Correlation between Antibodies to Bacterial Lipopolysaccharides and Barrier Proteins in Sera Positive for ASCA and ANCA




Abstract
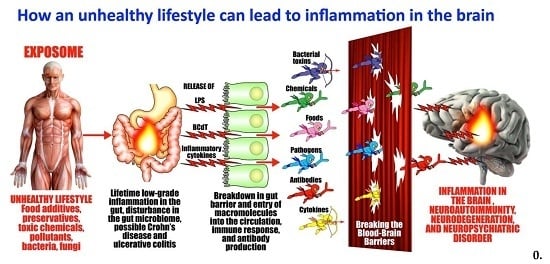
1. Introduction
1.1. Gut-Derived Lipopolysaccharides
1.2. Intestinal and Blood-Brain Barriers
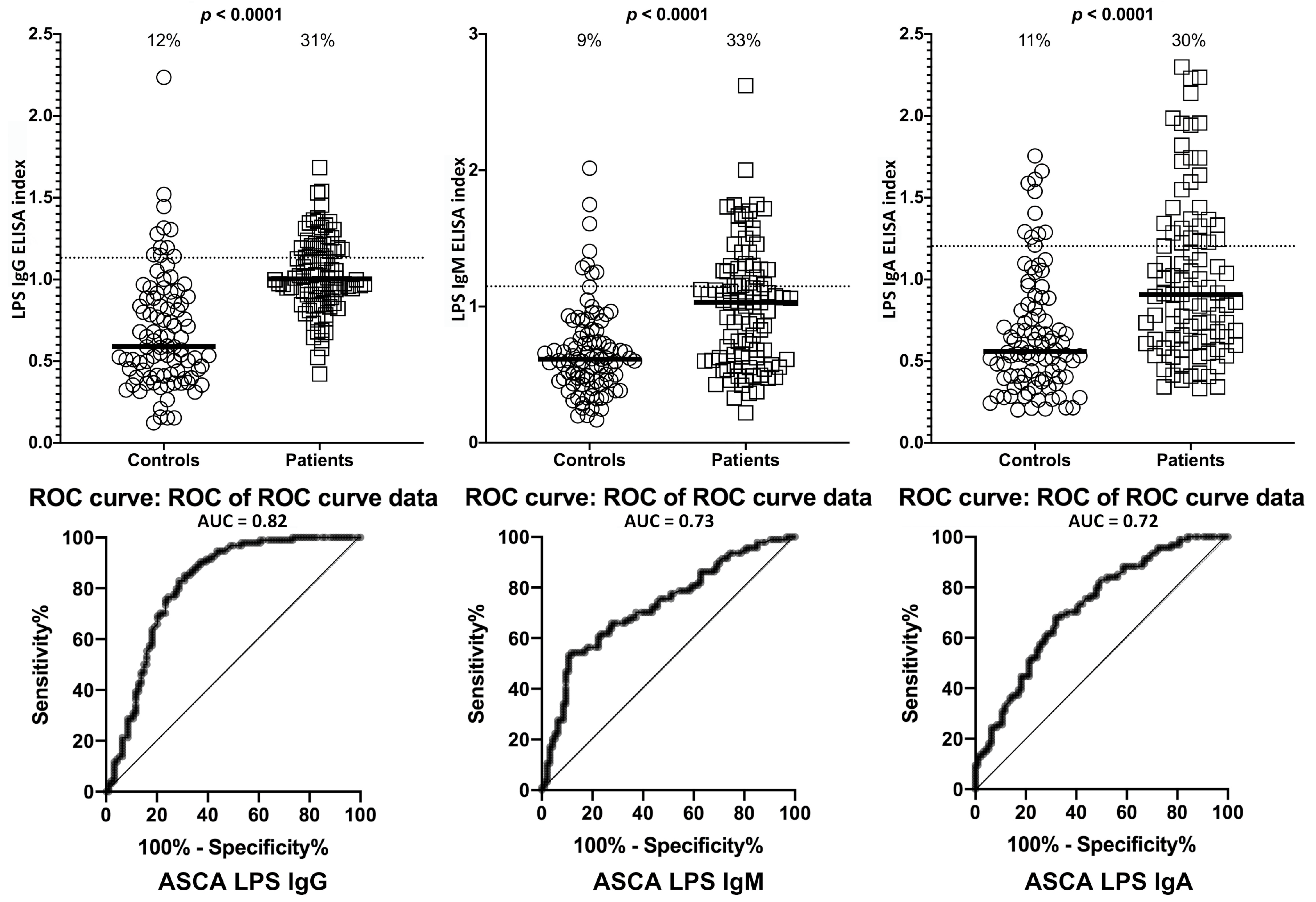
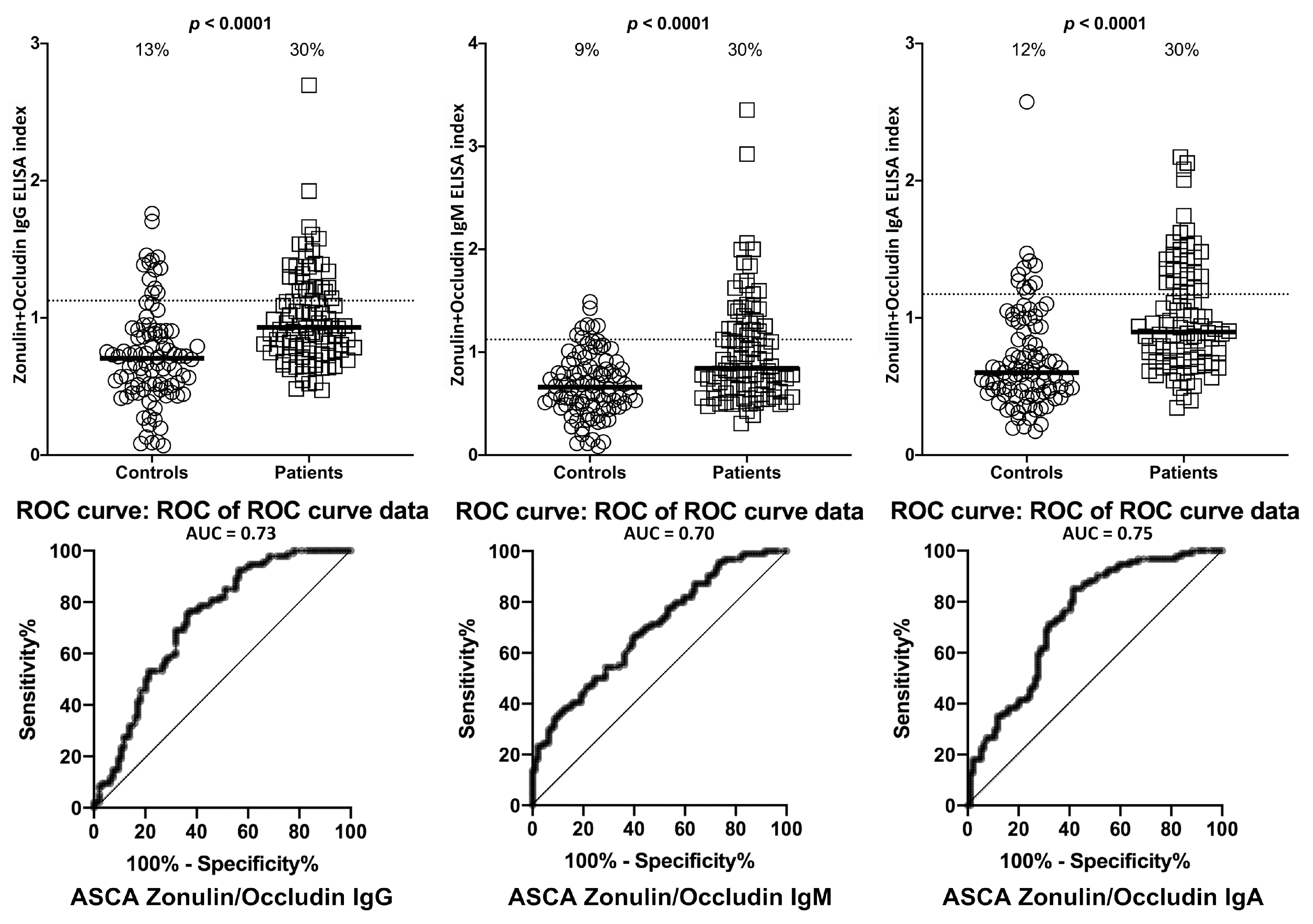
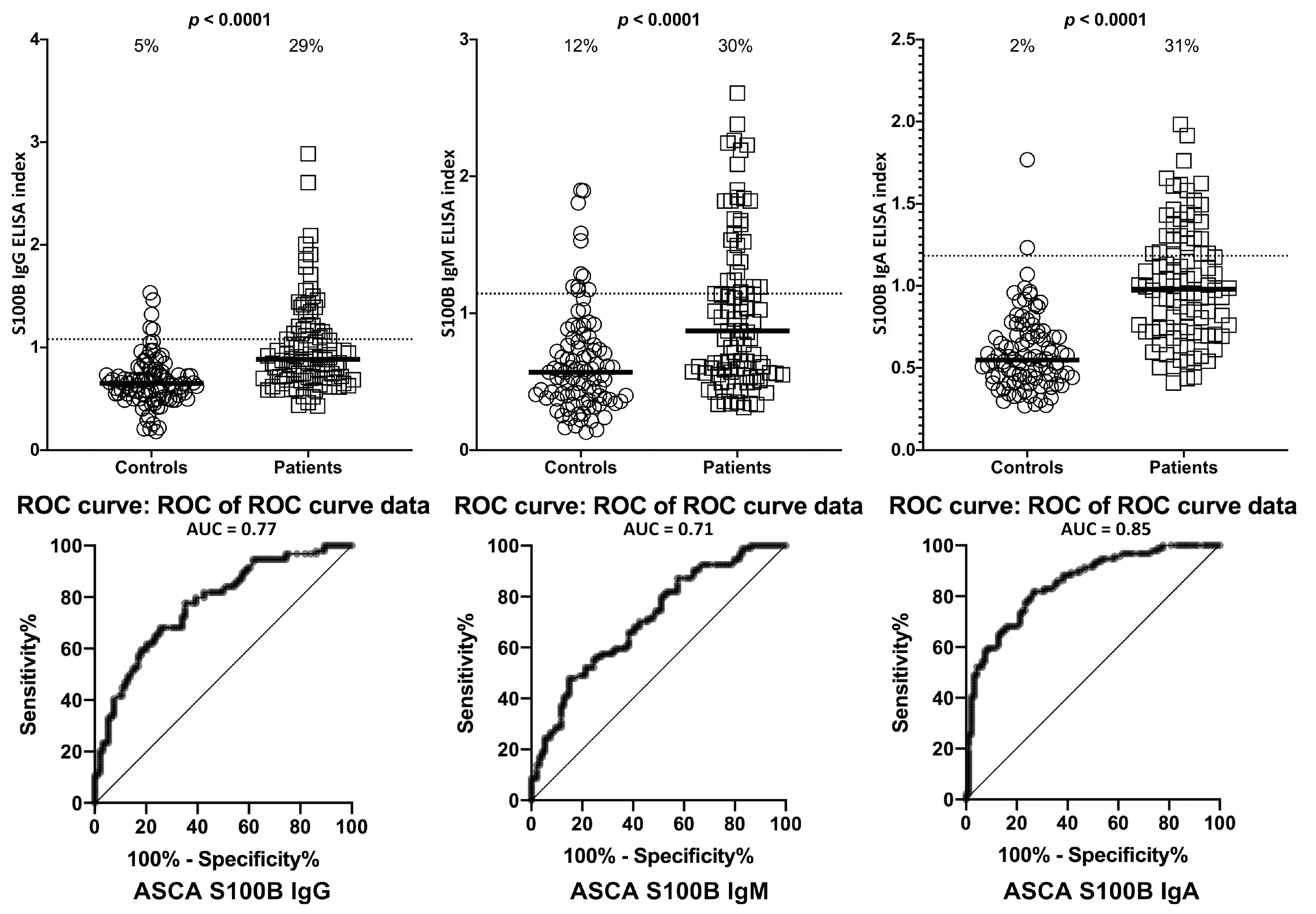
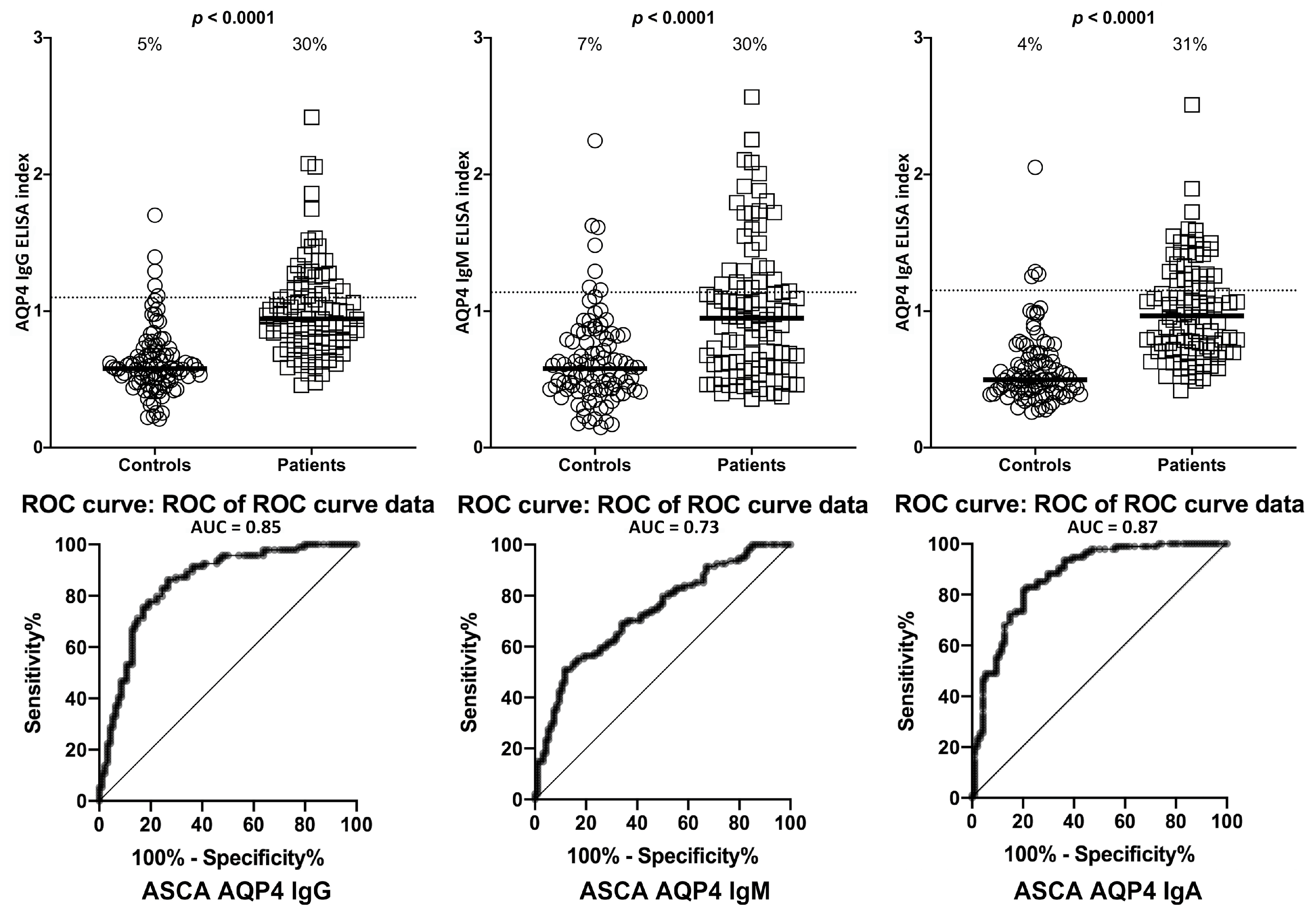
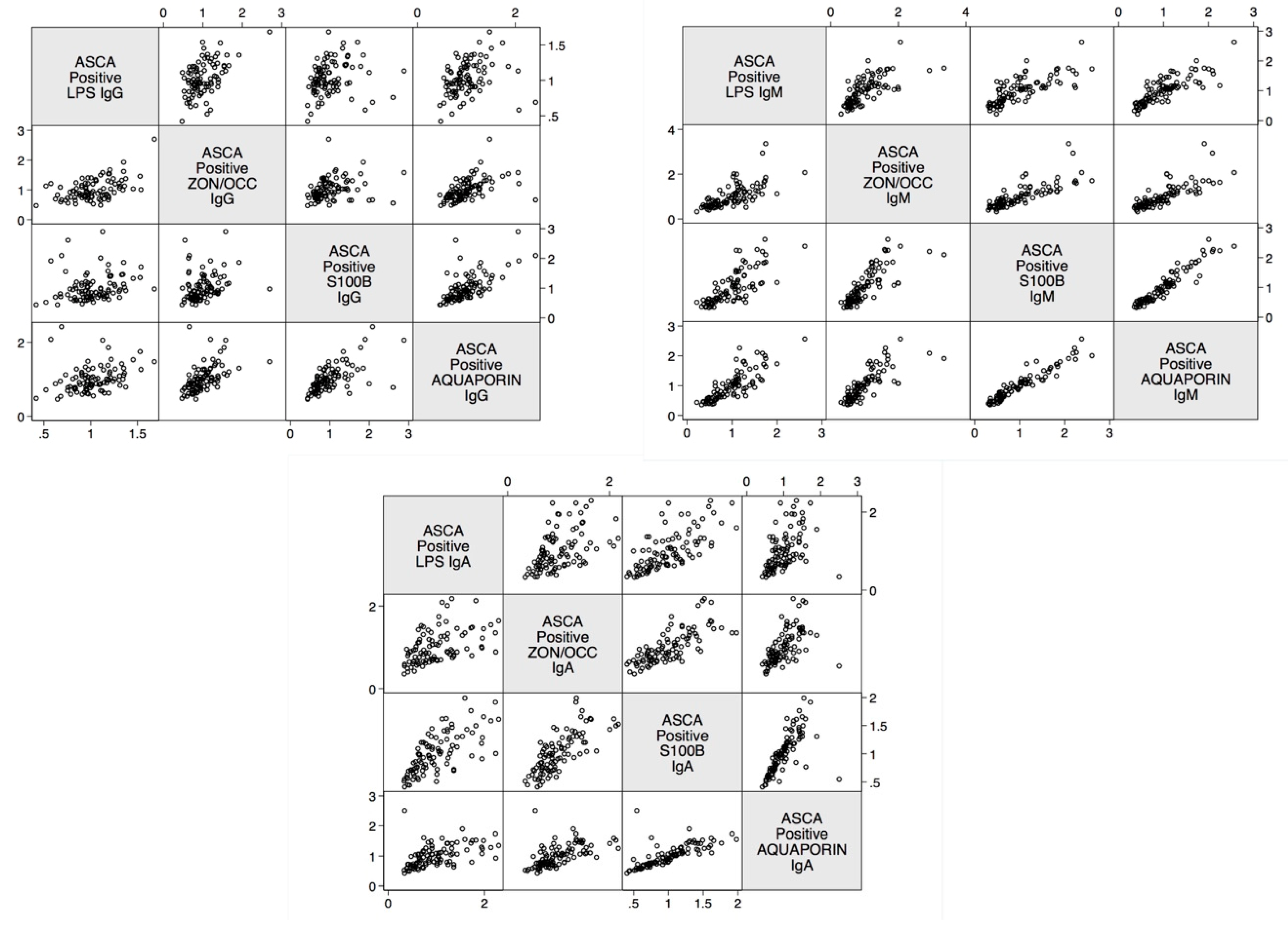
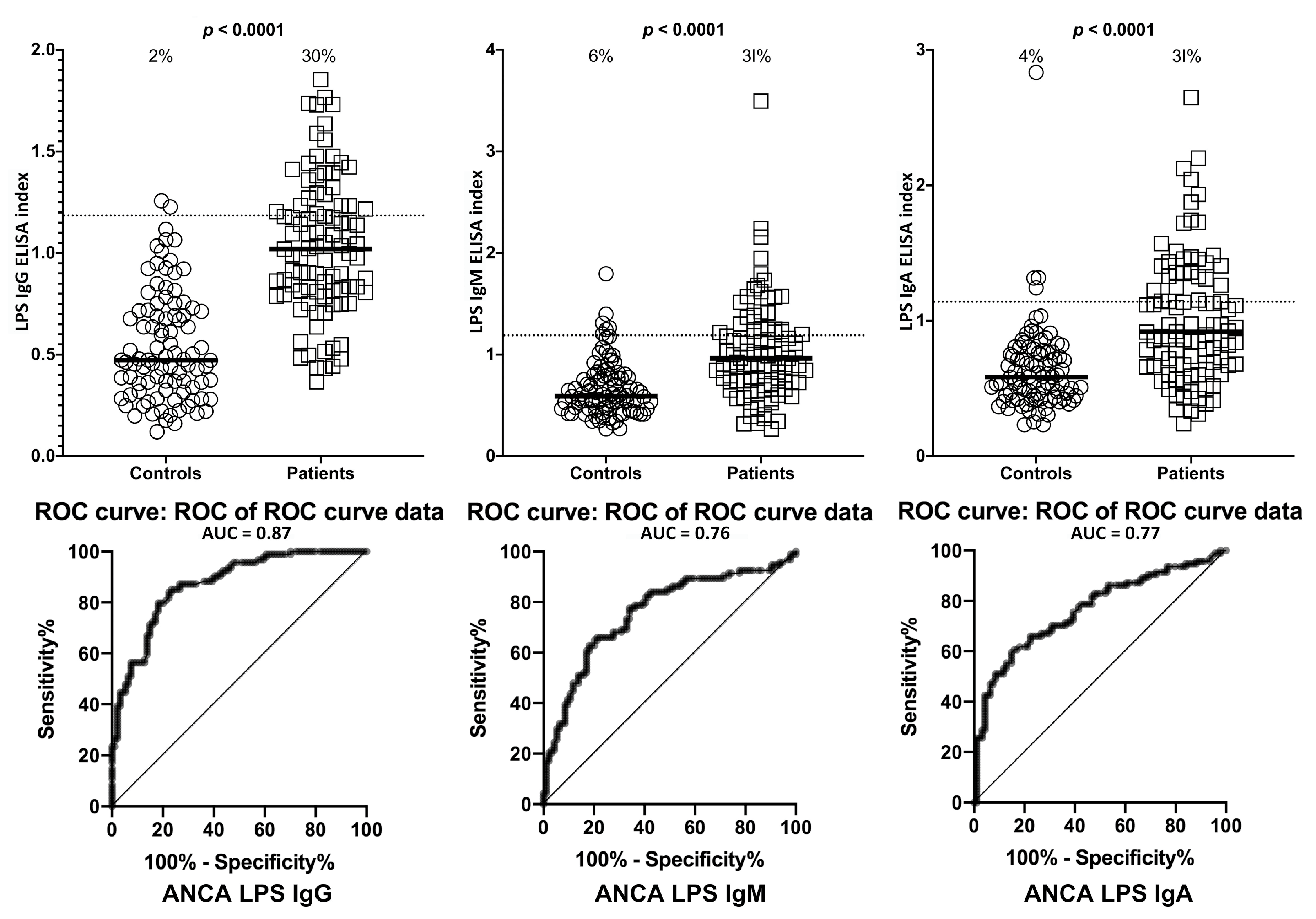
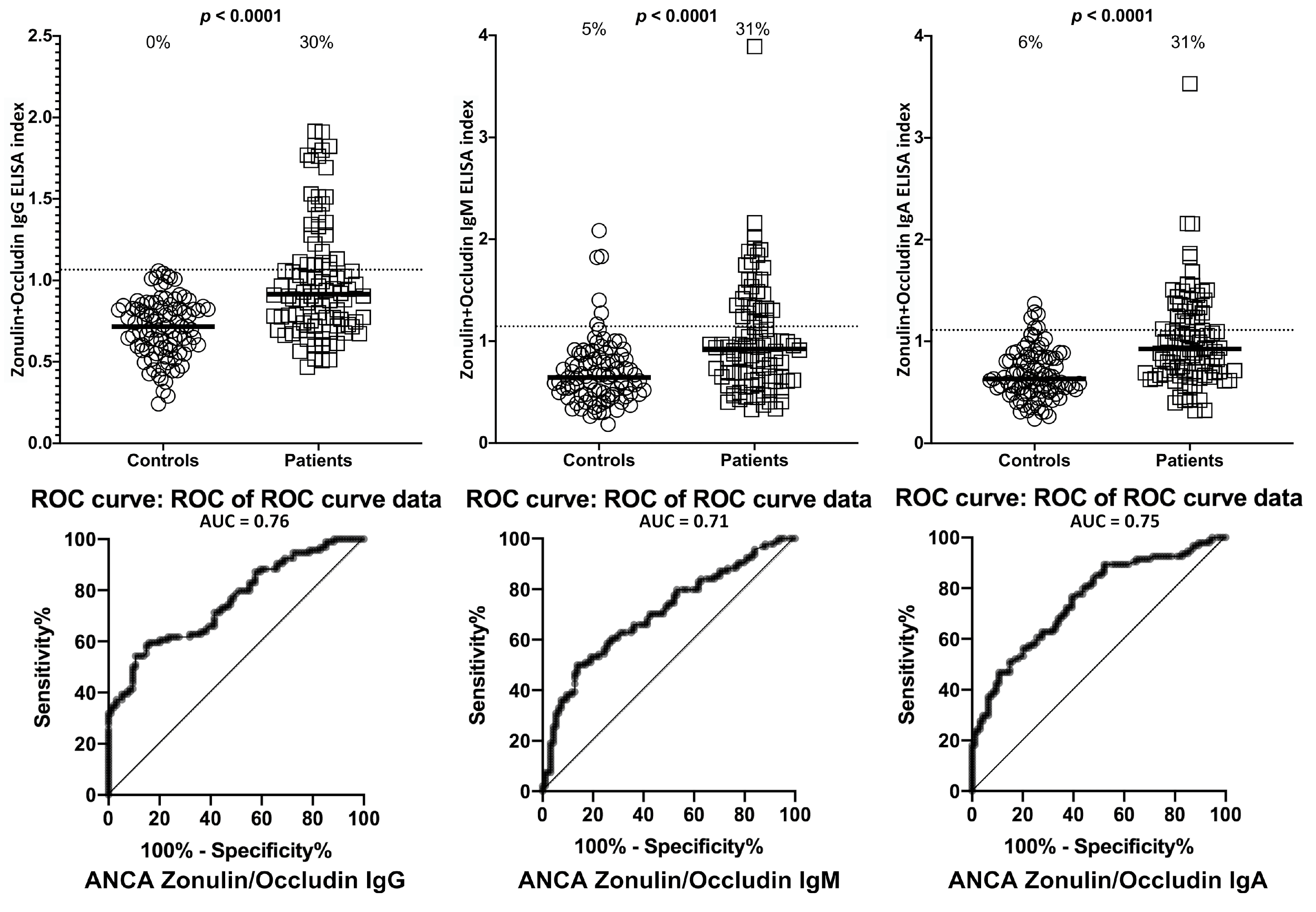
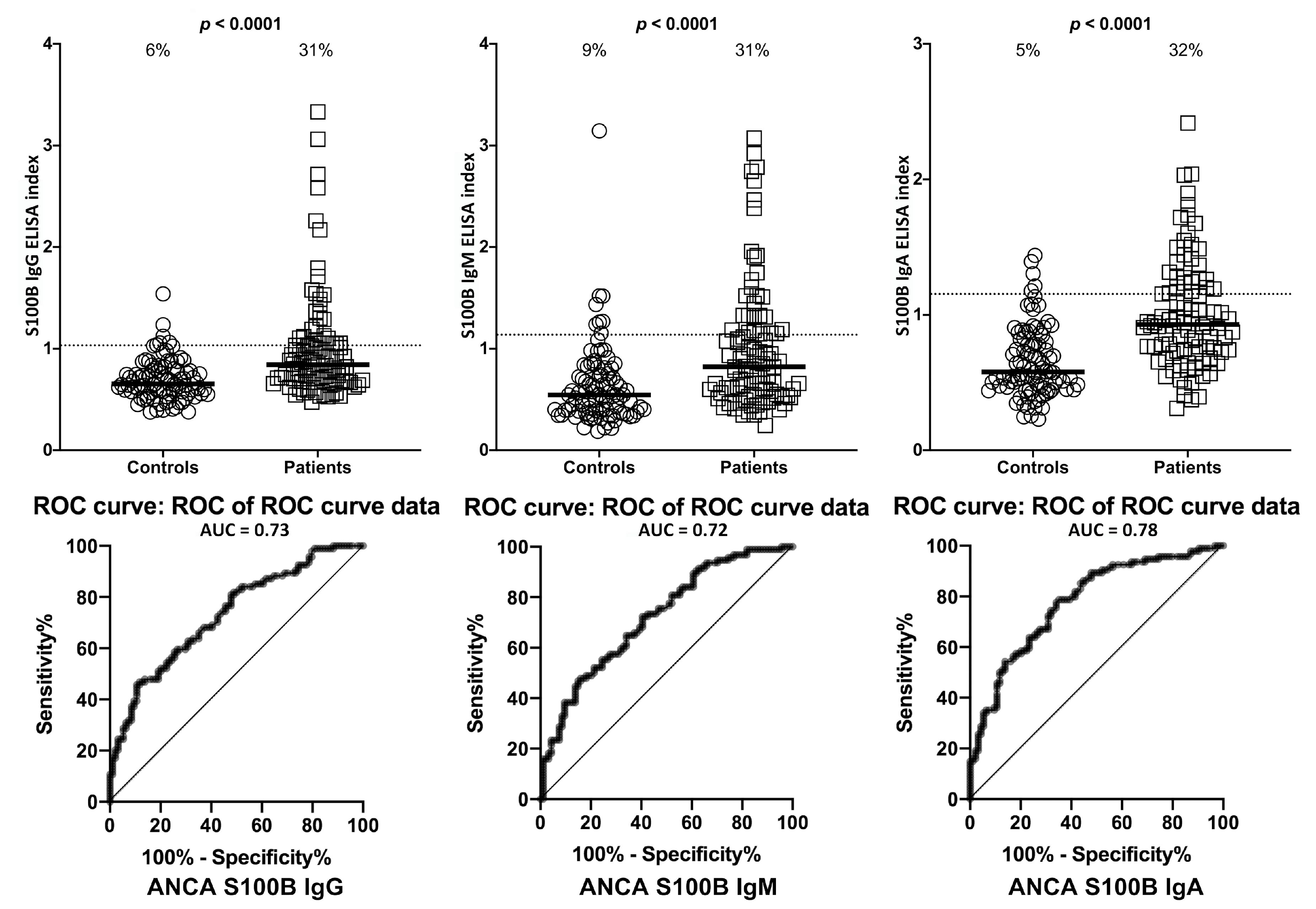
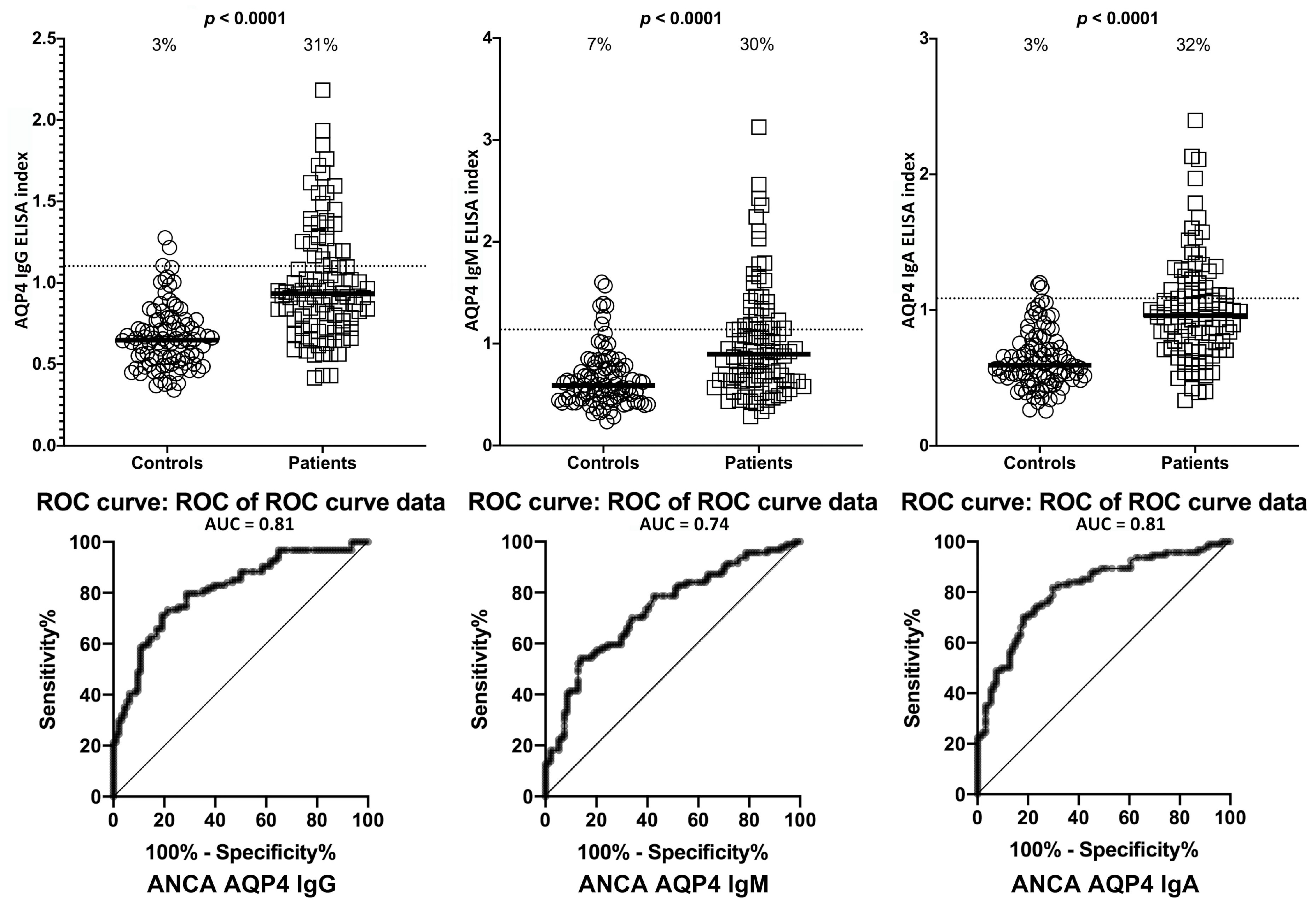
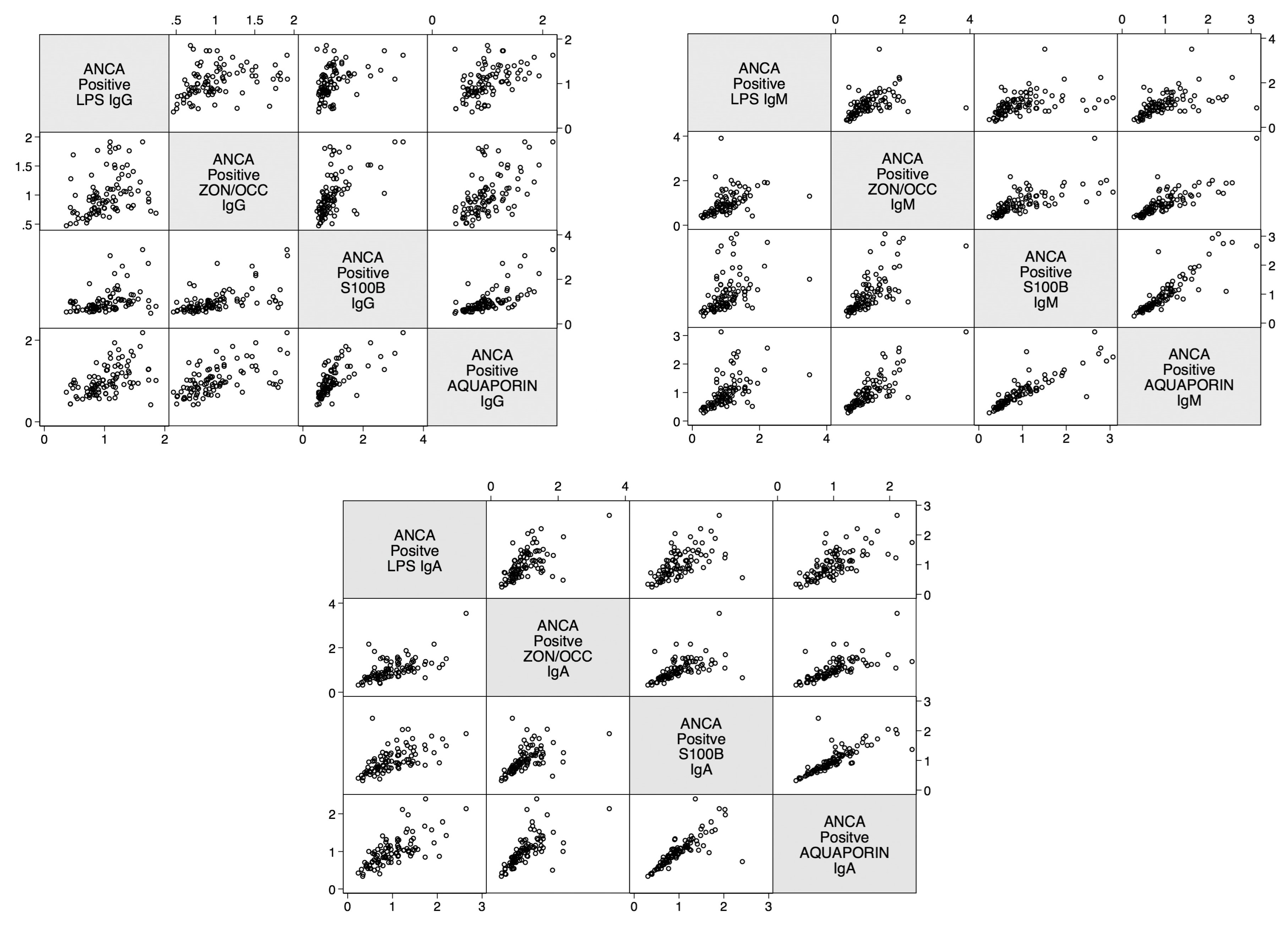
2. Results
3. Discussion
- We did not have any clinical information about our so-called healthy controls other than that they were screened based on a questionnaire and were negative for HIV and hepatitis-C antibodies. The additional information is important for the analysis of data from about 10% of controls who produced significant levels of antibodies against LPS as well as the barrier proteins.
- Likewise, other than their positivity or negativity when screened for ASCA or ANCA, we also did not have additional information about the clinical symptomatology of our ASCA- and ANCA-positive samples.
- We do not know whether the antibodies that we detected against AQP4 and S100B were made against these molecules in the brain, in the GI tract, or against both the gut and the brain glia and astrocytes.
- Due to our sample size and optical density variances, the confidence intervals were wide. This is due to the fact that only one-third of the ASCA- and ANCA-positive sera were highly reactive. However, there were still significant increased odds of 6- to 40-fold with the lowest tail of the confidence intervals.
4. Materials and Methods
4.1. Blood Samples and Antigens
4.2. Antibody Measurement
4.3. Statistical Analysis
4.4. Research Ethics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kuna, A.T. Serological markers of inflammatory bowel disease. Biochem. Med. (Zagreb) 2013, 23, 28–42. [Google Scholar] [CrossRef]
- What Is IBD? Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/ibd/what-is-IBD.htm (accessed on 17 February 2020).
- Lecis, P.; Germana, B.; Papa, N.; Bertiato, G.; Doglioni, C.; Galliani, E.; Biedo, F.C. p-ANCA and ASCA antibodies in the differential diagnosis between ulcerative rectocolitis and Crohn’s disease. Recenti. Prog. Med. 2002, 93, 308–313. [Google Scholar]
- Kvehaugen, A.S.; Aasbrenn, M.; Farup, P.G. Anti-Saccharomyces cerevisiae antibodies (ASCA) are associated with body fat mass and systemic inflammation, but not with dietary yeast consumption: A cross-ectional study. BMC Obes. 2017, 4, 28. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, J.; Qiao, Q.; Gao, M.; Cao, Q. Clinical significance of anti-Saccharomyces cerevisiae antibody in Crohn’s disease: A single-center study. Int. J. Clin. Exp. Pathol. 2016, 9, 11978–11983. [Google Scholar]
- Horn, M.P.; Peter, A.M.; Grunder, F.R.; Leichtle, A.B.; Spalinger, J.; Schibli, S.; Sokolik, C. PR3-ANCA and panel diagnostics in pediatric inflammatory bowel disease to distinguish ulcerative colitis from Crohn’s disease. PLoS ONE 2018, 13, e0208974. [Google Scholar] [CrossRef]
- Hilhorst, M.; van Paassen, P.; Tervaert, J.W.C. Proteinase 3-ANCA vasculitis versus myeloperoxidase-ANCA vasculitis. J. Am. Soc. Nephrol. 2015, 26, 2314–2327. [Google Scholar] [CrossRef]
- Yates, M.; Watts, R. ANCA-associated vasculitis. Clin. Med. (Lond.) 2017, 17, 60–64. [Google Scholar] [CrossRef]
- Suwanchote, S.; Rachayon, M.; Rodsaward, P.; Wongpiyabovorn, J.; Deekajorndech, T.; Wright, H.L.; Edwards, S.W.; Beresford, M.W.; Rerknimitr, P.; Chiewchengchoi, D. Anti-neutrophil cytoplasmic antibodies and their clinical significance. Clin. Rheumatol. 2018, 37, 875–884. [Google Scholar] [CrossRef]
- Michielan, A.; D’Incà, R. Intestinal permeability in inflammatory bowel disease: Pathogenesis, clinical evaluation, and therapy of leaky gut. Mediat. Inflamm. 2015, 2015, 628157. [Google Scholar] [CrossRef]
- Fukui, H. Increased intestinal permeability and decreased barrier function: Does it really influence the risk of inflammation? Inflamm. Intest. Dis. 2016, 1, 135–145. [Google Scholar] [CrossRef]
- Faisano, A. Zonulin and its regulation of intestinal barrier function: The biological door to inflammation, autoimmunity, and cancer. Physiol. Rev. 2011, 91, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A. For the assessment of intestinal permeability, size matters. Altern. Health. Med. 2013, 19, 12–24. [Google Scholar]
- Menard, S.; Cerf-Bensussan, N.; Heyman, M. Multiple facets of intestinal permeability and epithelial handling of dietary antigens. Mucosal Immunol. 2010, 3, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Harris, L.D.; Vinton, C.L.; Sung, H.; Briant, J.A.; Tabb, B.; Morcock, D.; McGinty, J.W.; Lifson, J.D.; Lafont, B.A.; et al. Compromised gastrointestinal integrity in pigtail macaques is associated with increased microbial translocation, immune activation and IL-17 production in the absence of SIV infection. Mucosal Immunol. 2010, 3, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Emi Aikawa, N.; de Carvalho, J.F.; Artur Almeida Silva, C.; Bonfá, E. Immunogenicity of anti-TNF-alpha agents in autoimmune diseases. Clin. Rev. Allergy Immunol. 2010, 38, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-Y.; Chen, Y.-G.; Lin, C.-L.; Huang, W.-S.; Kao, C.-H. Inflammatory bowel disease increases the risk of peripheral arterial disease. Medicine (Baltim.) 2015, 94, e2381. [Google Scholar] [CrossRef]
- Binus, A.M.; Han, J.; Qamar, A.A.; Mody, E.A.; Holt, E.W.; Qureshi, A.A. Associated comorbidities in psoriasis and inflammatory bowel disease. J. Eur. Acad. Derm. Venereol. 2012, 26, 644–650. [Google Scholar] [CrossRef]
- Vlachos, C.; Gaitanis, G.; Katsanos, K.; Christodoulou, D.; Tsianos, E.; Bassukas, I. Psoriasis and inflammatory bowel disease: Links and risks. Psoriasis: Targets Ther. 2016, 6, 73–92. [Google Scholar] [CrossRef]
- Scarpa, R.; Manguso, F.; D’Arienzo, A.; D’Armiento, F.P.; Astarita, C.; Mazzacca, G.; Ayala, F. Microscopic inflammatory changes in colon of patients with both active psoriasis and psoriatic arthritis without bowel symptoms. J. Rheumatol. 2000, 27, 1241–1246. [Google Scholar]
- Chan, J.; Sari, I.; Salonen, D.; Silverberg, M.S.; Haroon, N.; Inman, R.D. Prevalence of sacroiliitis in inflammatory bowel disease using a standardized computed tomography scoring system. Arthritis Care Res. (Hoboken) 2018, 70, 807–810. [Google Scholar] [CrossRef]
- Román, A.L.; Muñoz, F. Comorbidity in inflammatory bowel disease. World J. Gastroenterol. 2011, 17, 2723–2733. [Google Scholar] [CrossRef]
- Morís, G. Inflammatory bowel disease: An increased risk factor for neurologic complications. World J. Gastroenterol. 2014, 20, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef]
- Neisser, A.; Bernheimer, H.; Berger, T.; Moran, A.P.; Schwerer, B. Serum antibodies against gangliosides and Campylobacter jejuni lipopolysaccharides in Miller Fisher syndrome. Infect. Immun. 1997, 65, 4038–4042. [Google Scholar] [CrossRef] [PubMed]
- Poxton, I.R. Antibodies to lipopolysaccharide. J. Immunol. Methods 1995, 186, 1–15. [Google Scholar] [CrossRef]
- Sumazaki, R.; Fujita, T.; Kabashima, T.; Nishikaku, F.; Koyama, A.; Shibasaki, M.; Takita, H. Monoclonal antibody against bacterial lipopolysaccharide cross-reacts with DNA-histone. Clin. Exp. Immunol. 1986, 66, 103–110. [Google Scholar]
- Ziegler, T.R.; Luo, M.; Estívariz, C.F.; Moore, D.A., 3rd; Sitaraman, S.V.; Hao, L.; Bazargan, N.; Klapproth, J.M.; Tian, J.; Galloway, J.R.; et al. Detectable serum flagellin and lipopolysaccharide and upregulated anti-flagellin and lipopolysaccharide immunoglobulins in human short bowel syndrome. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R402–R410. [Google Scholar] [CrossRef] [PubMed]
- Hietbrink, F.; Besselink, M.G.; Renooij, W.; de Smet, M.B.; Draisma, A.; van der Hoeven, H.; Pickkers, P. Systemic inflammation increases intestinal permeability during experimental human endotoxemia. Shock 2009, 32, 374–378. [Google Scholar] [CrossRef]
- Walker, W.A.; Sanderson, I.R. Epithelial barrier function to antigens. Ann. N. Y. Acad. Sci. 2002, 664, 10–17. [Google Scholar] [CrossRef]
- Guo, S.; Nighot, M.; Al-Sadi, R.; Alhmoud, T.; Nighot, P.; Ma, T.Y. Lipopolysaccharide regulation of intestinal tight junction permeability is mediated by TLR4 signal transduction pathway activation of FAK and MyD88. J. Immunol. 2015, 195, 4999–5010. [Google Scholar] [CrossRef]
- Arican, N.; Kaya, M.; Kalayci, R.; Uzun, H.; Ahishali, B.; Bilgic, B.; Elmas, I.; Kucuk, M.; Gurses, C.; Uzun, M. Effects of lipopolysaccharide on blood–brain barrier permeability during pentylenetetrazole-induced epileptic seizures in rats. Life Sci. 2006, 79, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Shannon, K.M.; Kordower, J.H.; Voigt, R.M.; Shaikh, M.; Jaglin, J.A.; Estes, J.D.; Dodiya, H.B.; Keshavarzian, A. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS ONE 2011, 6, e28032. [Google Scholar] [CrossRef] [PubMed]
- Grin’kina, N.M.; Karnabi, E.E.; Damania, D.; Wadgaonkar, S.; Muslimov, I.A.; Wadgaonkar, R. Sphingosine kinase 1 deficiency exacerbates LPS-induced neuroinflammation. PLoS ONE 2012, 7, e36475. [Google Scholar] [CrossRef]
- Jeong, H.-K.; Jou, I.; Joe, E.-H. Systemic LPS administration induces brain inflammation but not dopaminergic neuronal death in the substantia nigra. Exp. Mol. Med. 2010, 42, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Tufekci, K.U.; Genc, S.; Genc, K. The endotoxin-induced neuroinflammation model of Parkinson’s disease. Parkinson’s Dis. 2011, 2011, 487450. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Lee, Y.K.; Yuk, D.Y.; Choi, D.Y.; Ban, S.B.; Oh, K.W.; Hong, J.T. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflamm. 2008, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Stamova, B.; Jin, L.-W.; DeCarrli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurologe 2016, 87, 2324–2332. [Google Scholar] [CrossRef]
- Zhao, Y.; Cong, L.; Jaber, V.; Lukiw, W.J. Microbiome-derived lipopolysaccharide enriched in the perinuclear region of Alzheimer’s disease brain. Front. Immunol. 2017, 8, 1064. [Google Scholar] [CrossRef]
- Candido, T.L.N.; Bressan, J.; Alfenas, R.C.G. Dysbiosis and metabolic endotoxemia induced by high-fat diet. Nutr. Hosp. 2018, 35, 1432–1440. [Google Scholar] [CrossRef]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological impllications. Exp. Mol. Med. 2018, 50, 103. [Google Scholar] [CrossRef]
- Geddes, K.; Ohilpott, D. A new role for intestinal alkaline phosphatase in gut barrier maintenance. Gastroenterolgy 2008, 135, 8–12. [Google Scholar] [CrossRef]
- Turner, J. Amgen Award Lecture. Molecular basis of epithelial barrier regulation, from basic mechanisms to clinical application. Am. J. Path. 2006, 169, 1901–1909. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Persidsky, Y.; Stins, M.; Way, D.; Witte, M.H.; Weinand, M.; Kim, K.S.; Bock, P.; Gendelman, H.E.; Fiala, M. A model for monocyte migration through the blood–brain barrier during HIV-1 encephalitis. J. Immunol. 1997, 158, 3499–3510. [Google Scholar]
- Dohgu, S.; Banks, W.A. Lipopolysaccharide-enhanced transcellular transport of HIV-1 across the blood–brain barrier is mediated by the p38 mitogen-activated protein kinase pathway. Exp. Neurol. 2008, 210, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Marchi, N.; Cavaglia, M.; Fazio, V.; Bhudia, S.; Hallene, K.; Janigro, D. Peripheral markers of blood-brain barrier damage. Clin. Chim. Acta 2004, 342, 1–12. [Google Scholar] [CrossRef]
- Hu, H.; Yao, H.T.; Zhang, W.P.; Zhang, L.; Ding, W.; Zhang, S.H.; Chen, Z.; Wei, E.Q. Increased expression of aquaporin-4 in human traumatic brain injury and brain tumors. J. Zhejiang Univ. Sci. B 2005, 6, 33–37. [Google Scholar] [CrossRef]
- Banks, W.A.; Gray, A.M.; Erickson, M.A.; Salameh, T.S.; Damodarasamy, M.; Sheibani, N.; Meabon, J.S.; Wing, E.E.; Morofuji, Y.; Cook, D.G.; et al. Lipopolysaccharide-induced blood-brain barrier disruption: Roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J. Neuroinflamm. 2015, 12, 223. [Google Scholar] [CrossRef]
- Kern, J.K.; Geier, D.A.; Sykes, L.K.; Geier, M.R. Relevance of neuroinflammation and encephalitis in autism. Front. Cell. Neurosci. 2016, 9, 519. [Google Scholar] [CrossRef]
- Jyonouchi, H. Immunological abnormalities in autism spectrum disorders. Adv. Neuroimmune Biol. 2013, 4, 141–159. [Google Scholar] [CrossRef]
- Desai, R.A.; Davies, A.L.; Tachrount, M.; Kasti, M.; Laulund, F.; Golay, X.; Smith, K.J. Cause and prevention of demyelination in a model multiple sclerosis lesion. Ann. Neurol. 2016, 79, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Felts, P.A.; Woolston, A.M.; Fernando, H.B.; Asquith, S.; Gregson, N.A.; Mizzi, O.J.; Smith, K.J. Inflammation and primary demyelination induced by the intraspinal injection of lipopolysaccharide. Brain 2005, 128, 1649–1666. [Google Scholar] [CrossRef] [PubMed]
- Nogai, A.; Siffrin, V.; Bonhagen, K.; Pfueller, C.F.; Hohnstein, T.; Volkmer-Engert, R.; Brück, W.; Stadelmann, C.; Kamradt, T. Lipopolysaccharide injection induces relapses of experimental autoimmune encephalomyelitis in nontransgenic mice via bystander activation of autoreactive CD4+ cells. J. Immunol. 2005, 175, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Noailles, A.; Maneu, V.; Campello, L.; Lax, P.; Cuenca, N. Systemic inflammation induced by lipopolysaccharide aggravates inherited retinal dystrophy. Cell Death Dis. 2018, 9, 350. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Vojdani, E.; Saidara, E.; Kharrazian, D. Reaction of amyloid-β peptide antibody with different infectious agents involved in Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 63, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Kubera, M.; Leunis, J.C. The gut-brain barrier in major depression: Intestinal mucosal dysfunction with an increased translocation of LPS from gram negative enterobacteria (leaky gut) plays a role in the inflammatory pathophysiology of depression. Neuro. Endocrinol. Lett. 2008, 29, 117–124. [Google Scholar]
- Maes, M.; Sirivichayakul, S.; Kanchanatawan, B.; Vojdani, A. Upregulation of the intestinal paracellular pathway with breakdown of tight and adherens junctions in deficit schizophrenia. Mol. Neurobiol. 2019, 56, 7056–7073. [Google Scholar] [CrossRef]
- Maes, M.; Vojdani, A.; Geffard, M.; Moreira, E.G.; Barbosa, D.S.; Michelin, A.P.; Semeão, L.O.; Sirivichayakul, S.; Kanchanatawan, B. Schizophrenia phenomenology comprises a bifactorial general severity and a single-group factor, which are differently associated with neurotoxic immune and immune-regulatory pathways. Biomol. Concepts 2019, 10, 209–225. [Google Scholar] [CrossRef]
- Maes, M.; Sirivichayakul, S.; Kanchanatawan, B. Breakdown of the paracellular tight and adherens junctions in the gut and blood brain barrier and damage to the vascular barrier in patients with deficit schizophrenia. Neurotox. Res. 2019, 36, 306–322. [Google Scholar] [CrossRef]
- Magro, D.O.; Kotze, P.G.; Martinez, C.A.R.; Camargo, M.G.; Guadagnini, D.; Calixto, A.R.; Vasques, A.C.J.; Ayrizono, M.L.S.; Geloneze, B.; Pareja, J.C.; et al. Changes in serum levels of lipopolysaccharides and CD26 in patients with Crohn’s disease. Intest. Res. 2017, 15, 352–357. [Google Scholar] [CrossRef]
- Caradonna, L.; Amati, L.; Magrone, T.; Pellegrino, N.M.; Jirillo, E.; Caccavo, D. Invited review: Enteric bacteria, lipopolysaccharides and related cytokines in inflammatory bowel disease: Biological and clinical significance. J. Endotoxin Res. 2000, 6, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Villarán, R.F.; Espinosa-Oliva, A.M.; Sarmiento, M.; De Pablos, R.M.; Argüelles, S.; Delgado-Cortés, M.J.; Sobrino, V.; Van Rooijen, N.; Venero, J.L.; Herrera, A.J.; et al. Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: Potential risk factor in Parkinson’s disease. J. Neurochem. 2010, 114, 1687–1700. [Google Scholar] [CrossRef] [PubMed]
- Chin, A.C.; Flynn, A.N.; Fedwick, J.P.; Buret, A.G. The role of caspase-3 in lipopolysaccharide-mediated disruption of intestinal epithelial tight junctions. Can. J. Physiol. Pharm. 2006, 84, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Berkes, J.; Viswanathan, K.; Savlovic, S.D.; Hecht, G. Intestinal epithelial responses to enteric pathogens: Effects on the tight junction barrier, ion transport, and inflammation. Gut 2003, 52, 439–451. [Google Scholar] [CrossRef]
- Lammers, K.M.; Lu, R.; Brownley, J.; Lu, B.; Gerard, C.; Thomas, K.; Rallabhandi, P.; Shea-Donohue, T.; Tamiz, A.; Alkan, S.; et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterologe 2008, 135, 194–204. [Google Scholar] [CrossRef]
- Scuron, M.D.; Boesze-Battaglia, K.; Dlakic, M.; Shenker, B.J. The cytolethal distending toxin contributes to microbial virulence and disease pathogenesis by acting as a tri-perditious toxin. Front. Cell. Infect. Microbiol. 2016, 6, 168. [Google Scholar] [CrossRef]
- Vojdani, A.; Vojdani, E. Reaction of antibodies to Campylobacter jejuni and cytolethal distending toxin B with tissues and food antigens. World J. Gastroenterol. 2019, 25, 1050–1066. [Google Scholar] [CrossRef]
- Stewart, L.; Edgar, J.D.M.; Blakely, G.; Patrick, S. Antigenic mimicry of ubiquitin by the gut bacterium Bacteroides fragilis: A potential link with autoimmune disease. Clin. Exp. Immunol. 2018, 194, 153–165. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; Macpherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental triggers of IBD: A review of progress and evidence. Nat. Rev. Gastroenter. Hepatol. 2018, 15, 39–49. [Google Scholar] [CrossRef]
- Nickerson, K.P.; McDonald, C. Crohn’s disease-associated adherent-invasive Escherichia coli adhesion is enhanced by exposure to the ubiquitous dietary polysaccharide maltodextrin. PLoS ONE 2012, 7, e52132. [Google Scholar] [CrossRef]
- Zuo, T.; Ng, S.C. The gut microbiota in the pathogenesis and therapeutics of inflammatory bowel disease. Front. Microbiol. 2018, 9, 2247. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Al-Sadi, R.; Said, H.M.; Ma, T.Y. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am. J. Pathol. 2013, 182, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Flynn, C.A.N.; Turner, J.R.; Buret, A.G. SGLT-1-mediated glucose uptake protects intestinal epithelial cells against LPS-induced apoptosis and barrier defects: A novel cellular rescue mechanism? FASEB J. 2005, 19, 1822–1835. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26. [Google Scholar] [CrossRef]
- Manco, M.; Putignani, L.; Bottazzo, G.F. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr. Rev. 2010, 31, 817–844. [Google Scholar] [CrossRef]
- Nymark, M.; Pussinen, P.J.; Tuomainen, A.M.; Forsblom, C.; Groop, P.H.; Lehto, M. FinnDiane Study Group Serum lipopolysaccharide activity is associated with the progression of kidney disease in Finnish patients with type 1 diabetes. Diabetes Care 2009, 32, 1689–1693. [Google Scholar] [CrossRef]
- Vélez, M.L.; Costamagna, E.; Kimura, E.T.; Fozzatti, L.; Pellizas, C.G.; Montesinos, M.M.; Lucero, A.M.; Coleoni, A.H.; Santisteban, P.; Masini-Repiso, A.M. Bacterial lipopolysaccharide stimulates the thyrotropin-dependent thyroglobulin gene expression at the transcriptional level by involving the transcription factors thyroid transcription factor-1 and paired box domain transcription factor 8. Endocrinology 2006, 147, 3260–3275. [Google Scholar] [CrossRef]
- Boelen, A.; Kwakkel, J.; Thijssen-Timmer, D.C.; Alkemade, A.; Fliers, E.; Wiersinga, W.M. Simultaneous changes in central and peripheral components of the hypothalamus-pituitary-thyroid axis in lipopolysaccharide-induced acute illness in mice. J. Endocrinol. 2004, 182, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Su, G.L. Lipopolysaccharides in liver injury: Molecular mechanisms of Kupffer cell activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G256–G265. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Bala, S. Alcoholic liver disease and the gut-liver axis. W. J. Gastroenterol. 2010, 16, 1321–1329. [Google Scholar] [CrossRef]
- Szeto, C.C.; Kwan, B.C.; Chow, K.M.; Lai, K.B.; Chung, K.Y.; Leung, C.B.; Li, P.K. Endotoxemia is related to systemic inflammation and atherosclerosis in peritoneal dialysis patients. Am. Soc. Nephrol. 2008, 3, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Niki, Y.; Kawasaki, T.; Takeda, Y.; Horiuchi, K.; Sasaki, A.; Okada, Y.; Umezawa, K.; Ikegami, H.; Toyama, Y.; et al. Enhanced susceptibility to lipopolysaccharide-induced arthritis and endotoxin shock in interleukin-32 alpha transgenic mice through induction of tumor necrosis factor alpha. Arthritis Res. 2012, 14, R120. [Google Scholar] [CrossRef]
- Yoshino, S.; Ohsawa, M. The role of lipopolysaccharide injected systemically in the reactivation of collagen-induced arthritis in mice. Br. J. Pharm. 2000, 129, 1309–1314. [Google Scholar] [CrossRef]
- Wilson, J.C.; Furlano, R.I.; Jick, S.S.; Meier, C.R. Inflammatory bowel disease and the risk of autoimmune diseases. J. Crohns Colitis 2016, 10, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Marrie, R.A.; Walld, R.; Bolton, J.M.; Sareen, J.; Walker, J.R.; Patten, S.B.; Singer, A.; Lix, L.M.; Hitchon, C.A.; El-Gabalawy, R.; et al. CIHR Team in Defining the Burden and Managing the Effects of Psychiatric Comorbidity in Chronic Immunoinflammatory Disease. Increased incidence of psychiatric disorders in immune-mediated inflammatory disease. J. Psychosom. Res. 2017, 101, 17–23. [Google Scholar] [CrossRef]
- Sajadinejad, M.S.; Asgari, K.; Molavi, H.; Kalantari, M.; Adibi, P. Psychological issues in inflammatory bowel disease: An overview. Gastroenterol. Res. Pr. 2012, 2012, 106502. [Google Scholar] [CrossRef] [PubMed]
- Gracie, D.J.; Ford, A.C. Irritable bowel syndrome-type symptoms are associated with psychological comorbidity, reduced quality of life, and health care use in patients with inflammatory bowel disease. Gastroenterology 2017, 153, 324–325. [Google Scholar] [CrossRef]
- Nemati, R.; Mehdizadeh, Z.; Salimipour, H.; Yaghoubi, E.; Alipour, Z.; Tabib, S.M.; Assadi, M. Neurological manifestations related to Crohn’s disease; a boon for the workforce. Gastroenterol. Rep. 2019, 7, 291–297. [Google Scholar] [CrossRef]
- Vojdani, A.; Vojdani, E.; Kharrazian, D. Fluctuation of zonulin levels in blood versus stability of antibodies. W. J. Gastroenterol. 2017, 23, 5669–5679. [Google Scholar] [CrossRef]
- Fasano, A.; Not, T.; Wang, W.; Uzzau, S.; Berti, I.; Tommasini, A.; Goldblum, S.E. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet 2000, 355, 1518–1519. [Google Scholar] [CrossRef]
- Yao, Z.; Mates, J.M.; Cheplowitz, A.M.; Hammer, L.P.; Maiseyeu, A.; Phillips, G.S.; Wewers, M.D.; Rajaram, M.V.; Robinson, J.M.; Anderson, C.L.; et al. Blood-borne lipopolysaccharide is rapidly eliminated by liver sinusoidal endothelial cells via high-density lipoprotein. J. Immunol. 2016, 197, 2390–2399. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.A.; Peters, M.A.; Beynon, H.L.; Walport, M.J. Immune complex processing in patients with systemic lupus erythematosus. In vivo imaging and clearance studies. J. Clin. Investig. 1992, 90, 2075–2083. [Google Scholar] [CrossRef] [PubMed]
- Ercole, A.; Thelin, E.; Holst, A.; Bellander, B.M.; Nelson, D.W. Kinetic modelling of serum S100B after traumatic brain injury. BMC Neurol. 2016, 16, 93. [Google Scholar] [CrossRef]
- Michetti, F.; Ambrosi, N.; Toesca, A.; Puglisi, M.A.; Serrano, A.; Marchese, E.; Corvino, V.; Geloso, M.C. The S100B story: From biomarker to active factor in neural injury. J. Neurochem. 2019, 148, 168–187. [Google Scholar] [CrossRef]
- Zonner, S.W.; Ejima, K.; Bevilacqua, Z.W.; Huibregtse, M.E.; Charleston, C.; Fulgar, C.; Kawata, K. Association of increased serum S100B levels with high school football subconcussive head impacts. Front. Neurol. 2019, 10, 327. [Google Scholar] [CrossRef]
- Laforenza, U. Water channel proteins in the gastrointestinal tract. Mol. Asp. Med. 2012, 33, 642–650. [Google Scholar] [CrossRef]
- Vojdani, A.; Mukherjee, P.S.; Berookhim, J.; Kharrazian, D. Detection of antibodies against human and plant aquaporins in patients with multiple sclerosis. Autoimmune Dis. 2015, 2015, 905208. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Wang, Y.; Duan, T.; Patel, J.; Liggett, T.; Loda, E.; Brahma, S.; Goswami, R.; Grouse, C.; Byrne, R.; et al. Cross-immunoreactivity between bacterial aquaporin-Z and human aquaporin-4: Potential relevance to neuromyelitis optica. J. Immunol. 2012, 189, 4602–4611. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, Y.; Chen, Z.; Xu, Z.; Xu, H. Knockdown of aquaporin 3 is involved in intestinal barrier integrity impairment. FEBS Lett. 2011, 585, 3113–3119. [Google Scholar] [CrossRef]
- Morgan, E.; Peplowski, M.; MacNaughton, W. Aquaporin 3 promotes intestinal epithelial proliferation and inhibits cytokine-induced apoptosis. FASEB 2015, 29, 766.11. [Google Scholar] [CrossRef]
- Cirillo, C.; Sarnelli, G.; Esposito, G.; Turco, F.; Steardo, L.; Cuomo, R. S100B protein in the gut: The evidence for enteroglial-sustained intestinal inflammation. W. J. Gastroenterol. 2011, 17, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Thi, M.M.; Spray, D.C.; Hanani, M. Aquaporin-4 water channels in enteric neurons. J. Neurosci. Res. 2008, 86, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Ribaldone, D.G.; Pellicano, R.; Actis, G.C. Inflammation in gastrointestinal disorders: Prevalent socioeconomic factors. Clin. Exp. Gastroenterol. 2019, 12, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Caviglia, G.P.; Dughera, F.; Ribaldone, D.G.; Rosso, C.; Abate, M.L.; Pellicano, R.; Bresso, F.; Smedile, A.; Saracco, G.M.; Astegiano, M. Serum zonulin in patients with inflammatory bowel disease: A pilot study. Minerva Med. 2019, 110, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Hagmeyer, S.; Romao, M.A.; Cristovao, J.S.; Vilella, A.; Zoli, M.; Gomes, C.M.; Grabrucker, A.M. Distribution and relative abundance of S100 proteins in the brain of the APP23 Alzheimer’s disease model mice. Front. Neurosci. 2019, 13, 640. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Hanley, N.; Campbell, M. Claudin-5: Gatekeeper of neurological function. Fluids Barriers CNS 2019, 16, 3. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Barrier Protein Antibodies | Odds Ratio | Confidence Interval | p-Value |
|---|---|---|---|
| S100B IgA | 36 | 11–110 | <0.0001 |
| S100B IgG | 65 | 21–191 | <0.0001 |
| S100B IgM | 5 | 3–10 | <0.0001 |
| Aquaporin-4 IgA | 149 | 40–533 | <0.0001 |
| Aquaporin-4 IgG | 96 | 30–305 | <0.0001 |
| Aquaporin-4 IgM | 9 | 4–20 | <0.0001 |
| Zonulin+Occludin IgA | 20 | 8–56 | <0.0001 |
| Zonulin+Occludin IgG | 13 | 6–29 | <0.0001 |
| Zonulin+Occludin IgM | 8 | 4–17 | <0.0001 |
| Lipopolysaccharides IgA | 107 | 34–340 | <0.0001 |
| Lipopolysaccharides IgG | 8 | 4–16 | <0.0001 |
| Lipopolysaccharides IgM | 11 | 5–24 | <0.0001 |
| Correlations | r-Value | p-Value |
|---|---|---|
| Zonulin+Occludin IgA and S100B IgA | 0.8 | <0.0001 |
| Zonulin+Occludin IgG and S100B IgG | 0.3 | <0.0001 |
| Zonulin+Occludin IgM and S100B IgM | 0.8 | <0.0001 |
| Lipopolysaccharide IgA and S100B IgA | 0.7 | <0.0001 |
| Lipopolysaccharide IgG and S100B IgG | 0.2 | <0.0001 |
| Lipopolysaccharide IgM and S100B IgM | 0.8 | <0.0001 |
| Zonulin+Occludin IgA and Aquaporin-4 IgA | 0.6 | <0.0001 |
| Zonulin+Occludin IgG and Aquaporin-4 IgG | 0.5 | <0.0001 |
| Zonulin+Occludin IgM and Aquaporin-4 IgM | 0.8 | <0.0001 |
| Lipopolysaccharide IgA and Aquaporin-4 IgA | 0.5 | <0.0001 |
| Lipopolysaccharide IgG and Aquaporin-4 IgG | 0.3 | <0.0001 |
| Lipopolysaccharide IgM and Aquaporin-4 IgM | 0.8 | <0.0001 |
| Aquaporin-4 IgA and S100B IgA | 0.7 | <0.0001 |
| Aquaporin-4 IgG and S100B IgG | 0.6 | <0.0001 |
| Aquaporin-4 IgM and S100B IgM | 0.9 | <0.0001 |
| Lipopolysaccharide IgA and Zonulin+Occludin IgA | 0.5 | <0.0001 |
| Lipopolysaccharide IgG and Zonulin+Occludin IgG | 0.5 | <0.0001 |
| Lipopolysaccharide IgM and Zonulin+Occludin IgM | 0.7 | <0.0001 |
| Antigens | ASCA- (Controls) | ASCA+ | ANCA- (Controls) | ANCA+ | |||||
|---|---|---|---|---|---|---|---|---|---|
| LPS | IgG | 11/94 | 12% | 29/94 | 31% | 2/94 | 2% | 28/94 | 30% |
| IgM | 8/94 | 9% | 31/94 | 33% | 6/94 | 6% | 29/94 | 31% | |
| IgA | 10/94 | 11% | 28/94 | 30% | 4/94 | 4% | 29/94 | 31% | |
| Zonulin + Occludin | IgG | 12/94 | 13% | 28/94 | 30% | 0/94 | 0% | 28/94 | 30% |
| IgM | 8/94 | 9% | 28/94 | 30% | 5/94 | 5% | 29/94 | 31% | |
| IgA | 11/94 | 12% | 28/94 | 30% | 6/94 | 6% | 29/94 | 31% | |
| S100B | IgG | 5/94 | 5% | 27/94 | 29% | 6/94 | 6% | 29/94 | 31% |
| IgM | 11/94 | 12% | 28/94 | 30% | 8/94 | 9% | 29/94 | 31% | |
| IgA | 2/94 | 2% | 29/94 | 31% | 5/94 | 5% | 30/94 | 32% | |
| AQP4 | IgG | 5/94 | 5% | 28/94 | 30% | 3/94 | 3% | 29/94 | 31% |
| IgM | 7/94 | 7% | 28/94 | 30% | 7/94 | 7% | 28/94 | 30% | |
| IgA | 4/94 | 4% | 29/94 | 31% | 3/94 | 3% | 30/94 | 32% | |
| Barrier Protein Antibodies | Odds Ratio | Confidence Interval | p-Value |
|---|---|---|---|
| S100B IgA | 47 | 16–134 | <0.0001 |
| S100B IgG | 27 | 9–86 | <0.0001 |
| S100B IgM | 9 | 4–20 | <0.0001 |
| Aquaporin-4 IgA | 87 | 27–281 | <0.0001 |
| Aquaporin-4I gG | 95 | 27–326 | <0.0001 |
| Aquaporin-4 IgM | 9 | 4–20 | <0.0001 |
| Zonulin+Occludin IgA | 11 | 5–26 | <0.0001 |
| Zonulin+Occludin IgG | 16 | 7–42 | <0.0001 |
| Zonulin+Occludin IgM | 8 | 4–16 | <0.0001 |
| Lipopolysaccharides IgA | 9 | 4–19 | <0.0001 |
| Lipopolysaccharides IgG | 38 | 15–98 | <0.0001 |
| Lipopolysaccharides IgM | 13 | 6–30 | <0.0001 |
| Correlations | r-Value | p-Value |
|---|---|---|
| Zonulin+Occludin IgA and S100B IgA | 0.6 | <0.0001 |
| Zonulin+Occludin IgG and S100B IgG | 0.6 | <0.0001 |
| Zonulin+Occludin IgM and S100B IgM | 0.7 | <0.0001 |
| Lipopolysaccharide IgA and S100B IgA | 0.6 | <0.0001 |
| Lipopolysaccharide IgG and S100B IgG | 0.4 | <0.0001 |
| Lipopolysaccharide IgM and S100B IgM | 0.4 | <0.0001 |
| Zonulin+Occludin IgA and Aquaporin-4 IgA | 0.7 | <0.0001 |
| Zonulin+Occludin IgG and Aquaporin-4 IgG | 0.6 | <0.0001 |
| Zonulin+Occludin IgM and Aquaporin-4 IgM | 0.8 | <0.0001 |
| Lipopolysaccharide IgA and Aquaporin-4 IgA | 0.7 | <0.0001 |
| Lipopolysaccharide IgG and Aquaporin-4 IgG | 0.5 | <0.0001 |
| Lipopolysaccharide IgM and Aquaporin-4 IgM | 0.5 | <0.0001 |
| Aquaporin-4 IgA and S100B IgA | 0.8 | <0.0001 |
| Aquaporin-4 IgG and S100B IgG | 0.7 | <0.0001 |
| Aquaporin-4 IgM and S100B IgM | 0.9 | <0.0001 |
| Lipopolysaccharide IgA and Zonulin+Occludin IgA | 0.6 | <0.0001 |
| Lipopolysaccharide IgG and Zonulin+Occludin IgG | 0.3 | 0.001 |
| Lipopolysaccharide IgM and Zonulin+Occludin IgM | 0.4 | <0.0001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vojdani, A.; Vojdani, E.; Herbert, M.; Kharrazian, D. Correlation between Antibodies to Bacterial Lipopolysaccharides and Barrier Proteins in Sera Positive for ASCA and ANCA. Int. J. Mol. Sci. 2020, 21, 1381. https://doi.org/10.3390/ijms21041381
Vojdani A, Vojdani E, Herbert M, Kharrazian D. Correlation between Antibodies to Bacterial Lipopolysaccharides and Barrier Proteins in Sera Positive for ASCA and ANCA. International Journal of Molecular Sciences. 2020; 21(4):1381. https://doi.org/10.3390/ijms21041381
Chicago/Turabian StyleVojdani, Aristo, Elroy Vojdani, Martha Herbert, and Datis Kharrazian. 2020. "Correlation between Antibodies to Bacterial Lipopolysaccharides and Barrier Proteins in Sera Positive for ASCA and ANCA" International Journal of Molecular Sciences 21, no. 4: 1381. https://doi.org/10.3390/ijms21041381
APA StyleVojdani, A., Vojdani, E., Herbert, M., & Kharrazian, D. (2020). Correlation between Antibodies to Bacterial Lipopolysaccharides and Barrier Proteins in Sera Positive for ASCA and ANCA. International Journal of Molecular Sciences, 21(4), 1381. https://doi.org/10.3390/ijms21041381