A Curcumin Derivative Activates TFEB and Protects Against Parkinsonian Neurotoxicity in Vitro

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
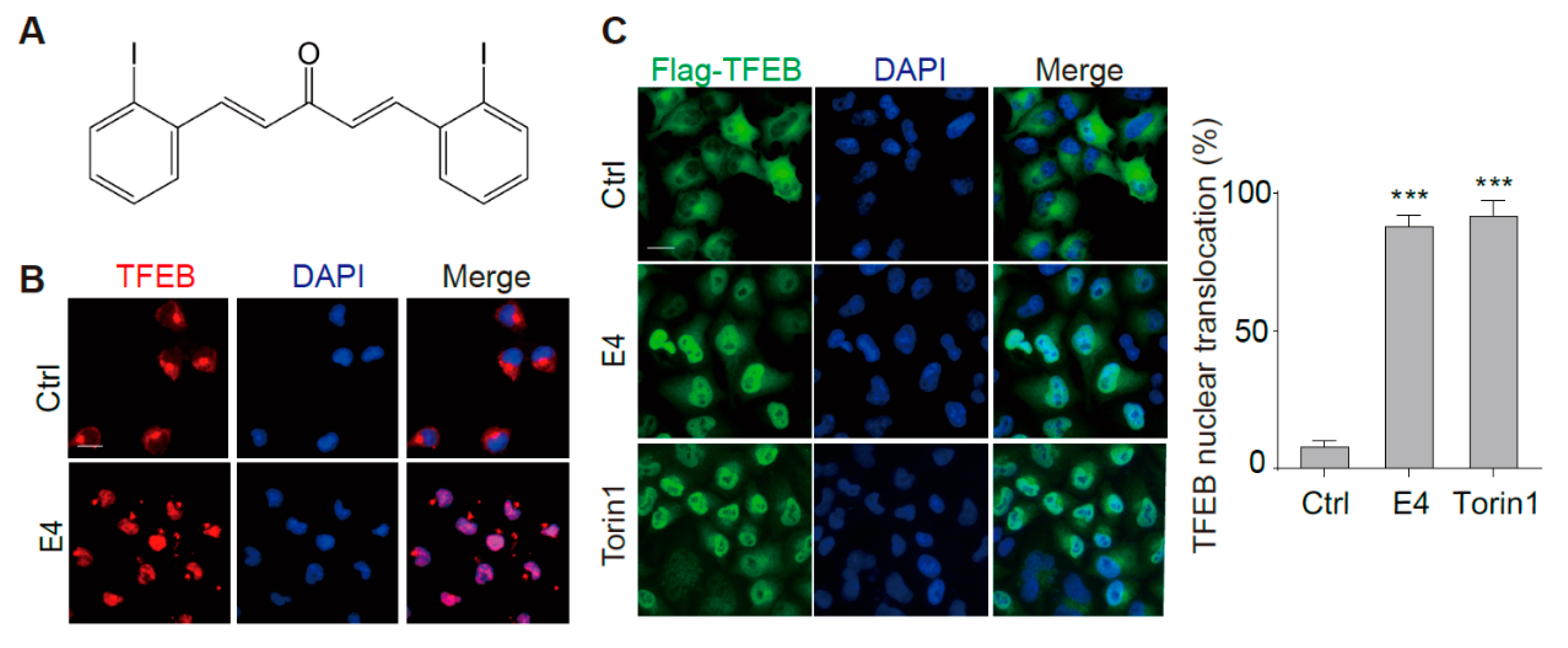
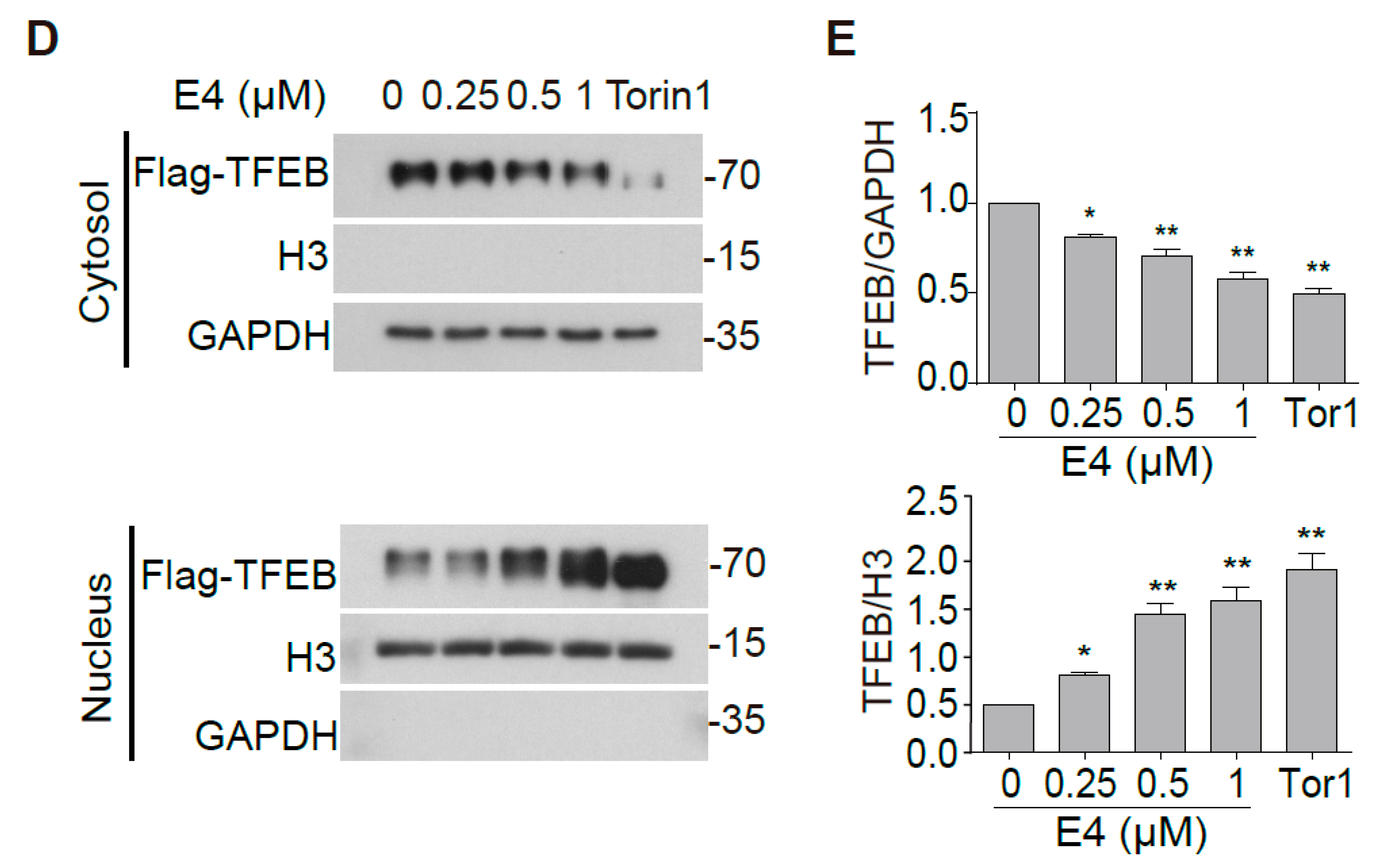
2.1. Compound E4 Promoted the Nuclear Translocation of Endogenous and Exogenous TFEB in Vitro
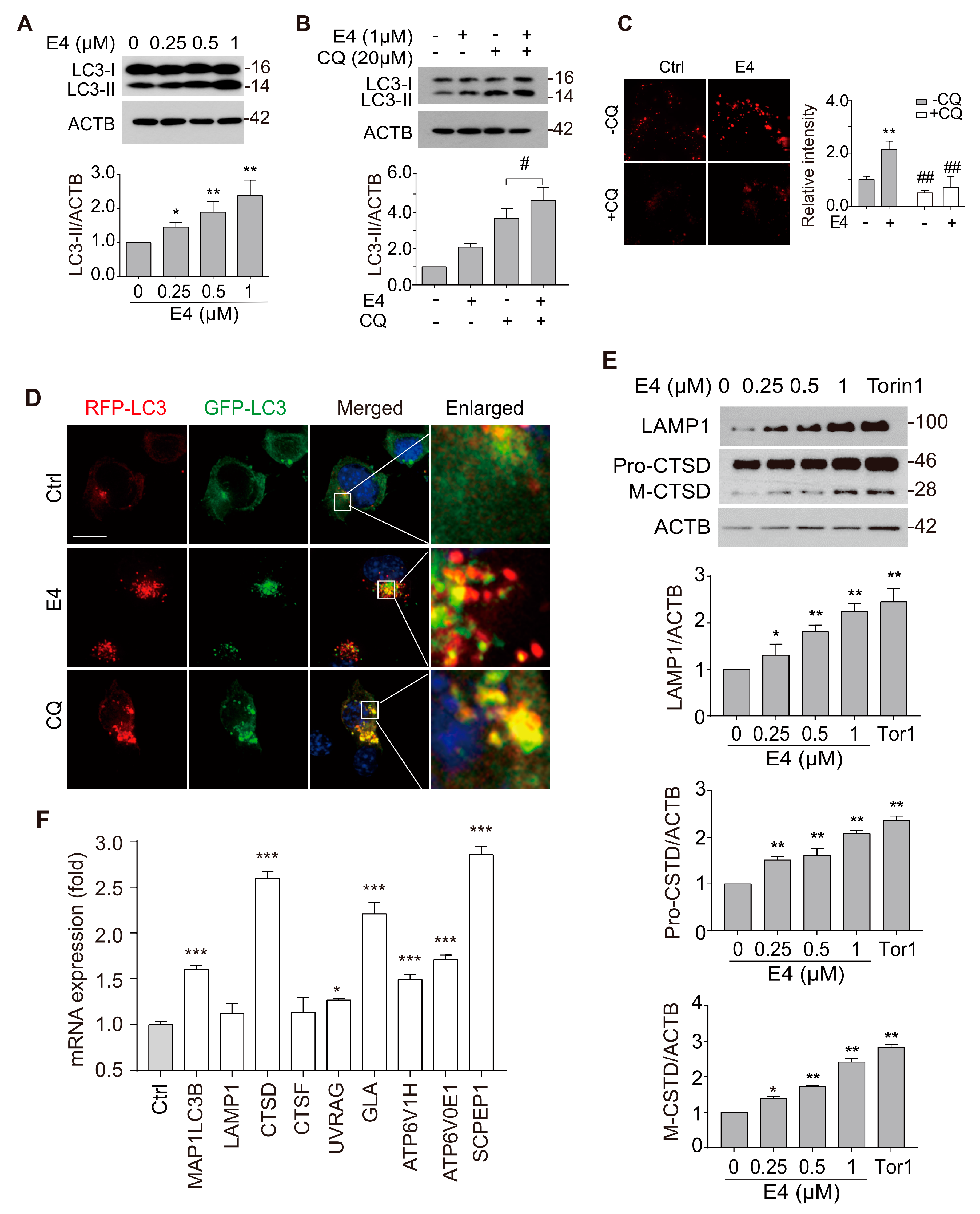
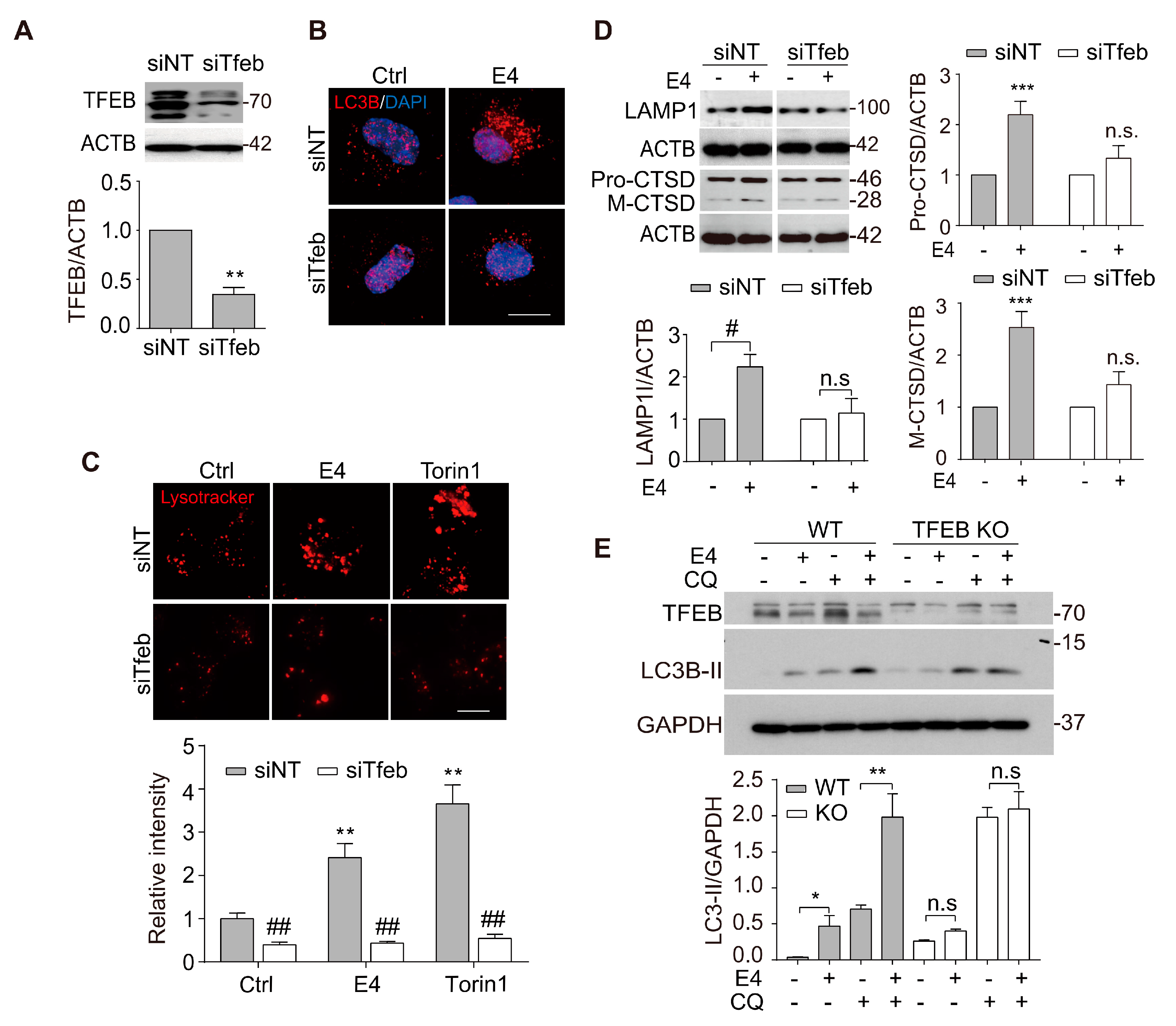
2.2. Compound E4 Promoted Autophagy Flux and Lysosomal Biogenesis Depending on TFEB
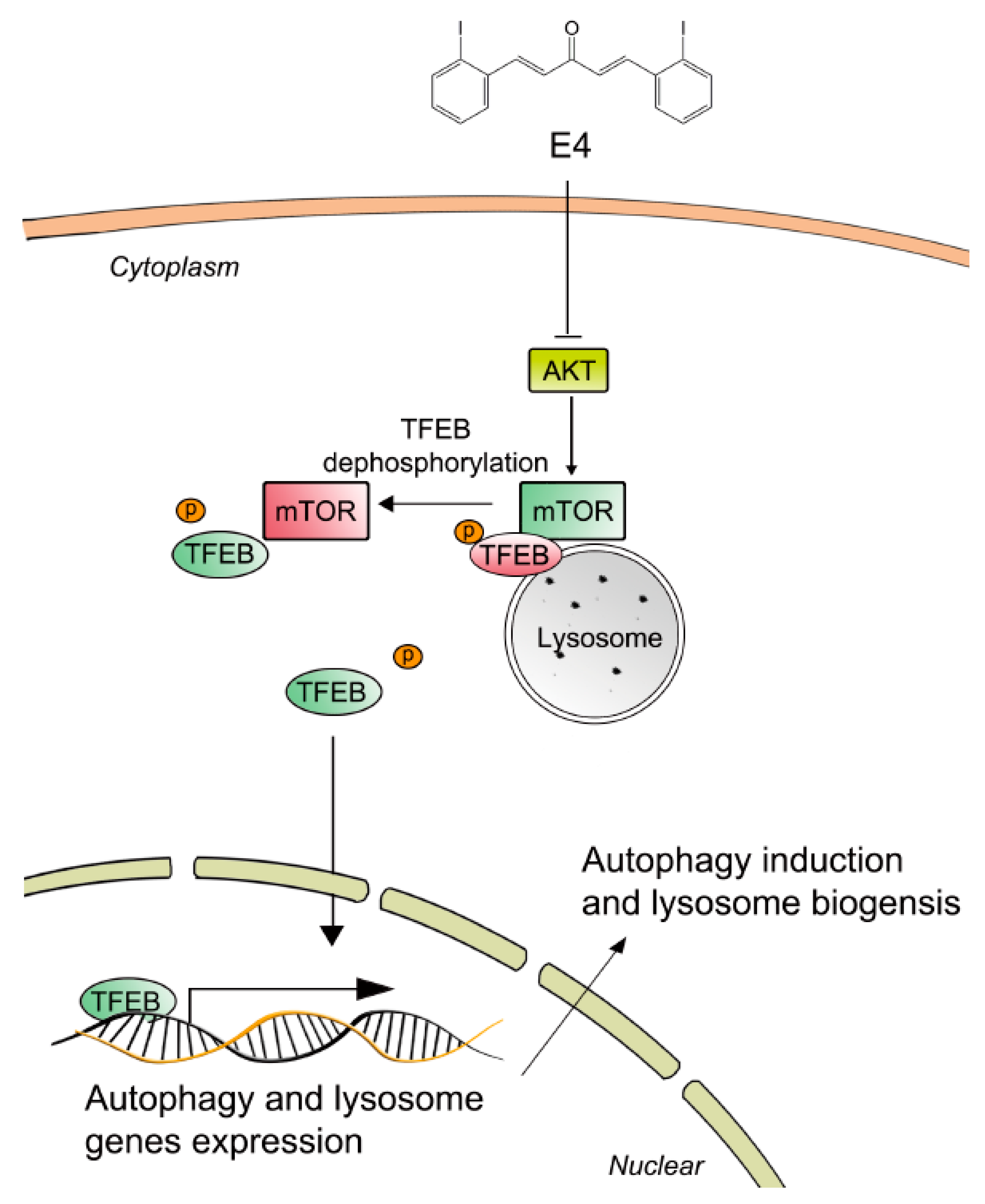
2.3. Compound E4 Activates TFEB Via Inhibiting AKT-MTORC1 Pathway
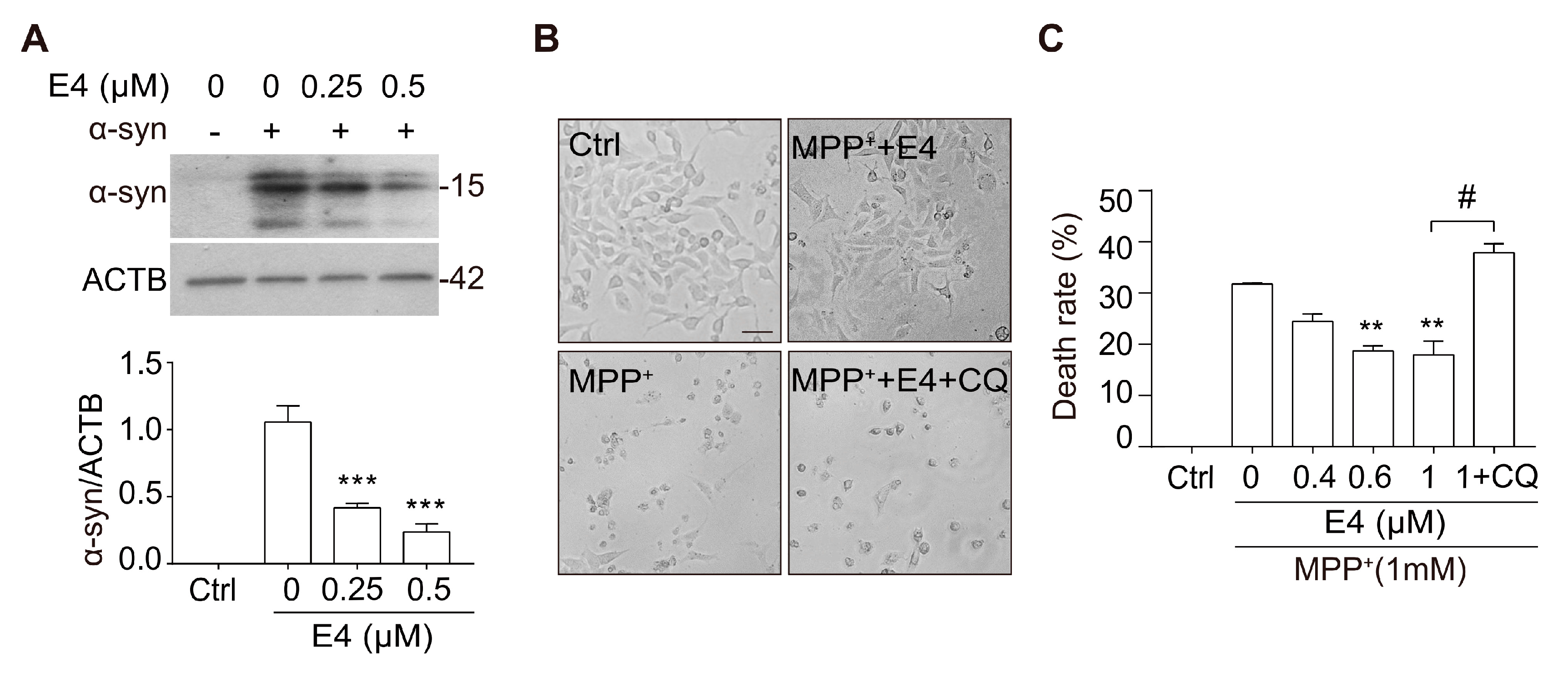
2.4. Compound E4 Reduces α-syn Levels and Protects Against MPP+ Toxicity in Vitro
3. Discussion
4. Material and Methods
4.1. Materials
4.2. Cell Culture and Drug Treatment
4.3. Cell Transfection and Gene Knockdown Assay
4.4. Quantitative Real-Time PCR
4.5. Alamar Blue Assay
4.6. Isolation of the Cytosol and the Nucleus Fractions
4.7. Western Blot
4.8. Immunocytochemistry
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Davie, C.A. A review of Parkinson’s disease. Br. Med. Bull. 2008, 86, 109–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Pillay, V.; Choonara, Y.E. Advances in the treatment of Parkinson’s disease. Prog. Neurobiol. 2007, 81, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Lynch-Day, M.A.; Mao, K.; Wang, K.; Zhao, M.; Klionsky, D.J. The role of autophagy in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Vicente, M.; Cuervo, A.M. Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurol. 2007, 6, 352–361. [Google Scholar] [CrossRef]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.Y.; Liu, J.Y.; Yang, C.B.; Malampati, S.; Huang, Y.Y.; Li, M.X.; Li, M.; Song, J.X. Neuroprotective Natural Products for the Treatment of Parkinson’s Disease by Targeting the Autophagy-Lysosome Pathway: A Systematic Review. Phytother. Res. 2017, 31, 1119–1127. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [Green Version]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [Green Version]
- Torra, A.; Parent, A.; Cuadros, T.; Rodriguez-Galvan, B.; Ruiz-Bronchal, E.; Ballabio, A.; Bortolozzi, A.; Vila, M.; Bove, J. Overexpression of TFEB Drives a Pleiotropic Neurotrophic Effect and Prevents Parkinson’s Disease-Related Neurodegeneration. Mol. Ther. 2018, 26, 1552–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arotcarena, M.L.; Bourdenx, M.; Dutheil, N.; Thiolat, M.L.; Doudnikoff, E.; Dovero, S.; Ballabio, A.; Fernagut, P.O.; Meissner, W.G.; Bezard, E.; et al. Transcription factor EB overexpression prevents neurodegeneration in experimental synucleinopathies. JCI Insight 2019, 4, e129719. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Wahlster, L. Restoring impaired protein metabolism in Parkinson’s disease--TFEB-mediated autophagy as a novel therapeutic target. Mov. Disord. 2013, 28, 1346. [Google Scholar] [CrossRef] [PubMed]
- Decressac, M.; Bjorklund, A. TFEB: Pathogenic role and therapeutic target in Parkinson disease. Autophagy 2013, 9, 1244–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.X.; Sun, Y.R.; Peluso, I.; Zeng, Y.; Yu, X.; Lu, J.H.; Xu, Z.; Wang, M.Z.; Liu, L.F.; Huang, Y.Y.; et al. A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy 2016, 12, 1372–1389. [Google Scholar] [CrossRef] [PubMed]
- Song, J.X.; Malampati, S.; Zeng, Y.; Durairajan, S.S.K.; Yang, C.B.; Tong, B.C.; Iyaswamy, A.; Shang, W.B.; Sreenivasmurthy, S.G.; Zhu, Z.; et al. A small molecule transcription factor EB activator ameliorates beta-amyloid precursor protein and Tau pathology in Alzheimer’s disease models. Aging Cell 2019, 19, e13069. [Google Scholar] [CrossRef] [Green Version]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.L.; Lee, E.X.; Gordon, K.L.; Paz, E.A.; Shen, W.C.; Ohnishi, K.; Meisenhelder, J.; Hunter, T.; La Spada, A.R. MAP4K3 mediates amino acid-dependent regulation of autophagy via phosphorylation of TFEB. Nat. Commun. 2018, 9, 942. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Kaizuka, T.; Morishita, H.; Hama, Y.; Tsukamoto, S.; Matsui, T.; Toyota, Y.; Kodama, A.; Ishihara, T.; Mizushima, T.; Mizushima, N. An Autophagic Flux Probe that Releases an Internal Control. Mol. Cell 2016, 64, 835–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef] [PubMed]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.; Wu, Y.T.; Chen, B.; Zhou, J.; Shen, H.M. Impaired autophagy due to constitutive mTOR activation sensitizes TSC2-null cells to cell death under stress. Autophagy 2011, 7, 1173–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [Green Version]
- Li, D.W.; Li, G.R.; Lu, Y.; Liu, Z.Q.; Chang, M.; Yao, M.; Cheng, W.; Hu, L.S. alpha-lipoic acid protects dopaminergic neurons against MPP+-induced apoptosis by attenuating reactive oxygen species formation. Int. J. Mol. Med. 2013, 32, 108–114. [Google Scholar] [CrossRef]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Hung, K.C.; Huang, H.J.; Lin, M.W.; Lei, Y.P.; Lin, A.M. Roles of autophagy in MPP+-induced neurotoxicity in vivo: The involvement of mitochondria and alpha-synuclein aggregation. PLoS ONE 2014, 9, e91074. [Google Scholar] [CrossRef] [Green Version]
- Miyara, M.; Kotake, Y.; Tokunaga, W.; Sanoh, S.; Ohta, S. Mild MPP(+) exposure impairs autophagic degradation through a novel lysosomal acidity-independent mechanism. J. Neurochem. 2016, 139, 294–308. [Google Scholar] [CrossRef] [Green Version]
- Dagda, R.K.; Das Banerjee, T.; Janda, E. How Parkinsonian toxins dysregulate the autophagy machinery. Int. J. Mol. Sci. 2013, 14, 22163–22189. [Google Scholar] [CrossRef]
- Dehay, B.; Bove, J.; Rodriguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalivendi, S.V.; Cunningham, S.; Kotamraju, S.; Joseph, J.; Hillard, C.J.; Kalyanaraman, B. Alpha-synuclein up-regulation and aggregation during MPP+-induced apoptosis in neuroblastoma cells: Intermediacy of transferrin receptor iron and hydrogen peroxide. J. Biol. Chem. 2004, 279, 15240–15247. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Yang, C.; Liu, J.; Chun-Kit Tong, B.; Zhu, Z.; Malampati, S.; Gopalkrishnashetty Sreenivasmurthy, S.; Cheung, K.-H.; Iyaswamy, A.; Su, C.; et al. A Curcumin Derivative Activates TFEB and Protects Against Parkinsonian Neurotoxicity in Vitro. Int. J. Mol. Sci. 2020, 21, 1515. https://doi.org/10.3390/ijms21041515
Wang Z, Yang C, Liu J, Chun-Kit Tong B, Zhu Z, Malampati S, Gopalkrishnashetty Sreenivasmurthy S, Cheung K-H, Iyaswamy A, Su C, et al. A Curcumin Derivative Activates TFEB and Protects Against Parkinsonian Neurotoxicity in Vitro. International Journal of Molecular Sciences. 2020; 21(4):1515. https://doi.org/10.3390/ijms21041515
Chicago/Turabian StyleWang, Ziying, Chuanbin Yang, Jia Liu, Benjamin Chun-Kit Tong, Zhou Zhu, Sandeep Malampati, Sravan Gopalkrishnashetty Sreenivasmurthy, King-Ho Cheung, Ashok Iyaswamy, Chengfu Su, and et al. 2020. "A Curcumin Derivative Activates TFEB and Protects Against Parkinsonian Neurotoxicity in Vitro" International Journal of Molecular Sciences 21, no. 4: 1515. https://doi.org/10.3390/ijms21041515
APA StyleWang, Z., Yang, C., Liu, J., Chun-Kit Tong, B., Zhu, Z., Malampati, S., Gopalkrishnashetty Sreenivasmurthy, S., Cheung, K. -H., Iyaswamy, A., Su, C., Lu, J., Song, J., & Li, M. (2020). A Curcumin Derivative Activates TFEB and Protects Against Parkinsonian Neurotoxicity in Vitro. International Journal of Molecular Sciences, 21(4), 1515. https://doi.org/10.3390/ijms21041515