Autoantibody Biomarkers in Rheumatic Diseases


Abstract
:
1. Introduction
2. Autoantibodies in Rheumatoid Arthritis
2.1. Pathogenic Roles of RFs
2.2. Pathogenic Roles of ACPAs
2.2.1. Fc Receptor Binding
2.2.2. Complement Activation
2.2.3. NET Formation
2.2.4. Osteoclastogenesis
2.2.5. Pain Induction
2.3. Assays to Detect ACPAs
2.4. RFs and ACPAs as Biomarkers
3. Autoantibodies in Myositis
3.1. Pathogenic Roles of MSAs
3.2. Assays to Detect MSAs
3.3. MSAs as a Biomarker
3.3.1. Anti-Jo-1 and Other Anti-ARS
3.3.2. Anti-MDA5
3.3.3. Anti-Mi2
3.3.4. Anti-Transcription Intermediary Factor 1 (Anti-TIF1)
3.3.5. Anti-Nuclear Matrix Protein 2 (Anti-NXP2)
3.3.6. Anti-Small Ubiquitin-Like Modifier Activating Enzyme (Anti-SAE)
3.3.7. Anti-SRP and Anti-HMGCR
4. Autoantibodies in SSc
4.1. Pathogenic Roles of SSc Specific Autoantibodies
4.2. Assays to Detect SSc Specific Autoantibodies
4.3. SSc specific Autoantibodies as Biomarkers
4.3.1. Classic SSc Specific ANAs
ACA
ATA
Anti-RNAP III
Anti-U3 RNP (Anti-Fibrillarin)
Anti-Th/To
Anti-U11/U12 RNP
Anti-Eukaryotic Initiation Factor 2B
4.3.2. Functional Antibodies
5. Autoantibodies in ANCA-Associated Vasculitis
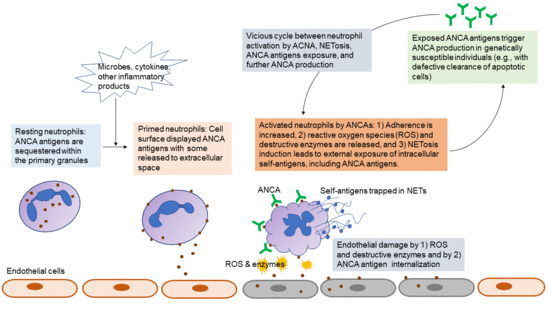
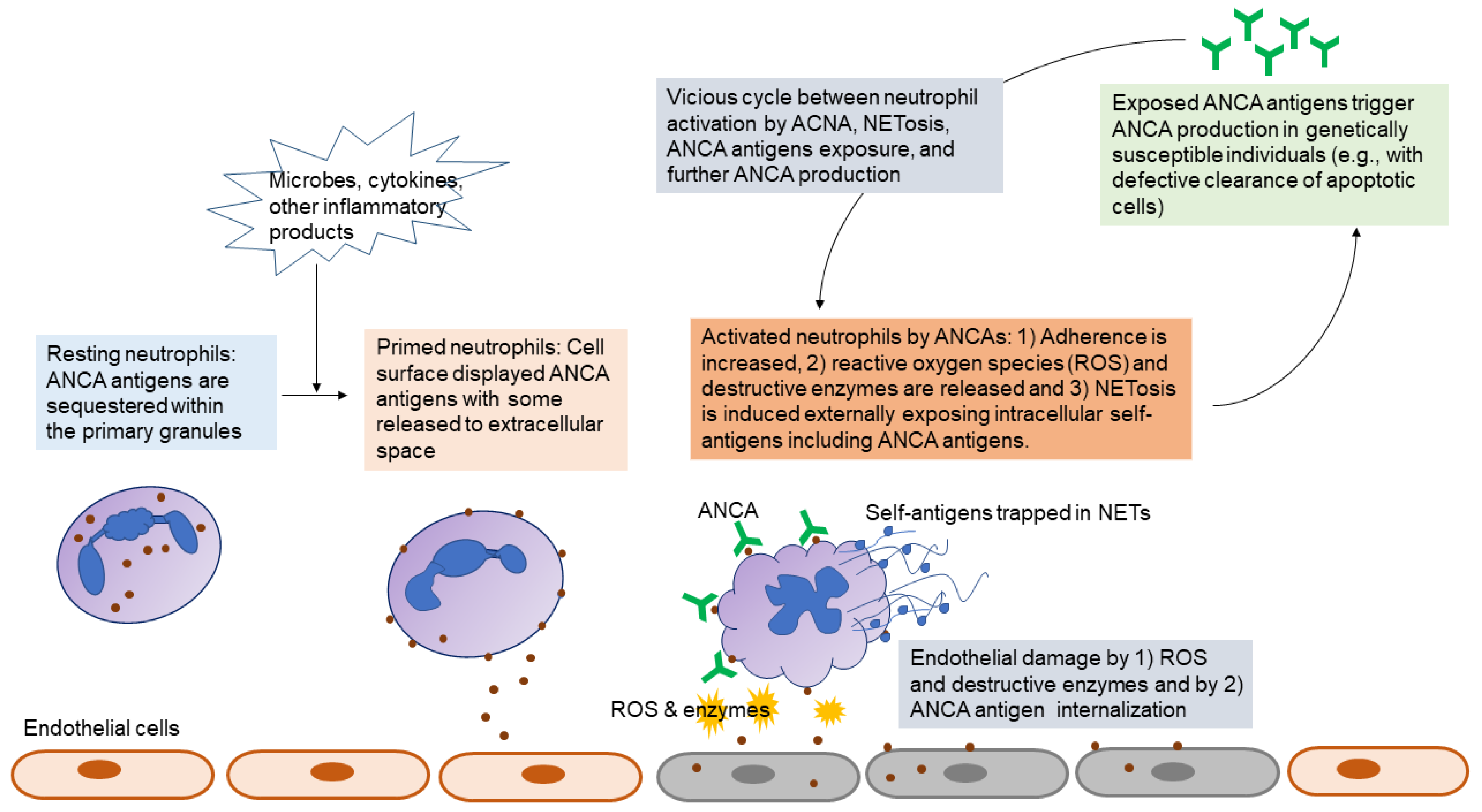
5.1. Pathogenic Roles of ANCA
5.2. Assays to Detect ANCAs
5.3. ANCA as Biomarkers
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bläss, S.; Engel, J.M.; Burmester, G.R. The immunologic homunculus in rheumatoid arthritis. Arthritis Rheum 1999, 42, 2499–2506. [Google Scholar] [CrossRef]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., III; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann. Rheum. Dis. 2010, 69, 1580–1588. [Google Scholar] [CrossRef]
- Tan, E.M.; Smolen, J.S. Historical observations contributing insights on etiopathogenesis of rheumatoid arthritis and role of rheumatoid factor. J. Exp. Med. 2016, 213, 1937–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.W.; Kang, E.H. Autoantibodies in rheumatoid arthritis: Rheumatoid factors and anticitrullinated protein antibodies. QJM 2010, 103, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernick, R.M.; Lipsky, P.E.; Marban-Arcos, E.; Maliakkal, J.J.; Edelbaum, D.; Ziff, M. IgG and IgM rheumatoid factor synthesis in rheumatoid synovial membrane cell cultures. Arthritis Rheum. 1985, 28, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Jones, V.; Taylor, P.C.; Jacoby, R.K.; Wallington, T.B. Synovial synthesis of rheumatoid factors and immune complex constituents in early arthritis. Ann. Rheum. Dis. 1984, 43, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Nell, V.P.; Machold, K.P.; Stamm, T.A.; Eberl, G.; Heinzl, H.; Uffmann, M.; Smolen, J.S.; Steiner, G. Autoantibody profiling as early diagnostic and prognostic tool for rheumatoid arthritis. Ann Rheum Dis 2005, 64, 1731–1736. [Google Scholar] [CrossRef]
- Børretzen, M.; Chapman, C.; Natvig, J.B.; Thompson, K.M. Differences in mutational patterns between rheumatoid factors in health and disease are related to variable heavy chain family and germ-line gene usage. Eur. J. Immunol. 1997, 27, 735–741. [Google Scholar] [CrossRef]
- Jónsson, T.; Thorsteinsson, J.; Kolbeinsson, A.; Jónasdóttir, E.; Sigfússon, N.; Valdimarsson, H. Population study of the importance of rheumatoid factor isotypes in adults. Ann. Rheum. Dis. 1992, 51, 863–868. [Google Scholar] [CrossRef] [Green Version]
- Anquetil, F.; Clavel, C.; Offer, G.; Serre, G.; Sebbag, M. IgM and IgA rheumatoid factors purified from rheumatoid arthritis sera boost the Fc receptor- and complement-dependent effector functions of the disease-specific anti-citrullinated protein autoantibodies. J. Immunol. 2015, 194, 3664–3674. [Google Scholar] [CrossRef] [Green Version]
- Sokolove, J.; Johnson, D.S.; Lahey, L.J.; Wagner, C.A.; Cheng, D.; Thiele, G.M.; Michaud, K.; Sayles, H.; Reimold, A.M.; Caplan, L.; et al. Rheumatoid factor as a potentiator of anti-citrullinated protein antibody-mediated inflammation in rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Hecht, C.; Englbrecht, M.; Rech, J.; Schmidt, S.; Araujo, E.; Engelke, K.; Finzel, S.; Schett, G. Additive effect of anti-citrullinated protein antibodies and rheumatoid factor on bone erosions in patients with RA. Ann. Rheum. Dis. 2015, 74, 2151–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.W.; Kang, E.H. The pathological role of rheumatoid factor in rheumatoid arthritis. Int. J. Clin. Rheumatol. 2010, 5, 651–658. [Google Scholar] [CrossRef]
- Dekkers, J.; Toes, R.E.; Huizinga, T.W.; van der Woude, D. The role of anticitrullinated protein antibodies in the early stages of rheumatoid arthritis. Curr. Opin. Rheumatol. 2016, 28, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, K.A.; Kulik, L.; Tomooka, B.; Braschler, K.J.; Arend, W.P.; Robinson, W.H.; Holers, V.M. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J. Clin. Investig. 2006, 116, 961–973. [Google Scholar] [CrossRef]
- Uysal, H.; Bockermann, R.; Nandakumar, K.S.; Sehnert, B.; Bajtner, E.; Engström, A.; Serre, G.; Burkhardt, H.; Thunnissen, M.M.; Holmdahl, R. Structure and pathogenicity of antibodies specific for citrullinated collagen type II in experimental arthritis. J. Exp. Med. 2009, 206, 449–462. [Google Scholar] [CrossRef]
- Bournazos, S.; Ravetch, J.V. Fcγ Receptor Function and the Design of Vaccination Strategies. Immunity 2017, 47, 224–233. [Google Scholar] [CrossRef]
- Sokolove, J.; Zhao, X.; Chandra, P.E.; Robinson, W.H. Immune complexes containing citrullinated fibrinogen costimulate macrophages via toll-like receptor 4 and Fcgamma receptor. Arthritis Rheum. 2011, 63, 53–62. [Google Scholar] [CrossRef]
- Clavel, C.; Nogueira, L.; Laurent, L.; Iobagiu, C.; Vincent, C.; Sebbag, M.; Serre, G. Induction of macrophage secretion of tumor necrosis factor alpha through Fcgamma receptor IIa engagement by rheumatoid arthritis-specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Rheum. 2008, 58, 678–688. [Google Scholar] [CrossRef]
- Laurent, L.; Clavel, C.; Lemaire, O.; Anquetil, F.; Cornillet, M.; Zabraniecki, L.; Nogueira, L.; Fournié, B.; Serre, G.; Sebbag, M. Fcγ receptor profile of monocytes and macrophages from rheumatoid arthritis patients and their response to immune complexes formed with autoantibodies to citrullinated proteins. Ann. Rheum. Dis. 2011, 70, 1052–1059. [Google Scholar] [CrossRef]
- Goëb, V.; Thomas-L’Otellier, M.; Daveau, R.; Charlionet, R.; Fardellone, P.; Le Loët, X.; Tron, F.; Gilbert, D.; Vittecoq, O. Candidate autoantigens identified by mass spectrometry in early rheumatoid arthritis are chaperones and citrullinated glycolytic enzymes. Arthritis Res. Ther. 2009, 11, R38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Li, X.; Fang, S.; Zhang, X.; Wang, Y.; Zhang, T.; Li, Z.; Xu, Y.; Qu, S.; Liu, C.; et al. Anti-Citrullinated Protein Antibodies Induce Macrophage Subset Disequilibrium in RA Patients. Inflammation 2015, 38, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Trouw, L.A.; Pickering, M.C.; Blom, A.M. The complement system as a potential therapeutic target in rheumatic disease. Nat. Rev. Rheumatol. 2017, 13, 538–547. [Google Scholar] [CrossRef]
- Ji, H.; Ohmura, K.; Mahmood, U.; Lee, D.M.; Hofhuis, F.M.; Boackle, S.A.; Takahashi, K.; Holers, V.M.; Walport, M.; Gerard, C.; et al. Arthritis critically dependent on innate immune system players. Immunity 2002, 16, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Banda, N.K.; Hyatt, S.; Antonioli, A.H.; White, J.T.; Glogowska, M.; Takahashi, K.; Merkel, T.J.; Stahl, G.L.; Mueller-Ortiz, S.; Wetsel, R.; et al. Role of C3a receptors, C5a receptors, and complement protein C6 deficiency in collagen antibody-induced arthritis in mice. J. Immunol. 2012, 188, 1469–1478. [Google Scholar] [CrossRef]
- Sabharwal, U.K.; Vaughan, J.H.; Fong, S.; Bennett, P.H.; Carson, D.A.; Curd, J.G. Activation of the classical pathway of complement by rheumatoid factors. Assessment by radioimmunoassay for C4. Arthritis Rheum. 1982, 25, 161–167. [Google Scholar] [CrossRef]
- Trouw, L.A.; Haisma, E.M.; Levarht, E.W.; van der Woude, D.; Ioan-Facsinay, A.; Daha, M.R. Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum. 2009, 60, 1923–1931. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Kaplan, M.J.; Radic, M. Neutrophil extracellular traps: Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef] [Green Version]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra40. [Google Scholar] [CrossRef] [Green Version]
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Investig. 2012, 122, 1791–1802. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, A.; Joshua, V.; Haj Hensvold, A.; Jin, T.; Sun, M.; Vivar, N.; Ytterberg, A.J.; Engström, M.; Fernandes-Cerqueira, C.; Amara, K.; et al. Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann. Rheum. Dis. 2016, 75, 721–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeling, M.; Hillenhoff, U.; David, J.P.; Schett, G.; Tuckermann, J.; Lux, A.; Nimmerjahn, F. Inflammatory monocytes and Fcγ receptor IV on osteoclasts are critical for bone destruction during inflammatory arthritis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 10729–10734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigerblad, G.; Bas, D.B.; Fernades-Cerqueira, C.; Krishnamurthy, A.; Nandakumar, K.S.; Rogoz, K.; Kato, J.; Sandor, K.; Su, J.; Jimenez-Andrade, J.M.; et al. Autoantibodies to citrullinated proteins induce joint pain independent of inflammation via a chemokine-dependent mechanism. Ann. Rheum. Dis. 2016, 75, 730–738. [Google Scholar] [CrossRef] [Green Version]
- van Venrooij, W.J.; van Beers, J.J.; Pruijn, G.J. Anti-CCP antibodies: The past, the present and the future. Nat. Rev. Rheumatol. 2011, 7, 391–398. [Google Scholar] [CrossRef]
- van Gaalen, F.A.; Visser, H.; Huizinga, T.W. A comparison of the diagnostic accuracy and prognostic value of the first and second anti-cyclic citrullinated peptides (CCP1 and CCP2) autoantibody tests for rheumatoid arthritis. Ann Rheum Dis. 2005, 64, 1510–1512. [Google Scholar] [CrossRef]
- De Rycke, L.; Peene, I.; Hoffman, I.E.; Kruithof, E.; Union, A.; Meheus, L.; Lebeer, K.; Wyns, B.; Vincent, C.; Mielants, H.; et al. Rheumatoid factor and anticitrullinated protein antibodies in rheumatoid arthritis: Diagnostic value, associations with radiological progression rate, and extra-articular manifestations. Ann. Rheum. Dis. 2004, 63, 1587–1593. [Google Scholar] [CrossRef] [Green Version]
- Isaacs, J.D.; Cohen, S.B.; Emery, P.; Tak, P.P.; Wang, J.; Lei, G.; Williams, S.; Lal, P.; Read, S.J. Effect of baseline rheumatoid factor and anticitrullinated peptide antibody serotype on rituximab clinical response: A meta-analysis. Ann. Rheum. Dis. 2013, 72, 329–336. [Google Scholar] [CrossRef]
- Sokolove, J.; Schiff, M.; Fleischmann, R.; Weinblatt, M.E.; Connolly, S.E.; Johnsen, A.; Zhu, J.; Maldonado, M.A.; Patel, S.; Robinson, W.H. Impact of baseline anti-cyclic citrullinated peptide-2 antibody concentration on efficacy outcomes following treatment with subcutaneous abatacept or adalimumab: 2-year results from the AMPLE trial. Ann Rheum Dis. 2016, 75, 709–714. [Google Scholar] [CrossRef]
- Alten, R.; Nüßlein, H.G.; Mariette, X.; Galeazzi, M.; Lorenz, H.M.; Cantagrel, A.; Chartier, M.; Poncet, C.; Rauch, C.; Le Bars, M. Baseline autoantibodies preferentially impact abatacept efficacy in patients with rheumatoid arthritis who are biologic naïve: 6-month results from a real-world, international, prospective study. RMD Open. 2017, 3, e000345. [Google Scholar] [CrossRef]
- Lundberg, I.E.; Tjärnlund, A.; Bottai, M.; Werth, V.P.; Pilkington, C.; de Visser, M.; Alfredsson, L.; Amato, A.A.; Barohn, R.J.; Liang, M.H.; et al. International Myositis Classification Criteria Project Consortium, the Euromyositis Register, and the Juvenile Dermatomyositis Cohort Biomarker Study and Repository (UK and Ireland). 2017 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Adult and Juvenile Idiopathic Inflammatory Myopathies and Their Major Subgroups. Arthritis Rheumatol. 2017, 12, 2271–2282. [Google Scholar]
- Pinal-Fernandez, I.; Amici, D.R.; Parks, C.A.; Derfoul, A.; Casal-Dominguez, M.; Pak, K.; Yeker, R.; Plotz, P.; Milisenda, J.C.; Grau-Junyent, J.M.; et al. Myositis Autoantigen Expression Correlates With Muscle Regeneration but Not Autoantibody Specificity. Arthritis Rheumatol. 2019, 71, 1371–1376. [Google Scholar] [CrossRef]
- Casciola-Rosen, L.; Nagaraju, K.; Plotz, P.; Wang, K.; Levine, S.; Gabrielson, E.; Corse, A.; Rosen, A. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J. Exp. Med. 2005, 201, 591–601. [Google Scholar] [CrossRef]
- Howard, O.M.; Dong, H.F.; Yang, D.; Raben, N.; Nagaraju, K.; Rosen, A.; Casciola-Rosen, L.; Hartlein, M.; Kron, M.; Yiadom, K.; et al. Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes immature dendritic cells. J. Exp. Med. 2002, 196, 781–791. [Google Scholar] [CrossRef]
- Soejima, M.; Kang, E.H.; Gu, X.; Katsumata, Y.; Clemens, P.R.; Ascherman, D.P. Role of innate immunity in a murine model of histidyl-transfer RNA synthetase (Jo-1)-mediated myositis. Arthritis Rheum. 2011, 63, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Eloranta, M.L.; Barbasso Helmers, S.; Ulfgren, A.K.; Rönnblom, L.; Alm, G.V.; Lundberg, I.E. A possible mechanism for endogenous activation of the type I interferon system in myositis patients with anti-Jo-1 or anti-Ro 52/anti-Ro 60 autoantibodies. Arthritis Rheum. 2007, 56, 3112–3124. [Google Scholar] [CrossRef]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Mammen, A.L. Immune-Mediated Necrotizing Myopathy. Curr. Rheumatol. Rep. 2018, 20, 21. [Google Scholar] [CrossRef]
- Christopher-Stine, L.; Casciola-Rosen, L.A.; Hong, G.; Chung, T.; Corse, A.M.; Mammen, A.L. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. 2010, 62, 2757–2766. [Google Scholar] [CrossRef] [Green Version]
- Arouche-Delaperche, L.; Allenbach, Y.; Amelin, D.; Preusse, C.; Mouly, V.; Mauhin, W.; Tchoupou, G.D.; Drouot, L.; Boyer, O.; Stenzel, W.; et al. Pathogenic role of anti-signal recognition protein and anti-3-Hydroxy-3-methylglutaryl-CoA reductase antibodies in necrotizing myopathies: Myofiber atrophy and impairment of muscle regeneration in necrotizing autoimmune myopathies. Ann. Neurol. 2017, 81, 538–548. [Google Scholar] [CrossRef]
- Allenbach, Y.; Arouche-Delaperche, L.; Preusse, C.; Radbruch, H.; Butler-Browne, G.; Champtiaux, N. Necrosis in anti-SRP+ and anti-HMGCR+myopathies: Role of autoantibodies and complement. Neurology 2018, 90, e507–e517. [Google Scholar] [CrossRef]
- Bergua, C.; Chiavelli, H.; Allenbach, Y.; Arouche-Delaperche, L.; Arnoult, C.; Bourdenet, G.; Jean, L.; Zoubairi, R.; Guerout, N.; Mahler, M.; et al. In vivo pathogenicity of IgG from patients with anti-SRP or anti-HMGCR autoantibodies in immune-mediated necrotising myopathy. Ann. Rheum. Dis. 2019, 78, 131–139. [Google Scholar] [CrossRef]
- Espinosa-Ortega, F.; Holmqvist, M.; Alexanderson, H.; Storfors, H.; Mimori, T.; Lundberg, I.E.; Rönnelid, J. Comparison of autoantibody specificities tested by a line blot assay and immunoprecipitation-based algorithm in patients with idiopathic inflammatory myopathies. Ann. Rheum. Dis. 2019, 78, 858–860. [Google Scholar] [CrossRef]
- Richards, T.J.; Eggebeen, A.; Gibson, K.; Yousem, S.; Fuhrman, C.; Gochuico, B.R.; Fertig, N.; Oddis, C.V.; Kaminski, N.; Rosas, I.O.; et al. Characterization and peripheral blood biomarker assessment of anti-Jo-1 antibody-positive interstitial lung disease. Arthritis Rheum. 2009, 60, 2183–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamaguchi, Y.; Fujimoto, M.; Matsushita, T.; Kaji, K.; Komura, K.; Hasegawa, M.; Kodera, M.; Muroi, E.; Fujikawa, K.; Seishima, M.; et al. Common and distinct clinical features in adult patients with anti-aminoacyl-tRNA synthetase antibodies: Heterogeneity within the syndrome. PLoS ONE 2013, 8, e60442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hervier, B.; Devilliers, H.; Stanciu, R.; Meyer, A.; Uzunhan, Y.; Masseau, A.; Dubucquoi, S.; Hatron, P.Y.; Musset, L.; Wallaert, B.; et al. Hierarchical cluster and survival analyses of antisynthetase syndrome: Phenotype and outcome are correlated with anti-tRNA synthetase antibody specificity. Autoimmun. Rev. 2012, 12, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Hirakata, M.; Suwa, A.; Takada, T.; Sato, S.; Nagai, S.; Genth, E.; Song, Y.W.; Mimori, T.; Targoff, I.N. Clinical and immunogenetic features of patients with autoantibodies to asparaginyl-transfer RNA synthetase. Arthritis Rheum. 2007, 56, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, K.; Kawakami, A.; Kaji, K.; Fujimoto, M.; Kawashiri, S.; Iwamoto, N.; Aramaki, T.; Ichinose, K.; Tamai, M.; Kamachi, M.; et al. Association of distinct clinical subsets with myositis-specific autoantibodies towards anti-155/140-kDa polypeptides, anti-140-kDa polypeptides, and anti-aminoacyl tRNA synthetases in Japanese patients with dermatomyositis: A single-centre, cross-sectional study. Scand. J. Rheumatol. 2009, 38, 263–267. [Google Scholar]
- Labrador-Horrillo, M.; Martinez, M.A.; Selva-O’Callaghan, A.; Trallero-Araguas, E.; Balada, E.; Vilardell-Tarres, M.; Juárez, C. Anti-MDA5 antibodies in a large Mediterranean population of adults with dermatomyositis. J. Immunol. Res. 2014, 2014, 290797. [Google Scholar] [CrossRef]
- Sato, S.; Hoshino, K.; Satoh, T.; Fujita, T.; Kawakami, Y.; Fujita, T.; Kuwana, M. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009, 60, 2193–2200. [Google Scholar] [CrossRef]
- Kang, E.H.; Nakashima, R.; Mimori, T.; Kim, J.; Lee, Y.J.; Lee, E.B.; Song, Y.W. Myositis autoantibodies in Korean patients with inflammatory myositis: Anti-140-kDa polypeptide antibody is primarily associated with rapidly progressive interstitial lung disease independent of clinically amyopathic dermatomyositis. BMC Musculoskelet Disord. 2010, 11, 223. [Google Scholar] [CrossRef]
- Tsuji, H.; Nakashima, R.; Hosono, Y.; Imura, Y.; Yagita, M.; Yoshifuji, H.; Hirata, S.; Nojima, T.; Sugiyama, E.; Hatta, K.; et al. A Multicenter Prospective Study of the Efficacy and Safety of Combined Immunosuppressive Therapy with High-Dose Glucocorticoid, Tacrolimus, and Cyclophosphamide in Interstitial Lung Diseases Accompanied by Anti-Melanoma Differentiation-Associated Gene 5-Positive Dermatomyositis. Arthritis Rheumatol. 2019. [Google Scholar] [CrossRef]
- So, H.; Wong, V.T.L.; Lao, V.W.N.; Pang, H.T.; Yip, R.M.L. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin. Rheumatol. 2018, 37, 1983–1989. [Google Scholar] [CrossRef] [PubMed]
- Tansley, S.L.; Betteridge, Z.E.; Gunawardena, H.; Jacques, T.S.; Owens, C.M.; Pilkington, C.; Arnold, K.; Yasin, S.; Moraitis, E.; Wedderburn, L.R.; et al. UK Juvenile Dermatomyositis Research Group. Anti-MDA5 autoantibodies in juvenile dermatomyositis identify a distinct clinical phenotype: A prospective cohort study. Arthritis Res. Ther. 2014, 16, R138. [Google Scholar] [CrossRef] [Green Version]
- Lega, J.C.; Fabien, N.; Reynaud, Q.; Durieu, I.; Durupt, S.; Dutertre, M.; Cordier, J.F.; Cottin, V. The clinical phenotype associated with myositis-specific and associated autoantibodies: A meta-analysis revisiting the so-called antisynthetase syndrome. Autoimmun. Rev. 2014, 13, 883–891. [Google Scholar] [CrossRef]
- Fujimoto, M.; Hamaguchi, Y.; Kaji, K.; Matsushita, T.; Ichimura, Y.; Kodera, M.; Ishiguro, N.; Ueda-Hayakawa, I.; Asano, Y.; Ogawa, F.; et al. Myositis-specific anti-155/140 autoantibodies target transcription intermediary factor 1 family proteins. Arthritis Rheum. 2012, 64, 513–522. [Google Scholar] [CrossRef]
- Gunawardena, H.; Wedderburn, L.R.; North, J.; Betteridge, Z.; Dunphy, J.; Chinoy, H.; Davidson, J.E.; Cooper, R.G.; McHugh, N.J. Clinical associations of autoantibodies to a p155/140 kDa doublet protein in juvenile dermatomyositis. Rheumatology (Oxford) 2008, 47, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Targoff, I.N.; Mamyrova, G.; Trieu, E.P.; Perurena, O.; Koneru, B.; O’Hanlon, T.P.; Miller, F.W.; Rider, L.G. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. 2006, 54, 3682–3689. [Google Scholar] [CrossRef]
- Kaji, K.; Fujimoto, M.; Hasegawa, M.; Kondo, M.; Saito, Y.; Komura, K.; Matsushita, T.; Orito, H.; Hamaguchi, Y.; Yanaba, K.; et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: An association with malignancy. Rheumatology (Oxford) 2007, 46, 25–28. [Google Scholar] [CrossRef] [Green Version]
- Kang, E.H.; Lee, S.J.; Ascherman, D.P.; Lee, Y.J.; Lee, E.Y.; Lee, E.B.; Song, Y.W. Temporal relationship between cancer and myositis identifies two distinctive subgroups of cancers: Impact on cancer risk and survival in patients with myositis. Rheumatology (Oxford) 2016, 55, 1631–1641. [Google Scholar] [CrossRef] [Green Version]
- Trallero-Araguás, E.; Rodrigo-Pendás, J.Á.; Selva-O’Callaghan, A.; Martínez-Gómez, X.; Bosch, X.; Labrador-Horrillo, M.; Grau-Junyent, J.M.; Vilardell-Tarrés, M. Usefulness of anti-p155 autoantibody for diagnosing cancer-associated dermatomyositis: A systematic review and meta-analysis. Arthritis Rheum. 2012, 64, 523–532. [Google Scholar] [CrossRef]
- Ichimura, Y.; Matsushita, T.; Hamaguchi, Y.; Kaji, K.; Hasegawa, M.; Tanino, Y.; Inokoshi, Y.; Kawai, K.; Kanekura, T.; Habuchi, M.; et al. Anti-NXP2 autoantibodies in adult patients with idiopathic inflammatory myopathies: Possible association with malignancy. Ann. Rheum. Dis. 2012, 71, 710–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aouizerate, J.; De Antonio, M.; Bader-Meunier, B.; Barnerias, C.; Bodemer, C.; Isapof, A.; Quartier, P.; Melki, I.; Charuel, J.L.; Bassez, G.; et al. Muscle ischaemia associated with NXP2 autoantibodies: A severe subtype of juvenile dermatomyositis. Rheumatology (Oxford) 2018, 57, 873–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunawardena, H.; Wedderburn, L.R.; Chinoy, H.; Betteridge, Z.E.; North, J.; Ollier, W.E.; Cooper, R.G.; Oddis, C.V.; Ramanan, A.V.; Davidson, J.E.; et al. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthritis Rheum. 2009, 60, 1807–1814. [Google Scholar] [CrossRef]
- Tansley, S.L.; Betteridge, Z.E.; Shaddick, G. Calcinosis in juvenile dermatomyositis is influenced by both anti-NXP2 autoantibody status and age at disease onset. Rheumatology (Oxford) 2014, 53, 2204–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, A.; Chung, L.; Li, S.; Casciola-Rosen, L.; Fiorentino, D.F. Cutaneous and Systemic Findings Associated With Nuclear Matrix Protein 2 Antibodies in Adult Dermatomyositis Patients. Arthritis Care Res. (Hoboken) 2017, 69, 1909–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albayda, J.; Pinal-Fernandez, I.; Huang, W.; Parks, C.; Paik, J.; Casciola-Rosen, L.; Danoff, S.K.; Johnson, C.; Christopher-Stine, L.; Mammen, A.L. Antinuclear Matrix Protein 2 Autoantibodies and Edema, Muscle Disease, and Malignancy Risk in Dermatomyositis Patients. Arthritis Care Res. (Hoboken) 2017, 69, 1771–1776. [Google Scholar] [CrossRef]
- Fiorentino, D.F.; Chung, L.S.; Christopher-Stine, L.; Zaba, L.; Li, S.; Mammen, A.L.; Rosen, A.; Casciola-Rosen, L. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1γ. Arthritis Rheum. 2013, 65, 2954–2962. [Google Scholar] [CrossRef]
- Ceribelli, A.; Fredi, M.; Taraborelli, M. Anti-MJ/NXP-2 autoantibody specificity in a cohort of adult Italian patients with polymyositis/dermatomyositis. Arthritis Res. Ther. 2012, 14, R97. [Google Scholar] [CrossRef] [Green Version]
- Betteridge, Z.E.; Gunawardena, H.; Chinoy, H.; North, J.; Ollier, W.E.; Cooper, R.G.; McHugh, N.J. Clinical and human leucocyte antigen class II haplotype associations of autoantibodies to small ubiquitin-like modifier enzyme, a dermatomyositis-specific autoantigen target, in UK Caucasian adult-onset myositis. Ann. Rheum. Dis. 2009, 68, 1621–1625. [Google Scholar] [CrossRef]
- Fujimoto, M.; Matsushita, T.; Hamaguchi, Y. Autoantibodies to small ubiquitin-like modifier activating enzymes in Japanese patients with dermatomyositis: Comparison with a UK Caucasian cohort. Ann. Rheum. Dis. 2013, 72, 151–153. [Google Scholar] [CrossRef]
- Ge, Y.; Lu, X.; Shu, X. Clinical characteristics of anti-SAE antibodies in Chinese patients with dermatomyositis in comparison with different patient cohorts. Sci. Rep. 2017, 7, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattanaik, D.; Brown, M.; Postlethwaite, B.C.; Postlethwaite, A.E. Pathogenesis of Systemic Sclerosis. Front Immunol. 2015, 6, 272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, F.M.; Frommer, K.W.; Dinser, R.; Walker, U.A.; Czirjak, L.; Denton, C.P.; Allanore, Y.; Distler, O.; Riemekasten, G.; Valentini, G.; et al. Update on the profile of the EUSTAR cohort: An analysis of the EULAR Scleroderma Trials and Research group database. Ann. Rheum. Dis. 2012, 71, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P.; Krieg, T.; Guillevin, L.; Schwierin, B.; Rosenberg, D.; Silkey, M.; Zultak, M.; Matucci-Cerinic, M. Demographic, clinical and antibody characteristics of patients with digital ulcers in systemic sclerosis: Data from the DUO Registry. Ann. Rheum. Dis. 2012, 71, 718–721. [Google Scholar] [CrossRef]
- van den Hoogen, F.; Khanna, D.; Fransen, J.; Johnson, S.R.; Baron, M.; Tyndall, A.; Matucci-Cerinic, M.; Naden, R.P.; Medsger, T.A., Jr.; Carreira, P.E.; et al. 2013 classification criteria for systemic sclerosis: An American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013, 65, 2737–2747. [Google Scholar] [CrossRef] [Green Version]
- Joseph, C.G.; Darrah, E.; Shah, A.A.; Skora, A.D.; Casciola-Rosen, L.A.; Wigley, F.M.; Boin, F.; Fava, A.; Thoburn, C.; Kinde, I.; et al. Association of the autoimmune disease scleroderma with an immunologic response to cancer. Science 2014, 343, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.J.; Shah, A.A.; Li, M.Z.; Xu, Q.; Rosen, A.; Casciola-Rosen, L.; Elledge, S.J. Systematic autoantigen analysis identifies a distinct subtype of scleroderma with coincident cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E7526–E7534. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.A.; Xu, G.; Rosen, A.; Hummers, L.K.; Wigley, F.M.; Elledge, S.J.; Casciola-Rosen, L. Anti-RNPC-3 Antibodies As a Marker of Cancer-Associated Scleroderma. Arthritis Rheumatol. 2017, 69, 1306–1312. [Google Scholar] [CrossRef]
- Salojin, K.V.; Le Tonquèze, M.; Saraux, A.; Nassonov, E.L.; Dueymes, M.; Piette, J.C.; Youinou, P.Y. Antiendothelial cell antibodies: Useful markers of systemic sclerosis. Am. J. Med. 1997, 102, 178–185. [Google Scholar] [CrossRef]
- Sgonc, R.; Gruschwitz, M.S.; Boeck, G.; Sepp, N.; Gruber, J.; Wick, G. Endothelial cell apoptosis in systemic sclerosis is induced by antibody-dependent cell-mediated cytotoxicity via CD95. Arthritis Rheum. 2000, 43, 2550–2562. [Google Scholar] [CrossRef]
- Wolf, S.I.; Howat, S.; Abraham, D.J.; Pearson, J.D.; Lawson, C. Agonistic anti-ICAM-1 antibodies in scleroderma: Activation of endothelial pro-inflammatory cascades. Vascul Pharmacol. 2013, 59, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihai, C.; Tervaert, J.W. Anti-endothelial cell antibodies in systemic sclerosis. Ann. Rheum. Dis. 2010, 69, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Gunther, J.; Kill, A.; Becker, M.O.; Heiddecke, H.; Rademacher, J.; Siegert, E.; Radic, M.; Burmester, G.R.; Gabriela, R. Angiotensin receptor type 1 and endothelin receptor type A on immune cells mediate migration and the expression of IL-8 and CCL18 when stimulated by autoantibodies from systemic sclerosis patients. Arthritis Res. Ther. 2014, 16, R65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, M.O.; Kill, A.; Kutsche, M.; Gunther, J.; Rose, A.; Tabeling, C.; Witzenrath, M.; Kuhl, A.A.; Heidecke, H.; Ghofrani, H.A.; et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am. J. Respir. Crit. Care Med. 2014, 190, 808–817. [Google Scholar] [CrossRef]
- Avouac, J.; Riemekasten, G.; Meune, C. Autoantibodies against endothelin 1 type a receptor are strong predictors of digital ulcers in systemic sclerosis. J. Rheumatol 2015, 42, 1801–1807. [Google Scholar] [CrossRef]
- Riemekasten, G.; Philippe, A.; Näther, M.; Slowinski, T.; Müller, D.N.; Heidecke, H.; Matucci-Cerinic, M.; Czirják, L.; Lukitsch, I.; Becker, M.; et al. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 530–536. [Google Scholar] [CrossRef]
- Kill, A.; Tabeling, C.; Undeutsch, R.; Kühl, A.A.; Günther, J.; Radic, M.; Becker, M.O.; Heidecke, H.; Worm, M.; Witzenrath, M.; et al. Autoantibodies to angiotensin and endothelin receptors in systemic sclerosis induce cellular and systemic events associated with disease pathogenesis. Arthritis Res. Ther. 2014, 16, R2. [Google Scholar] [CrossRef] [Green Version]
- Moroncini, G.; Grieco, A.; Nacci, G.; Paolini, C.; Tonnini, C.; Pozniak, K.N.; Cuccioloni, M.; Mozzicafreddo, M.; Svegliati, S.; Angeletti, M.; et al. Epitope Specificity Determines Pathogenicity and Detectability of Anti-Platelet-Derived Growth Factor Receptor α Autoantibodies in Systemic Sclerosis. Arthritis Rheumatol. 2015, 67, 1891–1903. [Google Scholar] [CrossRef] [Green Version]
- Baroni, S.S.; Santillo, M.; Bevilacqua, F.; Luchetti, M.; Spadoni, T.; Mancini, M.; Fraticelli, P.; Sambo, P.; Fudrius, A.; Kazlauskas, A.; et al. Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N. Engl. J. Med. 2006, 354, 2667–2676. [Google Scholar] [CrossRef] [Green Version]
- Luchetti, M.M.; Moroncini, G.; Escamez, M.j.; Sveglati-Baroni, S.; Spadoni, T.; Grieco, A.; Paolini, C.; Funaro, A.; Avvedimento, E.V.; Larcher, F.; et al. Induction of scleroderma fibrosis in skin-humanized mice by administration of anti-platelet-derived growth factor receptor agonistic autoantibodies. Arthritis Rheumatol. 2016, 68, 2263–2273. [Google Scholar] [CrossRef]
- Zhou, X.; Tan, F.K.; Milewicz, D.M.; Guo, X.; Bona, C.A.; Arnett, F.C. Autoantibodies to fibrillin-1 activate normal human fibroblasts in culture through the TGF-beta pathway to recapitulate the “scleroderma phenotype”. J. Immunol. 2005, 175, 4555–4560. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.; Eaker, E.Y.; Sallustio, J.E.; Peebles, C.; Tan, E.M.; Williams, R.C.J.r. Antimyenteric neuronal antibodies in scleroderma. J. Clin. Investig. 1994, 94, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaker, E.Y.; Kuldau, J.G.; Verne, G.N.; Ross, S.O.; Sallustio, J.E. Myenteric neuronal antibodies in scleroderma: Passive transfer evokes alterations in intestinal myoelectric activity in a rat model. J. Lab. Clin. Med. 1999, 133, 551–556. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, J.; Kedika, R.; Mendoza, F.; Jimenez, S.A.; Blomain, E.S.; Dimarino, A.J.; Cohen, S.; Rattan, S. Role of muscarinic-3 receptor antibody in systemic sclerosis: Correlation with disease duration and effects of IVIG. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G1052–G1060. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.A.; Rosen, A.; Hummers, L.; Wigley, F.; Casciola-Rosen, L. Close temporal relationship between onset of cancer and scleroderma in patients with RNA polymerase I/III antibodies. Arthritis Rheum. 2010, 62, 2787–2795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, K.A.; Roberts-Thomson, P.J.; Lester, S.; Tan, J.A.; Hakendorf, P.; Rischmueller, M.; Zochling, J.; Sahhar, J.; Nash, P.; Roddy, J.; et al. Interpretation of an Extended Autoantibody Profile in a Well-Characterized Australian Systemic Sclerosis (Scleroderma) Cohort Using Principal Components Analysis. Arthritis Rheumatol. 2015, 67, 3234–3244. [Google Scholar] [CrossRef]
- Loizos, N.; Lariccia, L.; Weiner, J.; Griffith, H.; Boin, F.; Hummers, L.; Wigley, F.; Kussie, P. Lack of detection of agonist activity by antibodies to platelet-derived growth factor receptor alpha in a subset of normal and systemic sclerosis patient sera. Arthritis Rheum. 2009, 60, 1145–1151. [Google Scholar] [CrossRef]
- Domsic, R.T. Scleroderma: The roal of serum autoantibodies in defining specific clinical phenotypes and organ system involvement. Curr. Opin. Rheumatol. 2014, 26, 646–652. [Google Scholar] [CrossRef] [Green Version]
- Kallenberg, C.G.; Wouda, A.A.; Hoet, M.H.; van Venrooij, W.J. Development of connective tissue disease in patients presenting with Raynaud’s phenomenon: A six year follow up with emphasis on the predictive value of antinuclear antibodies as detected by immunoblotting. Ann. Rheum. Dis. 1988, 47, 634–641. [Google Scholar] [CrossRef] [Green Version]
- Moroi, Y.; Peebles, C.; Fritzler, M.J.; Steigerwald, J.; Tan, E.M. Autoantibody to centromere (kinetochore) in scleroderma sera. Proc. Natl. Acad. Sci. USA 1980, 77, 1627–1631. [Google Scholar] [CrossRef] [Green Version]
- Kuwana, M.; Kaburaki, J.; Okano, Y.; Tojo, T.; Homma, M. Clinical and prognostic associations based on serum antinuclear antibodies in Japanese patients with systemic sclerosis. Arthritis Rheum. 1994, 37, 75–83. [Google Scholar] [CrossRef]
- Stupi, A.M.; Steen, V.D.; Owens, G.R.; Barnes, E.L.; Rodnan, G.P.; Medsger, T.A., Jr. Pulmonary hypertension in the CREST syndrome variant of systemic sclerosis. Arthritis Rheum. 1986, 29, 515–524. [Google Scholar] [CrossRef]
- Coghlan, J.G.; Denton, C.P.; Grünig, E.; Bonderman, D.; Distler, O.; Khanna, D.; Müller-Ladner, U.; Pope, J.E.; Vonk, M.C.; Doelberg, M.; et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: The DETECT study. Ann. Rheum. Dis. 2014, 73, 1340–1349. [Google Scholar] [CrossRef] [Green Version]
- Komócsi, A.; Vorobcsuk, A.; Faludi, R.; Pintér, T.; Lenkey, Z.; Költo, G.; Czirják, L. The impact of cardiopulmonary manifestations on the mortality of SSc: A systematic review and meta-analysis of observational studies. Rheumatology (Oxford) 2012, 51, 1027–1036. [Google Scholar] [CrossRef] [Green Version]
- Steen, V.D. Autoantibodies in systemic sclerosis. Semin. Arthritis Rheum. 2005, 35, 35–42. [Google Scholar] [CrossRef]
- Cavazzana, I.; Ceribelli, A.; Airò, P.; Zingarelli, S.; Tincani, A.; Franceschini, F. Anti-RNA polymerase III antibodies: A marker of systemic sclerosis with rapid onset and skin thickening progression. Autoimmun Rev. 2009, 8, 580–584. [Google Scholar] [CrossRef]
- Sobanski, V.; Dauchet, L.; Lefèvre, G.; Lambert, M.; Morell-Dubois, S.; Sy, T.; Hachulla, E.; Hatron, P.Y.; Launay, D.; Dubucquoi, S. Prevalence of anti-RNA polymerase III antibodies in systemic sclerosis: New data from a French cohort and a systematic review and meta-analysis. Arthritis Rheumatol. 2014, 66, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Kuwana, M.; Kaburaki, J.; Mimori, T.; Tojo, T.; Homma, M. Autoantibody reactive with three classes of RNA polymerases in sera from patients with systemic sclerosis. J. Clin. Investig. 1993, 91, 1399–1404. [Google Scholar] [CrossRef] [Green Version]
- Ghrénassia, E.; Avouac, J.; Khanna, D.; Derk, C.T.; Distler, O.; Suliman, Y.A.; Airo, P.; Carreira, P.E.; Foti, R.; Granel, B.; et al. Prevalence, correlates and outcomes of gastric antral vascular ectasia in systemic sclerosis: A EUSTAR case-control study. J. Rheumatol. 2014, 41, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, R.; Lucas, M.; Fertig, N.; Oddis, C.V.; Medsger, T.A., Jr. Anti-U3 RNP autoantibodies in systemic sclerosis. Arthritis Rheum. 2009, 60, 1112–1118. [Google Scholar] [CrossRef]
- Tall, F.; Dechomet, M.; Riviere, S.; Cottin, V.; Ballot, E.; Tiev, K.P.; Montin, R.; Morin, C.; Chantran, Y.; Grange, C.; et al. The Clinical Relevance of Antifibrillarin (anti-U3-RNP) Autoantibodies in Systemic Sclerosis. Scand. J. Immunol. 2017, 85, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Arnett, F.C.; Reveille, J.D.; Goldstein, R.; Pollard, K.M.; Leaird, K.; Smith, E.A.; Leroy, E.C.; Fritzler, M.J. Autoantibodies to fibrillarin in systemic sclerosis (scleroderma). An immunogenetic, serologic, and clinical analysis. Arthritis Rheum. 1996, 39, 1151–1160. [Google Scholar] [CrossRef]
- Yang, J.M.; Hildebrandt, B.; Luderschmidt, C.; Pollard, K.M. Human scleroderma sera contain autoantibodies to protein components specific to the U3 small nucleolar RNP complex. Arthritis Rheum. 2003, 48, 210–217. [Google Scholar] [CrossRef]
- Mitri, G.M.; Lucas, M.; Fertig, N.; Steen, V.D.; Medsger, T.A., Jr. A comparison between anti-Th/To- and anticentromere antibody-positive systemic sclerosis patients with limited cutaneous involvement. Arthritis Rheum. 2003, 48, 203–209. [Google Scholar] [CrossRef]
- Ceribelli, A.; Cavazzana, I.; Franceschini, F.; Airò, P.; Tincani, A.; Cattaneo, R.; Pauley, B.A.; Chan, E.K.; Satoh, M. Anti-Th/To are common antinucleolar autoantibodies in Italian patients with scleroderma. J. Rheumatol. 2010, 37, 2071–2075. [Google Scholar] [CrossRef]
- Fertig, N.; Domsic, R.T.; Rodriguez-Reyna, T.; Kuwana, M.; Lucas, M.; Medsger, T.A., Jr.; Feghali-Bostwickm, C.A. Anti-U11/U12 RNP antibodies in systemic sclerosis: A new serologic marker associated with pulmonary fibrosis. Arthritis Rheum. 2009, 61, 958–965. [Google Scholar] [CrossRef] [Green Version]
- Betteridge, Z.E.; Woodhead, F.; Lu, H.; Shaddick, G.; Bunn, C.C.; Denton, C.P.; Abraham, D.J.; du Bois, R.M.; Lewis, M.; Wells, A.U.; et al. Anti-Eukaryotic Initiation Factor 2B Autoantibodies Are Associated With Interstitial Lung Disease in Patients With Systemic Sclerosis. Arthritis Rheumatol. 2016, 68, 2778–2783. [Google Scholar] [CrossRef] [Green Version]
- Elhai, M.; Boubaya, M.; Distler, O.; Smith, V.; Matucci-Cerinic, M.; Alegre Sancho, J.J.; Truchetet, M.E.; Braun-Moscovici, Y.; Iannone, F.; Novikov, P.I.; et al. Outcomes of patients with systemic sclerosis treated with rituximab in contemporary practice: A prospective cohort study. Ann. Rheum. Dis. 2019, 78, 979–987. [Google Scholar] [CrossRef]
- Sircar, G.; Goswami, R.P.; Sircar, D.; Ghosh, A.; Ghosh, P. Intravenous cyclophosphamide vs. rituximab for the treatment of early diffuse scleroderma lung disease: Open label, randomized, controlled trial. Rheumatology (Oxford) 2018, 57, 2106–2113. [Google Scholar] [CrossRef]
- Jordan, S.; Distler, J.H.; Maurer, B.; Huscher, D.; van Laar, J.M.; Allanore, Y.; Distler, O. Effects and safety of rituximab in systemic sclerosis: An analysis from the European Scleroderma Trial and Research (EUSTAR) group. Ann. Rheum. Dis. 2015, 74, 1188–1194. [Google Scholar] [CrossRef]
- Damoiseaux, J.; Csernok, E.; Rasmussen, N.; Moosig, F.; van Paassen, P.; Baslund, B.; Vermeersch, P.; Blockmans, D.; Cohen Tervaert, J.W.; Bossuyt, X. Detection of antineutrophil cytoplasmic antibodies (ANCAs): A multicentre European Vasculitis Study Group (EUVAS) evaluation of the value of indirect immunofluorescence (IIF) versus antigen-specific immunoassays. Ann. Rheum. Dis. 2017, 76, 647–665. [Google Scholar] [CrossRef]
- Jennette, J.C.; Falk, R.J. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat. Rev. Rheumatol. 2014, 10, 463–473. [Google Scholar] [CrossRef]
- Xiao, H.; Heeringa, P.; Hu, P.; Liu, Z.; Zhao, M.; Aratani, Y.; Maeda, N.; Falk, R.J.; Jennette, J.C. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J. Clin. Investig. 2002, 110, 955–963. [Google Scholar] [CrossRef]
- Little, M.A.; Al-Ani, B.; Ren, S.; Al-Nuaimi, H.; Leite, M., Jr.; Alpers, C.E.; Savage, C.O.; Duffield, J.S. Anti-proteinase 3 anti-neutrophil cytoplasm autoantibodies recapitulate systemic vasculitis in mice with a humanized immune system. PLoS ONE 2012, 7, e28626. [Google Scholar] [CrossRef]
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013, 65, 1–11. [Google Scholar] [CrossRef]
- Bossuyt, X.; Cohen Tervaert, J.W.; Arimura, Y.; Blockmans, D.; Flores-Suárez, L.F.; Guillevin, L.; Hellmich, B.; Jayne, D.; Jennette, J.C.; Kallenberg, C.G.M.; et al. Position paper: Revised 2017 international consensus on testing of ANCAs in granulomatosis with polyangiitis and microscopic polyangiitis. Nat. Rev. Rheumatol. 2017, 13, 683–692. [Google Scholar] [CrossRef]
- Lyons, P.A.; Rayner, T.F.; Trivedi, S.; Holle, J.U.; Watts, R.A.; Jayne, D.R.; Baslund, B.; Brenchley, P.; Bruchfeld, A.; Chaudhry, A.N.; et al. Genetically distinct subsets within ANCA-associated vasculitis. N. Engl. J. Med. 2012, 367, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Basu, N.; Watts, R.; Bajema, I.; Baslund, B.; Bley, T.; Boers, M.; Brogan, P.; Calabrese, L.; Cid, M.C.; Cohen-Tervaert, J.W.; et al. EULAR points to consider in the development of classification and diagnostic criteria in systemic vasculitis. Ann. Rheum. Dis. 2010, 69, 1744–1750. [Google Scholar] [CrossRef]
- Xiao, H.; Heeringa, P.; Liu, Z.; Huugen, D.; Hu, P.; Maeda, N.; Falk, R.J.; Jennette, J.C. The role of neutrophils in the induction of glomerulonephritis by anti-myeloperoxidase antibodies. Am. J. Pathol. 2005, 167, 39–45. [Google Scholar] [CrossRef]
- Korkmaz, B.; Jenne, D.E.; Gauthier, F. Relevance of the mouse model as a therapeutic approach for neutrophil proteinase 3 associated human diseases. Int. Immunopharmacol. 2013, 17, 1198–1205. [Google Scholar] [CrossRef]
- Geetha, D.; Jefferson, J.A. ANCA-Associated Vasculitis: Core Curriculum 2020. Am. J. Kidney Dis. 2020, 75, 124–137. [Google Scholar] [CrossRef] [Green Version]
- Charles, L.A.; Caldas, M.L.; Falk, R.J.; Terrell, R.S.; Jennette, J.C. Antibodies against granule proteins activate neutrophils in vitro. J. Leukoc. Biol. 1991, 50, 539–546. [Google Scholar] [CrossRef]
- Hess, C.; Sadallah, S.; Schifferli, J.A. Induction of neutrophil responsiveness to myeloperoxidase antibodies by their exposure to supernatant of degranulated autologous neutrophils. Blood 2000, 96, 2822–2827. [Google Scholar] [CrossRef]
- Csernok, E.; Ernst, M.; Schmitt, W.; Bainton, D.F.; Gross, W.L. Activated neutrophils express proteinase 3 on their plasma membrane in vitro and in vivo. Clin. Exp. Immunol. 1994, 95, 244–250. [Google Scholar] [CrossRef]
- Gilligan, H.M.; Bredy, B.; Brady, H.R.; Hébert, M.J.; Slayter, H.S.; Xu, Y.; Rauch, J.; Shia, M.A.; Koh, J.S.; Levine, J.S. Antineutrophil cytoplasmic autoantibodies interact with primary granule constituents on the surface of apoptotic neutrophils in the absence of neutrophil priming. J. Exp. Med. 1996, 184, 2231–2241. [Google Scholar] [CrossRef]
- Kantari, C.; Pederzoli-Ribeil, M.; Amir-Moazami, O.; Gausson-Dorey, V.; Moura, I.C.; Lecomte, M.C.; Benhamou, M.; Witko-Sarsat, V. Proteinase 3, the Wegener autoantigen, is externalized during neutrophil apoptosis: Evidence for a functional association with phospholipid scramblase 1 and interference with macrophage phagocytosis. Blood 2007, 110, 4086–4095. [Google Scholar] [CrossRef] [Green Version]
- Kettritz, R.; Jennette, J.C.; Falk, R.J. Crosslinking of ANCA antigens stimulates superoxide release by human neutrophils. J. Am. Soc. Nephrol. 1997, 8, 386–394. [Google Scholar]
- Porges, A.J.; Redecha, P.B.; Kimberly, W.T.; Csernok, E.; Gross, W.; Kimberly, R.P. Anti-neutrophil cytoplasmic antibodies engage and activate human neutrophils via FcgRIIa. J. Immunol. 1994, 153, 1271–1280. [Google Scholar]
- Falk, R.J.; Terrell, R.S.; Charles, L.A.; Jennette, J.C. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc. Natl. Acad. Sci. USA 1990, 87, 4115–4119. [Google Scholar] [CrossRef] [Green Version]
- Söderberg, D.; Segelmark, M. Neutrophil extracellular traps in ANCA-associated vasculitis. Front. Immunol. 2016, 7, 256. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Harper, L.; Cockwell, P.; Adu, D.; Savage, C.O. Neutrophil priming and apoptosis in anti-neutrophil cytoplasmic autoantibody-associated vasculitis. Kidney Int. 2001, 59, 1729–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbwachs, L.; Lesavre, P. Endothelium-Neutrophil Interactions in ANCA-Associated Diseases. J. Am. Soc. Nephrol. 2012, 23, 1449–1461. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.J.; Preston, G.A.; Pendergraft, W.F.; Segelmark, M.; Heeringa, P.; Hogan, S.L.; Jennette, J.C.; Falk, R.J. Internalization of proteinase 3 is concomitant with endothelial cell apoptosis and internalization of myeloperoxidase with generation of intracellular oxidants. Am. J. Pathol. 2001, 158, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Harper, L.; Radford, D.; Plant, T.; Drayson, M.; Adu, D.; Savage, C.O. IgG from myeloperoxidase-antineutrophil cytoplasmic antibody-positive patients stimulates greater activation of primed neutrophils than IgG from proteinase 3-antineutrophil cytosplasmic antibody-positive patients. Arthritis Rheum. 2001, 44, 921–930. [Google Scholar] [CrossRef]
- Csernok, E.; Moosig, F. Current and emerging techniques for ANCA detection in vasculitis. Nat. Rev. Rheumatol. 2014, 10, 494–501. [Google Scholar] [CrossRef]
- Holle, J.U.; Hellmich, B.; Backes, M.; Gross, W.L.; Csernok, E. Variations in performance characteristics of commercial enzyme immunoassay kits for detection of antineutrophil cytoplasmic antibodies: What is the optimal cut off? Ann. Rheum. Dis. 2005, 64, 1773–1779. [Google Scholar] [CrossRef] [Green Version]
- Csernok, E.; Holle, J.; Hellmich, B.; Willem, J.; Tervaert, C.; Kallenberg, C.G.; Limburg, P.C.; Niles, J.; Pan, G.; Specks, U.; et al. Evaluation of capture ELISA for detection of antineutrophil cytoplasmic antibodies directed against proteinase 3 in Wegener’s granulomatosis: First results from a multicentre study. Rheumatology (Oxford) 2004, 43, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Csernok, E.; Ahlquist, D.; Ullrich, S.; Gross, W.L. A critical evaluation of commercial immunoassays for antineutrophil cytoplasmic antibodies directed against proteinase 3 and myeloperoxidase in Wegener’s granulomatosis and microscopic polyangiitis. Rheumatology (Oxford) 2002, 41, 1313–1317. [Google Scholar] [CrossRef] [Green Version]
- Hellmich, B.; Csernok, E.; Fredenhagen, G.; Gross, W.L. A novel high sensitivity ELISA for detection of antineutrophil cytoplasm antibodies against proteinase-3. Clin. Exp. Rheumatol. 2007, 25, S1–S5. [Google Scholar]
- Roggenbuck, D.; Buettner, T.; Hoffmann, L.; Schmechta, H.; Reinhold, D.; Conrad, K. High-sensitivity detection of autoantibodies against proteinase-3 by a novel third-generation enzyme-linked immunosorbent assay. Ann. N. Y. Acad. Sci. 2009, 1173, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Holle, J.U.; Csernok, E.; Fredenhagen, G.; Backes, M.; Bremer, J.P.; Gross, W.L. Clinical evaluation of hsPR3-ANCA ELISA for detection of antineutrophil cytoplasmatic antibodies directed against proteinase 3. Ann. Rheum. Dis. 2010, 69, 468–469. [Google Scholar] [CrossRef] [PubMed]
- Savige, J.; Gillis, D.; Benson, E.; Davies, D.; Esnault, V.; Falk, R.J.; Hagen, E.C.; Jayne, D.; Jennette, J.C.; Paspaliaris, B.; et al. International Consensus Statement on Testing and Reporting of Antineutrophil Cytoplasmic Antibodies (ANCA). Am. J. Clin. Pathol. 1999, 111, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Lionaki, S.L.; Blyth, E.R.; Hogan, S.L.; Hu, Y.; Senior, B.A.; Jennette, C.E.; Nachman, P.H.; Jennette, J.C.; Falk, R.J. Classification of antineutrophil cytoplasmic autoantibody vasculitides: The role of antineutrophil cytoplasmic autoantibody specificity for myeloperoxidase or proteinase 3 in disease recognition and prognosis. Arthritis Rheum. 2012, 64, 3452–3462. [Google Scholar] [CrossRef] [Green Version]
- Stone, J.H.; Merkel, P.A.; Spiera, R.; Seo, P.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.M.; Clair, E.W.S.; Turkiewicz, A.; Tchao, N.K.; et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N. Engl. J. Med. 2000, 363, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Unizony, S.; Villarreal, M.; Miloslavsky, E.M.; Lu, N.; Merkel, P.A.; Spiera, R.; Seo, P.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.; et al. Clinical outcomes of treatment of anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis based on ANCA type. Ann. Rheum. Dis. 2016, 75, 1166–1169. [Google Scholar] [CrossRef]
- Mahr, A.; Katsahian, S.; Varet, H.; Guillevin, L.; Hagen, E.C.; Hoglund, P.; Merkel, P.A.; Pagnoux, C.; Rasmussen, N.; Westman, K.; et al. Revisiting the classification of clinical phenotypes of anti-neutrophil cytoplasmic antibody-associated vasculitis: A cluster analysis. Ann. Rheum. Dis. 2013, 72, 1003–1010. [Google Scholar] [CrossRef]
- Mukhtyar, C.; Flossmann, O.; Hellmich, B.; Bacon, P.; Cid, M.; Cohen-Tervaert, J.W.; Gross, W.L.; Guillevin, L.; Jayne, D.; Mahr, A.; et al. Outcomes from studies of antineutrophil cytoplasm antibody associated vasculitis: A systematic review by the European League Against Rheumatism systemic vasculitis task force. Ann. Rheum. Dis. 2008, 67, 1004–1010. [Google Scholar] [CrossRef] [Green Version]
- Weidner, S.; Geuss, S.; Hafezi-Rachti, S.; Wonka, A.; Rupprecht, H.D. ANCA-associated vasculitis with renal involvement: An outcome analysis. Nephrol. Dial. Transplant. 2004, 19, 1403–1411. [Google Scholar] [CrossRef]
- de Joode, A.A.; Sanders, J.S.; Stegeman, C.A. Renal survival in proteinase 3 and myeloperoxidase ANCA-associated systemic vasculitis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1709–1717. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, A.J.; Segelmark, M. A population-based study showing better renal prognosis for proteinase 3 antineutrophil cytoplasmic antibody (ANCA)-associated nephritis versus myeloperoxidase ANCA-associated nephritis. J. Rheumatol. 2014, 41, 1366–1373. [Google Scholar] [CrossRef] [PubMed]
- Puéchal, X.; Pagnoux, C.; Perrodeau, É.; Hamidou, M.; Boffa, J.J.; Kyndt, X.; Lifermann, F.; Papo, T.; Merrien, D.; Smail, A.; et al. Long-Term Outcomes Among Participants in the WEGENT Trial of Remission-Maintenance Therapy for Granulomatosis With Polyangiitis (Wegener’s) or Microscopic Polyangiitis. Arthritis Rheumatol. 2016, 68, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Tomasson, G.; Grayson, P.C.; Mahr, A.D.; Lavalley, M.; Merkel, P.A. Value of ANCA measurements during remission to predict a relapse of ANCA-associated vasculitis: A metaanalysis. Rheumatology 2012, 51, 100–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemna, M.J.; Damoiseaux, J.; Austen, J.; Winkens, B.; Peters, J.; van Paassen, P.; Cohen Tervaert, J.W. ANCA as a predictor of relapse: Useful in patients with renal involvement but not in patients with nonrenal disease. J. Am. Soc. Nephrol. 2015, 26, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Land, J.; Rutgers, A.; Kallenberg, C.G. Anti-neutrophil cytoplasmic autoantibody pathogenicity revisited: Pathogenic versus non-pathogenic anti-neutrophil cytoplasmic autoantibody. Nephrol. Dial. Transplant. 2014, 29, 739–745. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| MSAs | Autoantigens | Representative Physiologic Functions of Autoantigens | Prevalence in Adults | Associated Clinical Phenotypes |
|---|---|---|---|---|
| Traditional Autoantibodies | ||||
| Anti-ARS | Aminoacyl-tRNA-synthetase | Translation | 20%–40% | Anti-synthetase syndrome |
| anti-Jo-1 | histidyl-tRNA-synthetase | 15%–20% | ||
| anti-PL-7 | threonyl-tRNA-synthetase | <5% | ||
| anti-PL-12 | alanyl-tRNA-synthetase | <5% | ||
| anti-OJ | isoleucyl-tRNA-synthetase | <5% | ||
| anti-EJ | glycyl-tRNA-synthetase | <5% | ||
| anti-KS | asparaginyl-tRNA-synthetase | <5% | ||
| anti-Ha | tyrosyl-tRNA-synthetase | <5% | ||
| anti-Zo | phenylalanyl-tRNA-synthetase | <5% | ||
| Anti-SRP | Signal recognition particle | Translocation of newly synthesized proteins across endoplasmic reticulum | <10%, but some studies reporting 20% among myositis patients | Immune-mediated necrotizing myopathy or cardiac involvement |
| Anti-Mi2 | Nucleosome remodeling deacetylase | Chromatin remodeling | <10% | Rashes of DM |
| Recent or Newly Found Antibodies | ||||
| Anti-MDA5 | Melanoma differentiation-associated protein 5 | Pattern recognition receptor for viral RNA | 20%–50% in Asians 7%–13% in Caucasians | Rapidly progressive ILD in classic DM and CADM |
| Anti-Mi2 | Nucleosome remodeling deacetylase | Chromatin remodeling | <10% | Rashes of DM |
| Anti-TIF1 | Transcriptional intermediary factor 1 | Chromatin remodeling; ubiquitination | 10%–20% of adult DM (but higher in cancer-associated myositis) | Malignancy in adult DM |
| Anti-NXP2 | Nuclear matrix protein 2 | Chromatin remodeling; activation of p53 | <10% | Malignancy in adult DM; calcinosis and severe myositis in juvenile DM |
| Anti-HMGCR | 3-hydroxy-3-methylglutaryl-coenzyme A reductase | Cholesterol synthesis | <10% | Immune-mediated necrotizing myopathy; previous statin exposure |
| Anti-SAE | SUMO activating enzyme subunit 1 | Post-translational protein modification | <5% | Transition from CADM to overt myositis; dysphagia in both Caucasians and Asians but ILD only in Asians. |
| Autoantibody | Autoantigens | Representative Physiologic Functions of Autoantigens | Prevalence | Associated Clinical Phenotypes |
|---|---|---|---|---|
| Traditional SSc-Specific ANAs | ||||
| Anti-centromere | Centromeric protein | Contains histone H3 and involves epigenetic process | 10%–50% | Limited cutaneous SSc PAH CREST DU in late phase |
| Anti-topoisomerase I | Topoisomerase I | Enzyme that cuts, relaxes, and reanneals one of the two strands of double-stranded DNA. | 20%–30% | Diffuse cutaneous SSc ILD DU in early phase |
| Anti-RNA polymerase | RNA polymerase | Transcription | 5%–30% | Diffuse cutaneous SSc Rapid progression of skin-thickening, renal crisis, GAVE, and cancer |
| Novel SSc-Specific ANAs | ||||
| Anti-U3 RNP | Fibrillarin complexed with small nucleolar RNA U3 | Pre-rRNA processing localized in the fibrillar region of the nucleolus. | <10% | Both limited and diffuse cutaneous SSc ILD, PAH, renal crisis, and lower GI involvement in early phase |
| Anti-Th/To | Human RNase MRP complex | Mitochondrial RNA processing; Pre-rRNA processing | <10% | Limited cutaneous SSc ILD PAH |
| Anti-U11/U12 RNP | Small nucleolar RNA U11/U12 | Noncoding RNA in the minor spliceosome complex that activates the alternative splicing | <5% | Both limited and diffuse cutaneous SSc ILD |
| Novel Functional SSc Antibodies | ||||
| Anti-PDGFR | Platelet-derived growth factor receptor | PDGF receptor on fibroblasts | NA | Vasculopathy and ILD |
| Anti-M3R | Type 3 muscarinic receptor | Acetylcholine receptor on myenteric neurons and visceral smooth muscles | NA | GI dysmotility |
| Anti-ICAM-1 or AECA | ICAM-1 or endothelial cells | Adhesion molecules on endothelial cells | NA | Vasculopathy |
| Anti-AT1R | Angiotensin II type 1 receptor | Receptor for angiotensin on visceral smooth muscles | NA | Vasculopathy, ILD |
| Anti-ETAR | Endothelin-1 type A receptor | Receptor for endothelin A on visceral smooth muscles | NA | Vasculopathy, ILD |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, E.H.; Ha, Y.-J.; Lee, Y.J. Autoantibody Biomarkers in Rheumatic Diseases. Int. J. Mol. Sci. 2020, 21, 1382. https://doi.org/10.3390/ijms21041382
Kang EH, Ha Y-J, Lee YJ. Autoantibody Biomarkers in Rheumatic Diseases. International Journal of Molecular Sciences. 2020; 21(4):1382. https://doi.org/10.3390/ijms21041382
Chicago/Turabian StyleKang, Eun Ha, You-Jung Ha, and Yun Jong Lee. 2020. "Autoantibody Biomarkers in Rheumatic Diseases" International Journal of Molecular Sciences 21, no. 4: 1382. https://doi.org/10.3390/ijms21041382
APA StyleKang, E. H., Ha, Y.-J., & Lee, Y. J. (2020). Autoantibody Biomarkers in Rheumatic Diseases. International Journal of Molecular Sciences, 21(4), 1382. https://doi.org/10.3390/ijms21041382