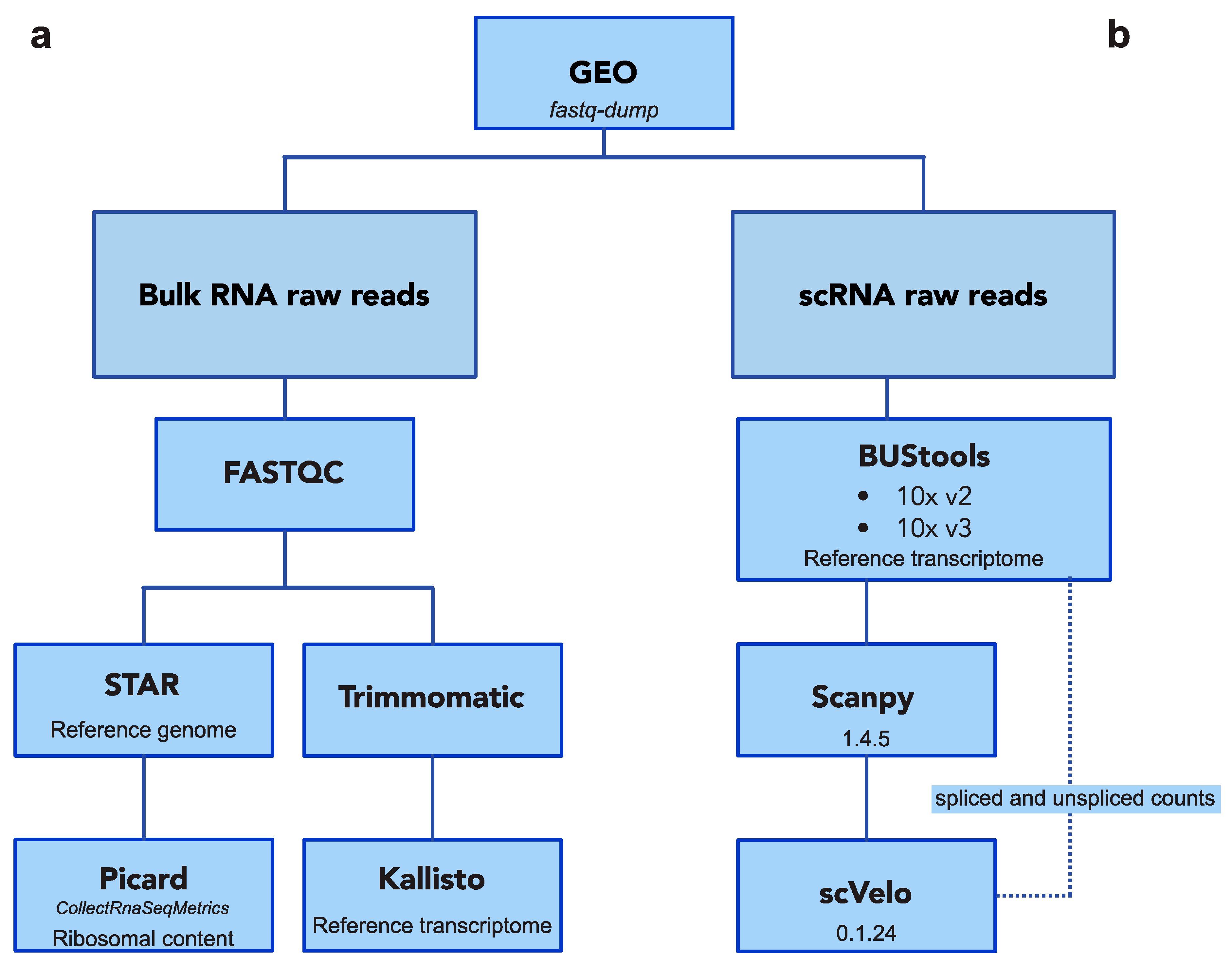
4.3.1. Bulk RNA-Seq Processing Pipeline
4.3.2. Single-Cell RNA-Seq Processing Pipeline
4.3.3. Mouse Embryo scRNA-Seq Analysis
4.3.4. Cell-Cycle and DSB Pathway Correlation in Bulk Time Course RNA-Seq
4.3.5. Comparison of Mammal Retina and iNGN
4.3.6. Organoid scRNA-Seq Analysis
4.4. Generation of hiPSC-Derived Retinal Organoids
4.5. Preparation and Sequencing of hiPSC-Derived Retinal Organoid Single-Cell cDNA Libraries
4.6. Retinal Organoid Immunocytochemistry
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IRD | Inherited retinal disease |
| DSB | Double-strand breaks |
| scRNA-seq | Single-cell RNA sequencing |
| PR | Photoreceptors |
| hiPSC | Human induced pluripotent stem cells |
| iNGN | Small molecule-inducible neurogenin hiPSC line |
| t-sne | t-distributed stochastic neighbor embedding |
| UMAP | Uniform manifold approximation and projection |
| TPM | Transcripts per million |
Appendix A




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell-Type | Markers |
|---|---|
| Stem cells | CCND1, FGF15, FOS, HES1, LHX2, NANOG, SFRP2, SOX2 |
| Neurogenic | ATOH7, HES6, NEUROG2, OLIG2 |
| PR-precursor | CRX, OTX2, NEUROD1, PRDM1 |
| Cones | ARR3, GNAT2, GUCA1C, GNGT2, GUCA1A, OPN1SW, OPN1MW, OPN1LW, PDE6H |
| Rods | CNGA1, GNAT1, GNGT1, GNGA1, NR2E3, NRL, PDE6A, PPEF2, RHO |
| RGC | GAP43, NEFL, NEFM, NRN1, POU4F2, RPBMS, SLC17A6, SNCG, THY |
| AC | CALB1, CHAT, C1QL2, GAD1, NRXN2, PAX6, TFAP2A, TFAP2B |
| Horizontal cells | LHX1, ONECUT1, ONECUT2 |
| Bipolar cells | CAMK2B, GRM6, PROX1, TMEM215, TRNP1, TRPM1, VSX1, VSX2 |
| Müller Glia | APOE, AQP4, CLU, GLUL, RLBP1 |
| Microglia | AIF1, C1QA, HLA-DPA1, HLA-DPB1, HLA-DRA, TMEM119 |
| Endothelial | ADAMTS9, RGS5 |
| Pericyte | VWF |
References
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa Prevalence and inheritance patterns. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010, 29, 335–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cideciyan, A.V.; Aleman, T.S.; Boye, S.L.; Schwartz, S.B.; Kaushal, S.; Roman, A.J.; Pang, J.-J.; Sumaroka, A.; Windsor, E.A.M.; Wilson, J.M.; et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc. Natl. Acad. Sci. USA 2008, 105, 15112–15117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziccardi, L.; Cordeddu, V.; Gaddini, L.; Matteucci, A.; Parravano, M.; Albedi, M.-; Varano, M.; Malchiodi-Albedi, F. Gene therapy in retinal dystrophies. Int. J. Mol. Sci. 2019, 20, 5722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, L.S.; Turunen, H.T.; Wassmer, S.J.; Luna-Velez, M.V.; Xiao, R.; Bennett, J.; Vandenberghe, L.H. Evaluating efficiencies of dual AAV approaches for retinal targeting. Front. Mol. Neurosci. 2017, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Trapani, I.; Colella, P.; Sommella, A.; Iodice, C.; Cesi, G.; de Simone, S.; Marrocco, E.; Rossi, S.; Giunti, M.; Palfi, A.; et al. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol. Med. 2014, 6, 194–211. [Google Scholar] [CrossRef] [PubMed]
- Parmeggiani, F.; Barbaro, V.; De Nadai, K.; Lavezzo, E.; Toppo, S.; Chizzolini, M.; Palù, G.; Parolin, C.; Di Iorio, E. Identification of novel X-linked gain-of-function RPGR-ORF15 mutation in Italian family with retinitis pigmentosa and pathologic myopia. Sci. Rep. 2016, 6, 39179. [Google Scholar] [CrossRef]
- Wilson, J.H.; Wensel, T.G. The Nature of Dominant Mutations of Rhodopsin and Implications for Gene Therapy. Mol. Neurobiol. 2003, 28, 149–158. [Google Scholar] [CrossRef]
- Schlegel, J.; Hoffmann, J.; Röll, D.; Müller, B.; Günther, S.; Zhang, W.; Janise, A.; Vössing, C.; Fühler, B.; Neidhardt, J.; et al. Toward genome editing in X-linked RP—development of a mouse model with specific treatment relevant features. Transl. Res. 2018, 203, 57–72. [Google Scholar] [CrossRef] [Green Version]
- Yanik, M.; Ponnam, S.P.G.; Wimmer, T.; Trimborn, L.; Müller, C.; Gambert, I.; Ginsberg, J.; Janise, A.; Domicke, J.; Wende, W.; et al. Development of a Reporter System to Explore MMEJ in the Context of Replacing Large Genomic Fragments. Mol. Ther. Nucleic Acids 2018, 11, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.D.; Richardson, C.D.; Corn, J.E. Review Article|SERIES Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol. 2019, 21, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Gersbach, C.A. Genome-editing technologies for gene and cell therapy. Mol. Ther. 2016, 24, 430–446. [Google Scholar] [CrossRef] [Green Version]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma. 2018, 127, 187–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA Double-Strand Breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyaoka, Y.; Berman, J.R.; Cooper, S.B.; Mayerl, S.J.; Chan, A.H.; Zhang, B.; Karlin-Neumann, G.A.; Conklin, B.R. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci. Rep. 2016, 6, 23549. [Google Scholar] [CrossRef]
- Tran, N.-T.; Bashir, S.; Li, X.; Rossius, J.; Chu, V.T.; Rajewsky, K.; Kühn, R. Enhancement of Precise Gene Editing by the Association of Cas9 With Homologous Recombination Factors. Front. Genet. 2019, 10, 365. [Google Scholar] [CrossRef] [Green Version]
- Aird, E.; Lovendahl, K.N.; Martin, A.S.; Harris, R.S.; Gordon, W. Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Commun. Biol. 2018, 1, 54. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Mookherjee, S.; Chaitankar, V.; Hiriyanna, S.; Kim, J.-W.; Brooks, M.; Ataeijannati, Y.; Sun, X.; Dong, L.; Li, T.; et al. Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat. Commun. 2017, 8, 14716. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Belmonte, J.C.I. In vivo genome editing via the HITI method as a tool for gene therapy. J. Hum. Genet. 2018, 63, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Giudice, Q.L.; Leleu, M.; la Manno, G.; Fabre, P.J. Single-cell transcriptional logic of cell-fate specification and axon guidance in early-born retinal neurons. Development 2019, 146, dev178103. [Google Scholar] [CrossRef] [Green Version]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Manno, G.; Soldatov, R.; Zeisel, A.; Braun, E.; Hochgerner, H.; Petukhov, V.; Lidschreiber, K.; Kastriti, M.E.; Lönnerberg, P.; Furlan, A.; et al. RNA velocity of single cells. Nature 2018, 560, 494–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haghverdi, L.; Büttner, M.; Wolf, F.A.; Buettner, F.; Theis, F. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 2016, 13, 845–848. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Ratnapriya, R.; Brooks, M.J.; Chaitankar, V.; Wilken, M.S.; Zhang, C.; Starostik, M.; Gieser, L.; La Torre, A.; Nishio, M.; et al. Molecular Anatomy of the Developing Human Retina. Dev. Cell 2017, 43, 763–779. [Google Scholar] [CrossRef] [Green Version]
- Kaewkhaw, R.; Kaya, K.D.; Brooks, M.; Homma, K.; Zou, J.; Chaitankar, V.; Rao, M.; Swaroop, A. Transcriptome Dynamics of Developing Photoreceptors in Three-Dimensional Retina Cultures Recapitulates Temporal Sequence of Human Cone and Rod Differentiation Revealing Cell Surface Markers and Gene Networks. Stem Cells 2015, 33, 3504–3518. [Google Scholar] [CrossRef]
- Busskamp, V.; Lewis, N.E.; Guye, P.; Ng, A.H.; Shipman, S.L.; Byrne, S.M.; E Sanjana, N.; Murn, J.; Li, Y.; Li, S.; et al. Rapid neurogenesis through transcriptional activation in human stem cells. Mol. Syst. Biol. 2014, 10, 760. [Google Scholar] [CrossRef]
- Kutsche, L.K.; Gysi, D.M.; Fallmann, J.; Lenk, K.; Petri, R.; Swiersy, A.; Klapper, S.D.; Pircs, K.; Khattak, S.; Stadler, P.F.; et al. Combined Experimental and System-Level Analyses Reveal the Complex Regulatory Network of miR-124 during Human Neurogenesis. Cell Syst. 2018, 7, 438–452. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.; Wernersson, R.; Jensen, L.J. Cyclebase 3.0: A multi-organism database on cell-cycle regulation and phenotypes. Nucleic Acids Res. 2015, 43, D1140–D1144. [Google Scholar] [CrossRef]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.-H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, B.S.; Stein-O’Brien, G.L.; Shiau, F.; Cannon, G.H.; Davis-Marcisak, E.; Sherman, T.; Santiago, C.P.; Hoang, T.V.; Rajaii, F.; James-Esposito, R.E.; et al. Single-Cell RNA-Seq Analysis of Retinal Development Identifies NFI Factors as Regulating Mitotic Exit and Late-Born Cell Specification. Neuron 2019, 102, 1111–1126. [Google Scholar] [CrossRef] [PubMed]
- Voigt, A.; Whitmore, S.S.; Flamme-Wiese, M.; Riker, M.; Wiley, L.; Tucker, B.; Stone, E.; Mullins, R.; Scheetz, T.E. Molecular characterization of foveal versus peripheral human retina by single-cell RNA sequencing. Exp. Eye Res. 2019, 184, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Lukowski, S.; Lo, C.Y.; A Sharov, A.; Nguyen, Q.; Fang, L.; Hung, S.S.; Zhu, L.; Zhang, T.; Grünert, U.; Nguyen, T.; et al. A single-cell transcriptome atlas of the adult human retina. EMBO J. 2019, 38, e100811. [Google Scholar] [CrossRef]
- Solovei, I.; Kreysing, M.; Lanctôt, C.; Kösem, S.; Peichl, L.; Cremer, T.; Guck, J.; Joffe, B. Nuclear Architecture of Rod Photoreceptor Cells Adapts to Vision in Mammalian Evolution. Cell 2009, 137, 356–368. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.E.O.; Enright, J.M.; Myers, C.A.; Shen, S.Q.; Corbo, J.C. Cell Type-Specific Epigenomic Analysis Reveals a Uniquely Closed Chromatin Architecture in Mouse Rod Photoreceptors. Sci. Rep. 2017, 7, 43184. [Google Scholar] [CrossRef] [Green Version]
- Frohns, A.; Frohns, F.; Naumann, S.C.; Layer, P.G.; Löbrich, M. Inefficient double-strand break repair in murine rod photoreceptors with inverted heterochromatin organization. Curr. Boil. 2014, 24, 1080–1090. [Google Scholar] [CrossRef] [Green Version]
- Mustafi, D.; Kevany, B.M.; Bai, X.; Golczak, M.; Adams, M.D.; Wynshaw-Boris, A.; Palczewski, K. Transcriptome analysis reveals rod/cone photoreceptor specific signatures across mammalian retinas. Hum. Mol. Genet. 2016, 25, 4376–4388. [Google Scholar] [CrossRef] [Green Version]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2017, 19, 1–9. [Google Scholar] [CrossRef]
- Vervoort, R.; Lennon, A.; Bird, A.C.; Tulloch, B.; Axton, R.; Miano, M.G.; Meindl, A.; Meitinger, T.; Ciccodicola, A.; Wright, A.F. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 2000, 25, 462–466. [Google Scholar] [CrossRef]
- Escribano-Diaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.; Tkac, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghezraoui, H.; Oliveira, C.; Becker, J.R.; Bilham, K.; Moralli, D.; Anzilotti, C.; Fischer, R.; Deobagkar-Lele, M.; Sanchiz-Calvo, M.; Fueyo-Marcos, E.; et al. 53BP1 cooperation with the REV7–shieldin complex underpins DNA structure-specific NHEJ. Nature 2018, 560, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.; Ellinwood, N.M.; Lorenz, B.; Stieger, K. Detection of DNA double strand breaks by γH2AX does not result in 53bp1 recruitment in mouse retinal tissues. Front. Neurosci. 2018, 12, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahaboglu, A.; Tanimoto, N.; Kaur, J.; Sancho-Pelluz, J.; Huber, G.; Fahl, A.; Arango-Gonzalez, B.; Zrenner, E.; Ekström, P.; Löwenheim, H.; et al. PARP1 gene knock-out increases resistance to retinal degeneration without affecting retinal function. PLoS ONE 2010, 5, e15495. [Google Scholar] [CrossRef] [Green Version]
- Dev, H.; Chiang, T.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef]
- Noordermeer, S.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Melsted, P.; Ntranos, V.; Pachter, L.; Birol, I. The barcode, UMI, set format and BUStools. Bioinformatics 2019, 35, 4472–4473. [Google Scholar] [CrossRef] [Green Version]
- Sultana, S.; Sarker, S.A.; Brüssow, H. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 2926–2934. [Google Scholar]
- Haghverdi, L.; Lun, A.T.L.; Morgan, M.D.; Marioni, J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018, 36, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Polański, K.; Young, M.D.; Miao, Z.; Meyer, K.B.; Teichmann, S.A.; Park, J.-E. BBKNN: Fast batch alignment of single cell transcriptomes. Bioinformatics 2020, 36, 964–965. [Google Scholar] [CrossRef] [PubMed]
- Blondel, V.D.; Guillaume, J.L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef] [Green Version]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.A.; Kwok, I.W.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019, 37, 38–47. [Google Scholar] [CrossRef]
- Bergen, V.; Lange, M.; Peidli, S.; Wolf, F.A.; Theis, F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. bioRxiv 2019. [Google Scholar] [CrossRef]
- Linta, L.; Stockmann, M.; Kleinhans, K.N.; Böckers, A.; Storch, A.; Zaehres, H.; Lin, Q.; Barbi, G.; Böckers, T.M.; Kleger, A.; et al. Rat Embryonic Fibroblasts Improve Reprogramming of Human Keratinocytes into Induced Pluripotent Stem Cells. Stem Cells Dev. 2011, 21, 965–976. [Google Scholar] [CrossRef]
- Frank, S.; Zhang, M.; Schöler, H.R.; Greber, B. Small Molecule-Assisted, Line-Independent Maintenance of Human Pluripotent Stem Cells in Defined Conditions. PLoS ONE 2012, 7, e41958. [Google Scholar] [CrossRef]
- Achberger, K.; Probst, C.; Haderspeck, J.; Bolz, S.; Rogal, J.; Chuchuy, J.; Nikolova, M.; Cora, V.; Antkowiak, L.; Haq, W.; et al. Merging organoid and organ-on-a-chip technology to generate complex multi-layer tissue models in a human retina-on-a-chip platform. Elife 2019, 8, 1–26. [Google Scholar] [CrossRef]

| Gene Symbol (Mouse Name) | Gene Name | Pathway | Function |
|---|---|---|---|
| XRCC5 | X-ray repair cross complementing 5 (Ku80) | NHEJ | determinant |
| XRCC6 | X-ray repair cross complementing 6 (Ku70) | NHEJ | determinant |
| TP53BP1 (Trp53bp1) | Tumor protein p53 binding protein 1 | NHEJ | protection |
| WRN | Werner syndrome RecQ-like helicase | NHEJ | protection |
| PARP1 | Poly(ADP-ribose) polymerase 1 | MMEJ | determinant |
| RBBP8 | RB binding protein 8, endonuclease (CtIP) | MMEJ | resection |
| MRE11 (Mre11a) | Meiotic recombination 11 homolog A | MMEJ | resection |
| NBN | Nijmegen breakage syndrome 1 | MMEJ | resection |
| RAD50 | Double-strand break repair protein | MMEJ | resection |
| BRCA1 | Breast cancer type 1 susceptibility protein | HDR | determinant |
| BRCA2 | Breast cancer type 2 susceptibility protein | HDR | determinant |
| RAD51 | BRCA1/BRCA2-containing complex, subunit 5 | HDR | determinant |
| PALB2 | Partner and localizer of BRCA2 | HDR | determinant |
| RPA1 | Replication protein A1 | HDR | SSA |
| RPA2 | Replication protein A2 | HDR | SSA |
| EXO1 | Exonuclease 1 | HDR | Add-resection |
| BLM | Bloom syndrome RecQ-like helicase | HDR | Add-resection |
| GEO ID | Species | Sample | Time Points (Days) | |
|---|---|---|---|---|
| GSE84930 | Homo sapiens | in vivo | Retina | Adult |
| GSE84927 | Mus musculus | in vivo | Retina | Adult |
| GSE84929 | Macaca fascicularis | in vivo | Retina | Adult |
| GSE84931 | Ictidomys tridecemlineatus | in vivo | Retina | Adult |
| GSE101986 | Homo sapiens | in vitro | Organoids CRX+ cells | d0, d37, d47, d67, d90 |
| GSE118307 | Homo sapiens | in vitro | Neurons | d0-4, d7, d14 |
| GSE101986 | Mus musculus | in vivo | Retina | E11-P28 |
| GEO ID | Species | Sample | Sequencing | Age | |
|---|---|---|---|---|---|
| GSE130636 | Homo sapiens | in vivo | Retina | 10x v3 | Adult |
| E-MTAB-74316 | Homo sapiens | in vivo | Retina | 10x v2 | Adult |
| GSE63473 | Mus musculus | in vivo | Retina | DropSeq | Young |
| GSE122566 | Mus musculus | in vivo | Retina | 10x v2 | Embryo (E15) |
| - | Homo sapiens | in vitro | Organoid | 10x v3 | d64, d106, d220, d330 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasquini, G.; Cora, V.; Swiersy, A.; Achberger, K.; Antkowiak, L.; Müller, B.; Wimmer, T.; Fraschka, S.A.-K.; Casadei, N.; Ueffing, M.; et al. Using Transcriptomic Analysis to Assess Double-Strand Break Repair Activity: Towards Precise in Vivo Genome Editing. Int. J. Mol. Sci. 2020, 21, 1380. https://doi.org/10.3390/ijms21041380
Pasquini G, Cora V, Swiersy A, Achberger K, Antkowiak L, Müller B, Wimmer T, Fraschka SA-K, Casadei N, Ueffing M, et al. Using Transcriptomic Analysis to Assess Double-Strand Break Repair Activity: Towards Precise in Vivo Genome Editing. International Journal of Molecular Sciences. 2020; 21(4):1380. https://doi.org/10.3390/ijms21041380
Chicago/Turabian StylePasquini, Giovanni, Virginia Cora, Anka Swiersy, Kevin Achberger, Lena Antkowiak, Brigitte Müller, Tobias Wimmer, Sabine Anne-Kristin Fraschka, Nicolas Casadei, Marius Ueffing, and et al. 2020. "Using Transcriptomic Analysis to Assess Double-Strand Break Repair Activity: Towards Precise in Vivo Genome Editing" International Journal of Molecular Sciences 21, no. 4: 1380. https://doi.org/10.3390/ijms21041380
APA StylePasquini, G., Cora, V., Swiersy, A., Achberger, K., Antkowiak, L., Müller, B., Wimmer, T., Fraschka, S. A.-K., Casadei, N., Ueffing, M., Liebau, S., Stieger, K., & Busskamp, V. (2020). Using Transcriptomic Analysis to Assess Double-Strand Break Repair Activity: Towards Precise in Vivo Genome Editing. International Journal of Molecular Sciences, 21(4), 1380. https://doi.org/10.3390/ijms21041380