Inherited Eye Diseases with Retinal Manifestations through the Eyes of Homeobox Genes


Abstract
1. Introduction
2. Retinal Organization
3. Homeobox Transcription Factors Expressed in Retina
3.1. Adnp
3.2. Alx Gene Family
3.3. CerS2
3.4. Crx
3.5. Hesx1
3.6. Hmx1
3.7. Lmx1B
3.8. Meis1
3.9. Msx2
3.10. Otx2
3.11. Pax2
3.12. Pax6
3.13. Rax
3.14. Rax2
3.15. Vax1
3.16. Vax2
3.17. Vsx1
3.18. Vsx2
4. Innovative Approaches of Modern Genomics and Cell Technology for IRDs Diagnostics
5. Gene-Based and Cellular Technologies in the Treatment of Inherited Retinal/Eye Diseases
6. Pharmacologic Neuroprotection and Activation of Endogenous Cell Potential as an Alternative to Genetic Engineering Methods
7. Conclusions/Insights
Author Contributions
Funding
Conflicts of Interest
References
- Fuhrmann, S. Eye morphogenesis and patterning of the optic vesicle. Curr. Top. Dev. Biol. 2010, 93, 61–84. [Google Scholar] [PubMed]
- Sinn, R.; Wittbrodt, J. An eye on eye development. Mech. Dev. 2013, 130, 347–358. [Google Scholar] [CrossRef]
- Fitzpatrick, D.R.; Van Heyningen, V. Developmental eye disorders. Curr. Opin. Genet. Dev. 2005, 15, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Zagozewski, J.L.; Zhang, Q.; Eisenstat, D.D. Genetic regulation of vertebrate eye development. Clin. Genet. 2014, 86, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Plaisancié, J.; Ceroni, F.; Holt, R.; Zazo Seco, C.; Calvas, P.; Chassaing, N.; Ragge, N.K. Genetics of anophthalmia and microphthalmia. Part 1: Non-syndromic anophthalmia/microphthalmia. Hum. Genet. 2019, 138, 799–830. [Google Scholar] [CrossRef]
- Zagozewski, J.L.; Zhang, Q.; Pinto, V.I.; Wigle, J.T.; Eisenstat, D.D. The role of homeobox genes in retinal development and disease. Dev. Biol. 2014, 393, 195–208. [Google Scholar] [CrossRef]
- Diakatou, M.; Manes, G.; Bocquet, B.; Meunier, I.; Kalatzis, V. Genome Editing as a Treatment for the Most Prevalent Causative Genes of Autosomal Dominant Retinitis Pigmentosa. Int. J. Mol. Sci. 2019, 20, 2542. [Google Scholar] [CrossRef]
- Khan, M.; Fadaie, Z.; Cornelis, S.S.; Cremers, F.P.M.; Roosing, S. Identification and Analysis of Genes Associated with Inherited Retinal Diseases. Methods Mol. Biol. 2019, 1834, 3–27. [Google Scholar]
- Yu, W.; Mookherjee, S.; Chaitankar, V.; Hiriyanna, S.; Kim, J.W.; Brooks, M.; Ataeijannati, Y.; Sun, X.; Dong, L.; Li, T.; et al. Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat. Commun. 2017, 8, 14716. [Google Scholar] [CrossRef]
- Mellough, C.B.; Bauer, R.; Collin, J.; Dorgau, B.; Zerti, D.; Dolan, D.W.P.; Jones, C.M.; Izuogu, O.G.; Yu, M.; Hallam, D.; et al. An integrated transcriptional analysis of the developing human retina. Development 2019, 146, dev169474. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, G.A.; Shalaev, E.; Karami, T.K.; Cunningham, J.; Slater, N.K.H.; Rivers, H.M. Pharmaceutical Development of AAV-Based Gene Therapy Products for the Eye. Pharm. Res. 2018, 36, 29. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H. Simple Anatomy of the Retina. In Webvision: The Organization of the Retina and Visual System [Internet]; Kolb, H., Fernandez, E., Nelson, R., Eds.; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 2005. [Google Scholar]
- Li, F.; Jiang, D.; Samuel, M.A. Microglia in the developing retina. Neural Dev. 2019, 14, 12. [Google Scholar] [CrossRef] [PubMed]
- Hoon, M.; Okawa, H.; Della Santina, L.; Wong, R.O. Functional architecture of the retina: Development and disease. Prog. Retin. Eye Res. 2014, 42, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, Y. Morphological Survey from Neurons to Circuits of the Mouse Retina. Methods Mol. Biol. 2018, 1753, 3–25. [Google Scholar]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef]
- Mazzolini, M.; Facchetti, G.; Andolfi, L.; Proietti Zaccaria, R.; Tuccio, S.; Treu, J.; Altafini, C.; Di Fabrizio, E.M.; Lazzarino, M.; Rapp, G.; et al. The phototransduction machinery in the rod outer segment has a strong efficacy gradient. Proc. Natl. Acad. Sci. USA 2015, 112, E2715–E2724. [Google Scholar] [CrossRef]
- Amram, B.; Cohen-Tayar, Y.; David, A.; Ashery-Padan, R. The retinal pigmented epithelium—from basic developmental biology research to translational approaches. Int. J. Dev Biol. 2017, 61, 225–234. [Google Scholar] [CrossRef]
- Sanes, J.R.; Masland, R.H. The types of retinal ganglion cells: Current status and implications for neuronal classification. Annu. Rev. Neurosci. 2015, 38, 221–246. [Google Scholar] [CrossRef]
- Kolb, H. Outer Plexiform Layer. In Webvision: The Organization of the Retina and Visual System [Internet]; Kolb, H., Fernandez, E., Nelson, R., Eds.; University of Utah Health Scisence. Center: Salt Lake City, UT, USA, 2005. [Google Scholar]
- Kolb, H. Inner Plexiform Layer. In Webvision: The Organization of the Retina and Visual System [Internet]; Kolb, H., Fernandez, E., Nelson, R., Eds.; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 2001. [Google Scholar]
- Subirada, P.V.; Paz, M.C.; Ridano, M.E.; Lorenc, V.E.; Vaglienti, M.V.; Barcelona, P.F.; Luna, J.D.; Sánchez, M.C. A journey into the retina: Müller glia commanding survival and death. Eur. J. Neurosci. 2018, 47, 1429–1443. [Google Scholar] [CrossRef]
- Kimelberg, H.K. Functions of mature mammalian astrocytes: A current view. Neuroscientist 2010, 16, 79–106. [Google Scholar] [CrossRef]
- Genini, S.; Beltran, W.A.; Stein, V.M.; Aguirre, G.D. Isolation and ex vivo characterization of the immunophenotype and function of microglia/macrophage populations in normal dog retina. Adv. Exp. Med. Biol. 2014, 801, 339–345. [Google Scholar] [PubMed]
- Selvam, S.; Kumar, T.; Fruttiger, M. Retinal vasculature development in health and disease. Prog. Retin. Eye Res. 2018, 63, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Vaz, J.; Bernardes, R.; Lobo, C. Blood-retinal barrier. Eur. J. Ophthalmol. 2011, 21 (Suppl. 6), S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Coránguez, M.; Ramos, C.; Antonetti, D.A. The inner blood-retinal barrier: Cellular basis and development. Vision Res. 2017, 139, 123–137. [Google Scholar] [CrossRef]
- Trost, A.; Bruckner, D.; Rivera, F.J.; Reitsamer, H.A. Pericytes in the Retina. Adv. Exp. Med. Biol. 2019, 1122, 1–26. [Google Scholar]
- Gregory-Evans, C.Y.; Wallace, V.A.; Gregory-Evans, K. Gene networks: Dissecting pathways in retinal development and disease. Prog. Retin. Eye Res. 2013, 33, 40–66. [Google Scholar] [CrossRef]
- Mann, I. The Development of the Human Eye, 2nd ed.; Grune and Stratton: New York, NY, USA, 1950; p. 312. [Google Scholar]
- Nishina, S.; Kohsaka, S.; Yamaguchi, Y.; Handa, H.; Kawakami, A.; Fujisawa, H.; Azuma, N. PAX6 expression in the developing human eye. Br. J. Ophthalmol. 1999, 83, 723–727. [Google Scholar] [CrossRef]
- Semina, E.V.; Brownell, I.; Mintz-Hittner, H.A.; Murray, J.C.; Jamrich, M. Mutations in the human forkhead transcription factor FOXE3 associated with anterior segment ocular dysgenesis and cataracts. Hum. Mol. Genet. 2001, 10, 231–236. [Google Scholar] [CrossRef]
- Markitantova, Y.V.; Smirnova, Y.A.; Panova, I.G.; Sukhikh, R.D.; Zinov’eva, V.I.; Mitashov, V.I. Analysis of expression of regulatory genes Pax6, Prox1, and Pitx2 in differentiating eye cells in human fetus. Biol. Bull. 2006, 33, 339–346. [Google Scholar] [CrossRef]
- Markitantova, Y.V.; Zinovieva, R.D. Intracellular localization of transcription factor PROX1 in the human retina in ontogeny. Biol. Bull. 2014, 41, 103–108. [Google Scholar] [CrossRef]
- Harding, P.; Moosajee, M. The Molecular Basis of Human Anophthalmia and Microphthalmia. J. Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.A.; Livesey, F.J.; Cepko, C.L.; Oliver, G. Prox1 function controls progenitor cell proliferation and horizontal cell genesis in the mammalian retina. Nat. Genet. 2003, 34, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Wigle, J.T.; Eisenstat, D.D. Homeobox genes in vertebrate forebrain development and disease. Clin. Genet. 2008, 73, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Giudetti, G.; Giannaccini, M.; Biasci, D.; Mariotti, S.; Degl’innocenti, A.; Perrotta, M.; Barsacchi, G.; Andreazzoli, M. Characterization of the Rx1-dependent transcriptome during early retinal development. Dev. Dyn. 2014, 243, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.T.; Ma, W.; Liang, L.; Wang, S.Z. bHLH genes and retinal cell fate specification. Mol. Neurobiol. 2005, 32, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, R.; Kageyama, R. Regulation of retinal cell fate specification by multiple transcription factors. Brain Res. 2008, 1192, 90–98. [Google Scholar] [CrossRef]
- Xiang, M. Intrinsic control of mammalian retinogenesis. Cell. Mol. Life Sci. 2013, 70, 2519–2532. [Google Scholar] [CrossRef]
- Cid, E.; Santos-Ledo, A.; Parrilla-Monge, M.; Lillo, C.; Arévalo, R.; Lara, J.M.; Aijón, J.; Velasco, A. Prox1 expression in rod precursors and Müller cells. Exp. Eye Res. 2010, 90, 267–276. [Google Scholar] [CrossRef]
- Heavner, W.; Pevny, L. Eye development and retinogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008391. [Google Scholar] [CrossRef]
- Buono, L.; Martinez-Morales, J.R. Retina Development in Vertebrates: Systems Biology Approaches to Understanding Genetic Programs: On the Contribution of Next-Generation Sequencing Methods to the Characterization of the Regulatory Networks Controlling Vertebrate Eye Development. Bioessays 2020, e1900187. [Google Scholar] [CrossRef]
- Miles, A.; Tropepe, V. Coordinating progenitor cell cycle exit and differentiation in the developing vertebrate retina. Neurogenesis (Austin) 2016, 3, e1161697. [Google Scholar] [CrossRef] [PubMed]
- Stanescu, D.; Iseli, H.P.; Schwerdtfeger, K.; Ittner, L.M.; Remé, C.E.; Hafezi, F. Continuous expression of the homeobox gene Pax6 in the ageing human retina. Eye (Lond) 2007, 21, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Yoshimoto, T.; Shimoda, M.; Kono, T.; Koizumi, M.; Yazumi, S.; Shimada, Y.; Doi, R.; Chiba, T.; Kubo, H. Loss of function of the candidate tumor suppressor prox1 by RNA mutation in human cancer cells. Neoplasia 2006, 8, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.T.; Kim, J.W. Compartmentalization of vertebrate optic neuroephithelium: External cues and transcription factors. Mol. Cells 2012, 33, 317–324. [Google Scholar] [CrossRef]
- Lamb, T.D.; Collin, S.P.; Pugh, E.N. Evolution of the vertebrate eye: Opsins, photoreceptors, retina and eye cup. Nat. Rev. Neurosci. 2007, 8, 960–976. [Google Scholar] [CrossRef]
- Wässle, H.; Puller, C.; Müller, F.; Haverkamp, S. Cone contacts, mosaics, and territories of bipolar cells in the mouse retina. J. Neurosci. 2009, 29, 106–117. [Google Scholar] [CrossRef]
- Farkas, M.H.; Grant, G.R.; White, J.A.; Sousa, M.E.; Consugar, M.B.; Pierce, E.A. Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 Mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genom. 2013, 14, 486. [Google Scholar] [CrossRef]
- Bibb, L.C.; Holt, J.K.; Tarttelin, E.E.; Hodges, M.D.; Gregory-Evans, K.; Rutherford, A.; Lucas, R.J.; Sowden, J.C.; Gregory-Evans, C.Y. Temporal and spatial expression patterns of the CRX transcription factor and its downstream targets. Critical differences during human and mouse eye development. Hum. Mol. Genet. 2001, 10, 1571–1579. [Google Scholar] [CrossRef]
- Hartl, D.; Krebs, A.R.; Jüttner, J.; Roska, B.; Schübeler, D. Cis-regulatory landscapes of four cell types of the retina. Nucleic Acids Res. 2017, 45, 11607–11621. [Google Scholar] [CrossRef]
- Hughes, A.E.; Enright, J.M.; Myers, C.A.; Shen, S.Q.; Corbo, J.C. Cell Type-Specific Epigenomic Analysis Reveals A Uniquely Closed Chromatin Architecture in Mouse Rod Photoreceptors Scientific Reports. Sci Rep. 2017, 7, 43184. [Google Scholar] [CrossRef]
- Ferda Percin, E.; Ploder, L.A.; Yu, J.J.; Arici, K.; Horsford, D.J.; Rutherford, A.; Bapat, B.; Cox, D.W.; Duncan, A.M.; Kalnins, V.I.; et al. Human microphthalmia associated with mutations in the retinal homeobox gene CHX10. Nat. Genet. 2000, 25, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Chowers, I.; Liu, D.; Farkas, R.H.; Gunatilaka, T.L.; Hackam, A.S.; Bernstein, S.L.; Campochiaro, P.A.; Parmigiani, G.; Zack, D.J. Gene expression variation in the adult human retina. Hum. Mol. Genet. 2003, 12, 2881–2893. [Google Scholar] [CrossRef] [PubMed]
- Voronina, V.A.; Kozhemyakina, E.A.; O’Kernick, C.M.; Kahn, N.D.; Wenger, S.L.; Linberg, J.V.; Schneider, A.S.; Mathers, P.H. Mutations in the human RAX homeobox gene in a patient with anophthalmia and sclerocornea. Hum. Mol. Genet. 2004, 13, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Hever, A.M.; Williamson, K.A.; Van Heyningen, V. Developmental malformations of the eye: The role of PAX6, SOX2 and OTX2. Clin. Genet. 2006, 69, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Westmoreland, J.J.; Kilic, G.; Sartain, C.; Sirma, S.; Blain, J.; Rehg, J.; Harvey, N.; Sosa-Pineda, B. Pancreas-specific deletion of Prox1 affects development and disrupts homeostasis of the exocrine pancreas. Gastroenterology 2012, 142, 999–1009. [Google Scholar] [CrossRef]
- Moisseiev, E.; Yiu, G. Retinal detachment in severe myopia. Lancet 2017, 389, 1133. [Google Scholar] [CrossRef]
- Zamostiano, R.; Pinhasov, A.; Gelber, E.; Steingart, R.A.; Seroussi, E.; Giladi, E.; Bassan, M.; Wollman, Y.; Eyre, H.J.; Mulley, J.C.; et al. Cloning and characterization of the human activity-dependent neuroprotective protein. J. Biol. Chem. 2001, 276, 708–714. [Google Scholar] [CrossRef]
- Furman, S.; Steingart, R.A.; Mandel, S.; Hauser, J.M.; Brenneman, D.E.; Gozes, I. Subcellular localization and secretion of activity-dependent neuroprotective protein in astrocytes. Neuron Glia Biol. 2004, 1, 193–199. [Google Scholar] [CrossRef]
- Teuchner, B.; Dimmer, A.; Humpel, C.; Amberger, A.; Fischer-Colbrie, R.; Nemeth, J.; Waschek, J.A.; Kieselbach, G.; Kralinger, M.; Schmid, E.; et al. VIP, PACAP-38, BDNF and ADNP in NMDA-induced excitotoxicity in the rat retina. Acta Ophthalmol. 2011, 7, 670–675. [Google Scholar] [CrossRef]
- Sragovich, S.; Merenlender-Wagner, A.; Gozes, I. ADNP Plays a Key Role in Autophagy: From Autism to Schizophrenia and Alzheimer’s Disease. Bioessays 2017, 39. [Google Scholar] [CrossRef]
- Jehle, T.; Dimitriu, C.; Auer, S.; Knoth, R.; Vidal-Sanz, M.; Gozes, I.; Lagrèze, W.A. The neuropeptide NAP provides neuroprotection against retinal ganglion cell damage after retinal ischemia and optic nerve crush. Graefes Arch. Clin. Exp. Ophthalmol. 2008, 246, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Guerri, C. The peptide NAP promotes neuronal growth and differentiation through extracellular signal-regulated protein kinase and Akt pathways, and protects neurons co-cultured with astrocytes damaged by ethanol. J. Neurochem. 2007, 103, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Helsmoortel, C.; Vulto-van Silfhout, A.T.; Coe, B.P.; Vandeweyer, G.; Rooms, L.; Van den Ende, J.; Schuurs-Hoeijmakers, J.H.; Marcelis, C.L.; Willemsen, M.H.; Vissers, L.E.; et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet. 2014, 46, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Gale, M.J.; Titus, H.E.; Harman, G.A.; Alabduljalil, T.; Dennis, A.; Wilson, J.L.; Koeller, D.M.; Finanger, E.; Blasco, P.A.; Chiang, P.W.; et al. Longitudinal ophthalmic findings in a child with Helsmoortel-Van der Aa Syndrome. Am. J. Ophthalmol. Case Rep. 2018, 10, 244–248. [Google Scholar] [CrossRef]
- Pascolini, G.; Agolini, E.; Majore, S.; Novelli, A.; Grammatico, P.; Digilio, M.C. Helsmoortel-Van der Aa Syndrome as emerging clinical diagnosis in intellectually disabled children with autistic traits and ocular involvement. Eur. J. Paediatr. Neurol. 2018, 22, 552–557. [Google Scholar] [CrossRef]
- Pinhasov, A.; Mandel, S.; Torchinsky, A.; Giladi, E.; Pittel, Z.; Goldsweig, A.M.; Servoss, S.J.; Brenneman, D.E.; Gozes, I. Activity-dependent neuroprotective protein: A novel gene essential for brain formation. Brain Res. Dev. Brain Res. 2003, 144, 83–90. [Google Scholar] [CrossRef]
- Sragovich, S.; Malishkevich, A.; Piontkewitz, Y.; Giladi, E.; Touloumi, O.; Lagoudaki, R.; Grigoriadis, N.; Gozes, I. The autism/neuroprotection-linked ADNP/NAP regulate the excitatory glutamatergic synapse. Transl. Psychiatry 2019, 9, 2. [Google Scholar] [CrossRef]
- Moriyama, S.; Iharada, M.; Omote, H.; Moriyama, Y.; Hiasa, M. Function and expression of a splicing variant of vesicular glutamate transporter 1. Biochim. Biophys. Acta Biomembr. 2017, 1859, 931–940. [Google Scholar] [CrossRef]
- McGonnell, I.M.; Graham, A.; Richardson, J.; Fish, J.L.; Depew, M.J.; Dee, C.T.; Holland, P.W.; Takahashi, T. Evolution of the Alx homeobox gene family: Parallel retention and independent loss of the vertebrate Alx3 gene. Evol. Dev. 2011, 13, 343–351. [Google Scholar] [CrossRef]
- Zhao, Q.; Behringer, R.R.; De Crombrugghe, B. Prenatal folic acid treatment suppresses acrania and meroanencephaly in mice mutant for the Cart1 homeobox gene. Nat. Genet. 1996, 13, 275–283. [Google Scholar] [CrossRef]
- Ten Berge, D.; Brouwer, A.; El Bahi, S.; Guénet, J.L.; Robert, B.; Meijlink, F. Mouse Alx3: An aristaless-like homeobox gene expressed during embryogenesis in ectomesenchyme and lateral plate mesoderm. Dev. Biol. 1998, 199, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Barabino, S.M.; Spada, F.; Cotelli, F.; Boncinelli, E. Inactivation of the zebrafish homologue of Chx10 by antisense oligonucleotides causes eye malformations similar to the ocular retardation phenotype. Mech. Dev. 1997, 63, 133–143. [Google Scholar] [CrossRef]
- Clark, B.S.; Stein-O’Brien, G.L.; Shiau, F.; Cannon, G.H.; Davis-Marcisak, E.; Sherman, T.; Santiago, C.P.; Hoang, T.V.; Rajaii, F.; James-Esposito, R.E. Single-Cell RNA-Seq Analysis of Retinal Development Identifies NFI Factors as Regulating Mitotic Exit and Late-Born Cell Specification. Neuron 2019, 102, 1111–1126.e5. [Google Scholar] [CrossRef] [PubMed]
- Uz, E.; Alanay, Y.; Aktas, D.; Vargel, I.; Gucer, S.; Tuncbilek, G.; Von Eggeling, F.; Yilmaz, E.; Deren, O.; Posorski, N.; et al. Disruption of ALX1 causes extreme microphthalmia and severe facial clefting: Expanding the spectrum of autosomal-recessive ALX-related frontonasal dysplasia. Am. J. Hum. Genet. 2010, 86, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Twigg, S.R.; Versnel, S.L.; Nürnberg, G.; Lees, M.M.; Bhat, M.; Hammond, P.; Hennekam, R.C.; Hoogeboom, A.J.; Hurst, J.A.; Johnson, D.; et al. Frontorhiny, a distinctive presentation of frontonasal dysplasia caused by recessive mutations in the ALX3 homeobox gene. Am. J. Hum. Genet. 2009, 84, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Kayserili, H.; Uz, E.; Niessen, C.; Vargel, I.; Alanay, Y.; Tuncbilek, G.; Yigit, G.; Uyguner, O.; Candan, S.; Okur, H.; et al. ALX4 dysfunction disrupts craniofacial and epidermal development. Hum. Mol. Genet. 2009, 18, 4357–4366. [Google Scholar] [CrossRef]
- Chen, B.; Chen, L.; Zhou, Y.; Mi, T.; Chen, D.Y.; Chen, L.; Yin, J.; Xue, Z.F. Multiple abnormalities due to a nonsense mutation in the Alx4 gene. Genet. Mol. Res. 2013, 12, 2771–2778. [Google Scholar] [CrossRef]
- Brouwer, A.; Ten Berge, D.; Wiegerinck, R.; Meijlink, F. The OAR/aristaless domain of the homeodomain protein Cart1 has an attenuating role in vivo. Mech. Dev. 2003, 120, 241–252. [Google Scholar] [CrossRef]
- Dee, C.T.; Szymoniuk, C.R.; Mills, P.E.; Takahashi, T. Defective neural crest migration revealed by a Zebrafish model of Alx1-related frontonasal dysplasia. Hum. Mol. Genet. 2013, 22, 239–251. [Google Scholar] [CrossRef]
- Lakhwani, S.; García-Sanz, P.; Vallejo, M. Alx3-deficient mice exhibit folic acid-resistant craniofacial midline and neural tube closure defects. Dev. Biol. 2010, 344, 869–880. [Google Scholar] [CrossRef]
- Beverdam, A.; Brouwer, A.; Reijnen, M.; Korving, J.; Meijlink, F. Severe nasal clefting and abnormal embryonic apoptosis in Alx3/Alx4 double mutant mice. Development 2001, 128, 3975–3986. [Google Scholar] [PubMed]
- Levy, M.; Futerman, A.H. Mammalian ceramide synthases. IUBMB Life 2010, 62, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Stiban, J.; Tidhar, R.; Futerman, A.H. Ceramide synthases: Roles in cell physiology and signaling. Adv. Exp. Med. Biol. 2010, 688, 60–71. [Google Scholar] [PubMed]
- Laviad, E.L.; Albee, L.; Pankova-Kholmyansky, I.; Epstein, S.; Park, H.; Merrill, A.H.; Futerman, A.H. Characterization of ceramide synthase 2: Tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J. Biol. Chem. 2008, 283, 5677–5684. [Google Scholar] [CrossRef]
- Brüggen, B.; Kremser, C.; Bickert, A.; Ebel, P.; Vom Dorp, K.; Schultz, K.; Dörmann, P.; Willecke, K.; Dedek, K. Defective ceramide synthases in mice cause reduced amplitudes in electroretinograms and altered sphingolipid composition in retina and cornea. Eur. J. Neurosci. 2016, 44, 1700–1713. [Google Scholar] [CrossRef] [PubMed]
- Kremser, C.; Klemm, A.L.; Van Uelft, M.; Imgrund, S.; Ginkel, C.; Hartmann, D.; Willecke, K. Cell-type-specific expression pattern of ceramide synthase 2 protein in mouse tissues. Histochem. Cell Biol. 2013, 140, 533–547. [Google Scholar] [CrossRef]
- Schenck, M.; Carpinteiro, A.; Grassmé, H.; Lang, F.; Gulbins, E. Ceramide: Physiological and pathophysiological aspects. Arch. Biochem. Biophys. 2007, 462, 171–175. [Google Scholar] [CrossRef]
- German, O.L.; Miranda, G.E.; Abrahan, C.E.; Rotstein, N.P. Ceramide is a mediator of apoptosis in retina photoreceptors. Invest. Ophthalmol. Vis. Sci. 2006, 47, 1658–1668. [Google Scholar] [CrossRef]
- Kirin, M.; Chandra, A.; Charteris, D.G.; Hayward, C.; Campbell, S.; Celap, I.; Bencic, G.; Vatavuk, Z.; Kirac, I.; Richards, A.J.; et al. Genome-wide association study identifies genetic risk underlying primary rhegmatogenous retinal detachment. Hum. Mol. Genet. 2013, 22, 3174–3185. [Google Scholar] [CrossRef]
- Barak, A.; Morse, L.S.; Goldkorn, T. Ceramide: A potential mediator of apoptosis in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2001, 42, 247–254. [Google Scholar]
- Kannan, R.; Jin, M.; Gamulescu, M.A.; Hinton, D.R. Ceramide-induced apoptosis: Role of catalase and hepatocyte growth factor. Free Radic. Biol. Med. 2004, 37, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Pujol-Lereis, L.M.; Liebisch, G.; Schick, T.; Lin, Y.; Grassmann, F.; Uchida, K.; Zipfel, P.F.; Fauser, S.; Skerka, C.; Weber, B.H.F. Evaluation of serum sphingolipids and the influence of genetic risk factors in age-related macular degeneration. PLoS ONE 2018, 13, e0200739. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tran, J.T.; Eckerd, A.; Huynh, T.P.; Elliott, M.H.; Brush, R.S.; Mandal, N.A. Inhibition of de novo ceramide biosynthesis by FTY720 protects rat retina from light-induced degeneration. J. Lipid Res. 2013, 54, 1616–1629. [Google Scholar] [CrossRef]
- Zarbin, M.A.; Green, W.R.; Moser, H.W.; Morton, S.J. Farber’s disease. Light and electron microscopic study of the eye. Arch. Ophthalmol. 1985, 103, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Zarbin, M.A.; Green, W.R.; Moser, A.B.; Tiffany, C. Increased levels of ceramide in the retina of a patient with Farber’s disease. Arch. Ophthalmol. 1988, 106, 1163. [Google Scholar] [CrossRef] [PubMed]
- Imgrund, S.; Hartmann, D.; Farwanah, H.; Eckhardt, M.; Sandhoff, R.; Degen, J.; Gieselmann, V.; Sandhoff, K.; Willecke, K. Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. J. Biol. Chem. 2009, 284, 33549–33560. [Google Scholar] [CrossRef]
- Freund, C.L.; Gregory-Evans, C.Y.; Furukawa, T.; Papaioannou, M.; Looser, J.; Ploder, L.; Bellingham, J.; Ng, D.; Herbrick, J.A.; Duncan, A.; et al. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 1997, 91, 543–553. [Google Scholar] [CrossRef]
- Furukawa, T.; Morrow, E.M.; Cepko, C.L. Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell 1997, 91, 531–541. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Q.L.; Nie, Z.; Sun, H.; Lennon, G.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Zack, D.J. Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron 1997, 19, 1017–1030. [Google Scholar] [CrossRef]
- Shen, Y.C.; Raymond, P.A. Zebrafish cone-rod (crx) homeobox gene promotes retinogenesis. Dev. Biol. 2004, 269, 237–251. [Google Scholar] [CrossRef]
- Livesey, F.J.; Furukawa, T.; Steffen, M.A.; Church, G.M.; Cepko, C.L. Microarray analysis of the transcriptional network controlled by the photoreceptor homeobox gene Crx. Curr. Biol. 2000, 10, 301–310. [Google Scholar] [CrossRef]
- Furukawa, A.; Koike, C.; Lippincott, P.; Cepko, C.L.; Furukawa, T. The mouse Crx 5’-upstream transgene sequence directs cell-specific and developmentally regulated expression in retinal photoreceptor cells. J. Neurosci. 2002, 22, 1640–1647. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Assawachananont, J.; Kim, S.Y.; Kaya, K.D.; Fariss, R.; Roger, J.E.; Swaroop, A. Cone-rod homeobox CRX controls presynaptic active zone formation in photoreceptors of mammalian retina. Hum. Mol. Genet. 2018, 27, 3555–3567. [Google Scholar] [CrossRef] [PubMed]
- Rivolta, C.; Berson, E.L.; Dryja, T.P. Dominant Leber congenital amaurosis, cone-rod degeneration, and retinitis pigmentosa caused by mutant versions of the transcription factor CRX. Hum. Mutat. 2001, 18, 488–498. [Google Scholar] [CrossRef]
- Kawamura, T.; Ohtsubo, M.; Mitsuyama, S.; Ohno-Nakamura, S.; Shimizu, N.; Minoshima, S. KMeyeDB: A graphical database of mutations in genes that cause eye diseases. Hum. Mutat. 2010, 31, 667–674. [Google Scholar] [CrossRef]
- Huang, L.; Xiao, X.; Li, S.; Jia, X.; Wang, P.; Guo, X.; Zhang, Q. CRX variants in cone-rod dystrophy and mutation overview. Biochem. Biophys. Res. Commun. 2012, 426, 498–503. [Google Scholar] [CrossRef]
- Ibrahim, M.T.; Alarcon-Martinez, T.; Lopez, I.; Fajardo, N.; Chiang, J.; Koenekoop, R.K. A complete, homozygous CRX deletion causing nullizygosity is a new genetic mechanism for Leber congenital amaurosis. Sci. Rep. 2018, 8, 5034. [Google Scholar] [CrossRef]
- Zhu, Y.; Tan, H.; Zeng, J.; Tao, D.; Ma, Y.; Liu, Y. A novel CRX variant (p.R98X) is identified in a Chinese family of Retinitis pigmentosa with atypical and mild manifestations. Genes Genom. 2019, 41, 359–366. [Google Scholar] [CrossRef]
- Kohl, S.; Kitiratschky, V.; Papke, M.; Schaich, S.; Sauer, A.; Wissinger, B. Genes and mutations in autosomal dominant cone and cone-rod dystrophy. Adv. Exp. Med. Biol. 2012, 723, 337–343. [Google Scholar]
- Dias, M.F.; Joo, K.; Kemp, J.A.; Fialho, S.L.; Da Silva Cunha, A.; Woo, S.J.; Kwon, Y.J. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retin. Eye Res. 2018, 63, 107–131. [Google Scholar] [CrossRef]
- Verbakel, S.K.; Van Huet, R.A.C.; Boon, C.J.F.; Den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog, Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Geller, A.M.; Sieving, P.A. Assessment of foveal cone photoreceptors in Stargardt’s macular dystrophy using a small dot detection task. Vision Res. 1993, 33, 1509–1524. [Google Scholar] [CrossRef][Green Version]
- Coppieters, F.; De Wilde, B.; Lefever, S.; De Meester, E.; De Rocker, N.; Van Cauwenbergh, C.; Pattyn, F.; Meire, F.; Leroy, B.P.; Hellemans, J.; et al. Massively parallel sequencing for early molecular diagnosis in Leber congenital amaurosis. Genet. Med. 2012, 14, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Weleber, R.G.; Francis, P.J.; Trzupek, K.M.; Beattie, C. Leber Congenital Amaurosis. In Source Gene Reviews® [Internet]; University of Washington: Seattle, WA, USA, 2004. [Google Scholar]
- Furukawa, T.; Morrow, E.M.; Li, T.; Davis, F.C.; Cepko, C.L. Retinopathy and attenuated circadian entrainment in Crx-deficient mice. Nat. Genet. 1999, 23, 466–470. [Google Scholar] [CrossRef]
- Morrow, E.M.; Furukawa, T.; Raviola, E.; Cepko, C.L. Synaptogenesis and outer segment formation are perturbed in the neural retina of Crx mutant mice. BMC Neurosci. 2005, 6, 5. [Google Scholar] [CrossRef]
- Ruzycki, P.A.; Tran, N.M.; Kefalov, V.J.; Kolesnikov, A.V.; Chen, S. Graded gene expression changes determine phenotype severity in mouse models of CRX-associated retinopathies. Genome Biol. 2015, 16, 171. [Google Scholar] [CrossRef]
- Kelberman, D.; Dattani, M.T. Genetics of septo-optic dysplasia. Pituitary 2007, 10, 393–407. [Google Scholar] [CrossRef]
- Dattani, M.T.; Martinez-Barbera, J.P.; Thomas, P.Q.; Brickman, J.M.; Gupta, R.; Martensson, I.L.; Toresson, H.; Fox, M.; Wales, J.K.; Hindmarsh, P.C.; et al. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat. Genet. 1998, 19, 125–133. [Google Scholar] [CrossRef]
- Fernández-Garre, P.; Rodríguez-Gallardo, L.; Gallego-Díaz, V.; Alvarez, I.S.; Puelles, L. Fate map of the chicken neural plate at stage 4. Development 2002, 129, 2807–2822. [Google Scholar]
- Thomas, P.Q.; Dattani, M.T.; Brickman, J.M.; McNay, D.; Warne, G.; Zacharin, M.; Cameron, F.; Hurst, J.; Woods, K.; Dunger, D.; et al. Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia. Hum. Mol. Genet. 2001, 10, 39–45. [Google Scholar] [CrossRef]
- Pozzi, S.; Bowling, S.; Apps, J.; Brickman, J.M.; Rodriguez, T.A.; Martinez-Barbera, J.P. Genetic Deletion of Hesx1 Promotes Exit from the Pluripotent State and Impairs Developmental Diapause. Stem Cell Rep. 2019, 13, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, R.; Qiao, N.; Peng, G.; Zhang, K.; Tang, K.; Han, J.J.; Jing, N. Transcriptome analysis reveals determinant stages controlling human embryonic stem cell commitment to neuronal cells. J. Biol. Chem. 2017, 292, 19590–19604. [Google Scholar] [CrossRef] [PubMed]
- Fish, M.B.; Nakayama, T.; Fisher, M.; Hirsch, N.; Cox, A.; Reeder, R.; Carruthers, S.; Hall, A.; Stemple, D.L.; Grainger, R.M. Xenopus mutant reveals necessity of rax for specifying the eye field which otherwise forms tissue with telencephalic and diencephalic character. Dev. Biol. 2014, 395, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; McNally, R.J.; Harrison, E.; Lloyd, I.C.; Clayton, P.E. Geographical distribution of optic nerve hypoplasia and septo-optic dysplasia in Northwest England. J. Pediatr. 2006, 148, 85–88. [Google Scholar] [CrossRef]
- Sajedi, E.; Gaston-Massuet, C.; Signore, M.; Andoniadou, C.L.; Kelberman, D.; Castro, S.; Etchevers, H.C.; Gerrelli, D.; Dattani, M.T.; Martinez-Barbera, J.P. Analysis of mouse models carrying the I26T and R160C substitutions in the transcriptional repressor HESX1 as models for septo-optic dysplasia and hypopituitarism. Dis. Model. Mech. 2008, 1, 241–254. [Google Scholar] [CrossRef]
- Andoniadou, C.L.; Signore, M.; Sajedi, E.; Gaston-Massuet, C.; Kelberman, D.; Burns, A.J.; Itasaki, N.; Dattani, M.; Martinez-Barbera, J.P. Lack of the murine homeobox gene Hesx1 leads to a posterior transformation of the anterior forebrain. Development 2007, 134, 1499–1508. [Google Scholar] [CrossRef]
- Schorderet, D.F.; Nichini, O.; Boisset, G.; Polok, B.; Tiab, L.; Mayeur, H.; Raji, B.; De la Houssaye, G.; Abitbol, M.M.; Munier, F.L. Mutation in the human homeobox gene NKX5-3 causes an oculo-auricular syndrome. Am. J. Hum. Genet. 2008, 82, 1178–1184. [Google Scholar] [CrossRef]
- Yoshiura, K.; Leysens, N.J.; Reiter, R.S.; Murray, J.C. Cloning, characterization, and mapping of the mouse homeobox gene Hmx1. Genomics 1998, 50, 61–68. [Google Scholar] [CrossRef]
- Wang, W.; Lo, P.; Frasch, M.; Lufkin, T. Hmx: An evolutionary conserved homeobox gene family expressed in the developing nervous system in mice and Drosophila. Mech. Dev. 2000, 99, 123–137. [Google Scholar] [CrossRef]
- Boisset, G.; Schorderet, D.F. Zebrafish hmx1 promotes retinogenesis. Exp. Eye Res. 2012, 105, 34–42. [Google Scholar] [CrossRef]
- Vaclavik, V.; Schorderet, D.F.; Borruat, F.X.; Munier, F.L. Retinal dystrophy in the oculo-auricular syndrome due to HMX1 mutation. Ophthalmic Genet. 2011, 32, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, G.M.H.; Abdel-Hamid, M.S.; Mehrez, M.I.; Kamal, A.M.; Taher, M.B.; Afifi, H.H. Further delineation of the oculoauricular syndrome phenotype: A new family with a novel truncating HMX1 mutation. Ophthalmic Genet. 2018, 39, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, R.L.; Urquhart, J.; Lovell, S.C.; Biswas, S.; Parry, N.R.; Schorderet, D.F.; Lloyd, I.C.; Clayton-Smith, J.; Black, G.C. Abrogation of HMX1 function causes rare oculoauricular syndrome associated with congenital cataract, anterior segment dysgenesis, and retinal dystrophy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Munroe, R.J.; Prabhu, V.; Acland, G.M.; Johnson, K.R.; Harris, B.S.; O’Brien, T.P.; Welsh, I.C.; Noden, D.M.; Schimenti, J.C. Mouse H6 Homeobox 1 (Hmx1) mutations cause cranial abnormalities and reduced body mass. BMC Dev. Biol. 2009, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Dunston, J.A.; Hamlington, J.D.; Zaveri, J.; Sweeney, E.; Sibbring, J.; Tran, C.; Malbroux, M.; O’Neill, J.P.; Mountford, R.; McIntosh, I. The human LMX1B gene: Transcription unit, promoter, and pathogenic mutations. Genomics 2004, 84, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, C.; Hu, X. LIM homeobox transcription factors, a novel subfamily which plays an important role in cancer (review). Oncol. Rep. 2014, 31, 1975–1985. [Google Scholar] [CrossRef]
- Pressman, C.L.; Chen, H.; Johnson, R.L. LMX1B, a LIM homeodomain class transcription factor, is necessary for normal development of multiple tissues in the anterior segment of the murine eye. Genesis 2000, 26, 15–25. [Google Scholar] [CrossRef]
- Bongers, E.M.; Huysmans, F.T.; Levtchenko, E.; De Rooy, J.W.; Blickman, J.G.; Admiraal, R.J.; Huygen, P.L.; Cruysberg, J.R.; Toolens, P.A.; Prins, J.B.; et al. Genotype-phenotype studies in nail-patella syndrome show that LMX1B mutation location is involved in the risk of developing nephropathy. Eur. J. Hum. Genet. 2005, 13, 935–946. [Google Scholar] [CrossRef]
- Millá, E.; Hernan, I.; Gamundi, M.J.; Martínez-Gimeno, M.; Carballo, M. Novel LMX1B mutation in familial nail-patella syndrome with variable expression of open angle glaucoma. Mol. Vis. 2007, 13, 639–648. [Google Scholar]
- Choquet, H.; Paylakhi, S.; Kneeland, S.C.; Thai, K.K.; Hoffmann, T.J.; Yin, J.; Kvale, M.N.; Banda, Y.; Tolman, N.G.; Williams, P.A.; et al. A multiethnic genome-wide association study of primary open-angle glaucoma identifies novel risk loci. Nat. Commun. 2018, 9, 2278. [Google Scholar] [CrossRef]
- Bongers, E.M.; De Wijs, I.J.; Marcelis, C.; Hoefsloot, L.H.; Knoers, N.V. Identification of entire LMX1B gene deletions in nail patella syndrome: Evidence for haploinsufficiency as the main pathogenic mechanism underlying dominant inheritance in man. Eur. J. Hum. Genet. 2008, 16, 1240–1244. [Google Scholar] [CrossRef]
- Cross, S.H.; Macalinao, D.G.; McKie, L.; Rose, L.; Kearney, A.L.; Rainger, J.; Thaung, C.; Keighren, M.; Jadeja, S.; West, K.; et al. A dominant-negative mutation of mouse Lmx1b causes glaucoma and is semi-lethal via LDB1-mediated dimerization. PLoS Genet. 2014, 10, e1004359. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.; Gestri, G.; Wilson, S.W.; Link, B.A. Lmx1b is essential for survival of periocular mesenchymal cells and influences Fgf-mediated retinal patterning in zebrafish. Dev. Biol. 2009, 332, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Catoire, H.; Dion, P.; Gaspar, C.; Lafrenière, R.G.; Girard, S.L.; Levchenko, A.; Rivière, J.B.; Fiori, L.; St-Onge, J.; et al. MEIS1 intronic risk haplotype associated with restless legs syndrome affects its mRNA and protein expression levels. Hum. Mol. Genet. 2009, 18, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Longobardi, E.; Penkov, D.; Mateos, D.; De Florian, G.; Torres, M.; Blasi, F. Biochemistry of the tale transcription factors PREP, MEIS, and PBX in vertebrates. Dev. Dyn. 2014, 243, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Schulte, D.; Geerts, D. MEIS transcription factors in development and disease. Development 2019, 146, dev174706. [Google Scholar] [CrossRef]
- Bessa, J.; Tavares, M.J.; Santos, J.; Kikuta, H.; Laplante, M.; Becker, T.S.; Gómez-Skarmeta, J.L.; Casares, F. Meis1 regulates cyclin D1 and c-myc expression; and controls the proliferation of the multipotent cells in the early developing zebrafish eye. Development 2008, 135, 799–803. [Google Scholar] [CrossRef]
- Heine, P.; Dohle, E.; Bumsted-O’Brien, K.; Engelkamp, D.; Schulte, D. Evidence for an evolutionary conserved role of homothorax/Meis1/2 during vertebrate retina development. Development 2008, 135, 805–811. [Google Scholar] [CrossRef]
- Erickson, T.; French, C.R.; Waskiewicz, A.J. Meis1 specifies positional information in the retina and tectum to organize the zebrafish visual system. Neural Dev. 2010, 5, 22. [Google Scholar] [CrossRef]
- Hisa, T.; Spence, S.E.; Rachel, R.A.; Fujita, M.; Nakamura, T.; Ward, J.M.; Devor-Henneman, D.E.; Saiki, Y.; Kutsuna, H.; Tessarollo, L.; et al. Hematopoietic, angiogenic and eye defects in Meis1 mutant animals. EMBO J. 2004, 23, 450–459. [Google Scholar] [CrossRef]
- Islam, M.M.; Li, Y.; Luo, H.; Xiang, M.; Cai, L. Meis1 regulates Foxn4 expression during retinal progenitor cell differentiation. Biol. Open. 2013, 2, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Marcos, S.; González-Lázaro, M.; Beccari, L.; Carramolino, L.; Martin-Bermejo, M.J.; Aarie, O.; Mateos-San Martín, D.; Torroja, C.; Bogdanović, O.; Doohan, R.; et al. Meis1 coordinates a network of genes implicated in eye development and microphthalmia. Development 2015, 142, 3009–3020. [Google Scholar] [CrossRef] [PubMed]
- Alappat, S.; Zhang, Z.Y.; Chen, Y.P. Msx homeobox gene family and craniofacial development. Cell Res. 2003, 13, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.; Robert, B. msh/Msx gene family in neural development. Trends Genet. 2005, 21, 624–632. [Google Scholar] [CrossRef]
- Trousse, F.; Esteve, P.; Bovolenta, P. Bmp4 mediates apoptotic cell death in the developing chick eye. J. Neurosci. 2001, 21, 1292–1301. [Google Scholar] [CrossRef]
- Wang, W.; Chen, X.; Xu, H.; Lufkin, T. Msx3: A novel murine homologue of the Drosophila msh homeobox gene restricted to the dorsal embryonic central nervous system. Mech. Dev. 1996, 58, 203–215. [Google Scholar] [CrossRef]
- Cheng, S.L.; Shao, J.S.; Charlton-Kachigian, N.; Loewy, A.P.; Towler, D.A. MSX2 promotes osteogenesis and suppresses adipogenic differentiation of multipotent mesenchymal progenitors. J. Biol. Chem. 2003, 278, 45969–45977. [Google Scholar] [CrossRef]
- Newberry, E.P.; Latifi, T.; Battaile, J.T.; Towler, D.A. Structure-function analysis of Msx2-mediated transcriptional suppression. Biochemistry 1997, 36, 10451–10462. [Google Scholar] [CrossRef]
- Jabs, E.W.; Müller, U.; Li, X.; Ma, L.; Luo, W.; Haworth, I.S.; Klisak, I.; Sparkes, R.; Warman, M.L.; Mulliken, J.B.; et al. A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell 1993, 75, 443–450. [Google Scholar] [CrossRef]
- Florisson, J.M.; Verkerk, A.J.; Huigh, D.; Hoogeboom, A.J.; Swagemakers, S.; Kremer, A.; Heijsman, D.; Lequin, M.H.; Mathijssen, I.M.; Van der Spek, P.J. Boston type craniosynostosis: Report of a second mutation in MSX2. Am. J. Med. Genet. A. 2013, 161A, 2626–2633. [Google Scholar] [CrossRef]
- Wilkie, A.O.; Tang, Z.; Elanko, N.; Walsh, S.; Twigg, S.R.; Hurst, J.A.; Wall, S.A.; Chrzanowska, K.H.; Maxson, R.E. Functional haploinsufficiency of the human homeobox gene MSX2 causes defects in skull ossification. Nat. Genet. 2000, 24, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Plaisancié, J.; Collet, C.; Pelletier, V.; Perdomo, Y.; Studer, F.; Fradin, M.; Schaefer, E.; Speeg-Schatz, C.; Bloch-Zupan, A.; Flori, E.; et al. MSX2 Gene Duplication in a Patient with Eye Development Defects. Ophthalmic Genet 2015, 36, 353–358. [Google Scholar]
- Satokata, I.; Ma, L.; Ohshima, H.; Bei, M.; Woo, I.; Nishizawa, K.; Maeda, T.; Takano, Y.; Uchiyama, M.; Heaney, S.; et al. Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat. Genet. 2000, 24, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Ichida, F.; Nishimura, R.; Hata, K.; Matsubara, T.; Ikeda, F.; Hisada, K.; Yatani, H.; Cao, X.; Komori, T.; Yamaguchi, A.; et al. Reciprocal roles of MSX2 in regulation of osteoblast and adipocyte differentiation. J. Biol. Chem. 2004, 279, 34015–34022. [Google Scholar] [CrossRef]
- Liu, Y.H.; Kundu, R.; Wu, L.; Luo, W.; Ignelzi, M.A.; Snead, M.L.; Maxson, R.E. Premature suture closure and ectopic cranial bone in mice expressing Msx2 transgenes in the developing skull. Proc. Natl. Acad. Sci. USA 1995, 92, 6137–6141. [Google Scholar] [CrossRef]
- Homon, J.A.; Gong, S.G. A statistical analysis of the overexpression of the msx2 RNA in Xenopus laevis. Arch. Oral. Biol. 1999, 44, 795–803. [Google Scholar] [CrossRef]
- Wu, L.Y.; Li, M.; Hinton, D.R.; Guo, L.; Jiang, S.; Wang, J.T.; Zeng, A.; Xie, J.B.; Snead, M.; Shuler, C.; et al. Microphthalmia resulting from MSX2-induced apoptosis in the optic vesicle. Invest. Ophthalmol. Vis. Sci. 2003, 44, 2404–2412. [Google Scholar] [CrossRef]
- Foerst-Potts, L.; Sadler, T.W. Disruption of Msx-1 and Msx-2 reveals roles for these genes in craniofacial, eye, and axial development. Dev. Dyn. 1997, 209, 70–84. [Google Scholar] [CrossRef]
- Jiang, S.Y.; Wang, J.T. Msx2 alters the timing of retinal ganglion cells fate commitment and differentiation. Biochem. Biophys. Res. Commun. 2010, 395, 524–529. [Google Scholar] [CrossRef]
- Holme, R.H.; Thomson, S.J.; Davidson, D.R. Ectopic expression of Msx2 in chick retinal pigmented epithelium cultures suggests a role in patterning the optic vesicle. Mech. Dev. 2000, 91, 175–187. [Google Scholar] [CrossRef]
- Dateki, S.; Kosaka, K.; Hasegawa, K.; Tanaka, H.; Azuma, N.; Yokoya, S.; Muroya, K.; Adachi, M.; Tajima, T.; Motomura, K.; et al. Heterozygous orthodenticle homeobox 2 mutations are associated with variable pituitary phenotype. J. Clin. Endocrinol. Metab. 2010, 95, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Beby, F.; Lamonerie, T. The homeobox gene Otx2 in development and disease. Exp. Eye Res. 2013, 111, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Baas, D.; Bumsted, K.M.; Martinez, J.A.; Vaccarino, F.M.; Wikler, K.C.; Barnstable, C.J. The subcellular localization of Otx2 is cell-type specific and developmentally regulated in the mouse retina. Mol. Brain Res. 2000, 78, 26–37. [Google Scholar] [CrossRef]
- Fossat, N.; Le Greneur, C.; Béby, F.; Vincent, S.; Godement, P.; Chatelain, G.; Lamonerie, T. A new GFP-tagged line reveals unexpected Otx2 protein localization in retinal photoreceptors. BMC Dev. Biol. 2007, 7, 122. [Google Scholar] [CrossRef]
- Ragge, N.K.; Brown, A.G.; Poloschek, C.M.; Lorenz, B.; Henderson, R.A.; Clarke, M.P.; Russell-Eggitt, I.; Fielder, A.; Gerrelli, D.; Martinez-Barbera, J.P.; et al. Heterozygous mutations of OTX2 cause severe ocular malformations. Am. J. Hum. Genet. 2005, 76, 1008–1022. [Google Scholar] [CrossRef]
- Wyatt, A.; Bakrania, P.; Bunyan, D.J.; Osborne, R.J.; Crolla, J.A.; Salt, A.; Ayuso, C.; Newbury-Ecob, R.; Abou-Rayyah, Y.; Collin, J.R.; et al. Novel heterozygous OTX2 mutations and whole gene deletions in anophthalmia; microphthalmia and coloboma. Hum. Mutat. 2008, 29, E278–E283. [Google Scholar] [CrossRef]
- Ashkenazi-Hoffnung, L.; Lebenthal, Y.; Wyatt, A.W.; Ragge, N.K.; Dateki, S.; Fukami, M.; Ogata, T.; Phillip, M.; Gat-Yablonski, G. A novel loss-of-function mutation in OTX2 in a patient with anophthalmia and isolated growth hormone deficiency. Hum. Genet. 2010, 12, 721–729. [Google Scholar] [CrossRef]
- Catania, A.; Legati, A.; Peverelli, L.; Nanetti, L.; Marchet, S.; Zanetti, N.; Lamperti, C.; Ghezzi, D. Homozygous variant in OTX2 and possible genetic modifiers identified in a patient with combined pituitary hormone deficiency, ocular involvement, myopathy, ataxia, and mitochondrial impairment. Am. J. Med. Genet. A 2019, 179, 827–831. [Google Scholar] [CrossRef]
- Abdalla-Elsayed, M.E.; Schatz, P.; Neuhaus, C.; Khan, A.O. Heterozygous mutation in OTX2 associated with early-onset retinal dystrophy with atypical maculopathy. Mol. Vis. 2017, 23, 778–784. [Google Scholar]
- Schilter, K.F.; Schneider, A.; Bardakjian, T.; Soucy, J.F.; Tyler, R.C.; Reis, L.M.; Semina, E.V. OTX2 microphthalmia syndrome: Four novel mutations and delineation of a phenotype. Clin. Genet. 2011, 79, 158–168. [Google Scholar] [CrossRef]
- Acampora, D.; Mazan, S.; Lallemand, Y.; Avantaggiato, V.; Maury, M.; Simeone, A.; Brûlet, P. Forebrain and midbrain regions are deleted in Otx2−/− mutants due to a defective anterior neuroectoderm specification during gastrulation. Development 1995, 121, 3279–3290. [Google Scholar] [PubMed]
- Matsuo, I.; Kuratani, S.; Kimura, C.; Takeda, N.; Aizawa, S. Mouse Otx2 functions in the formation and patterning of rostral head. Genes. Dev. 1995, 9, 2646–2658. [Google Scholar] [CrossRef] [PubMed]
- Nishida, A.; Furukawa, A.; Koike, C.; Tano, Y.; Aizawa, S.; Matsuo, I.; Furukawa, T. Otx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland development. Nat. Neurosci. 2003, 6, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Koike, C.; Nishida, A.; Ueno, S.; Saito, H.; Sanuki, R.; Sato, S.; Furukawa, A.; Aizawa, S.; Matsuo, I.; Suzuki, N.; et al. Functional roles of Otx2 transcription factor in postnatal mouse retinal development. Mol. Cell. Biol. 2007, 27, 8318–8329. [Google Scholar] [CrossRef]
- Béby, F.; Housset, M.; Fossat, N.; Le Greneur, C.; Flamant, F.; Godement, P.; Lamonerie, T. Otx2 gene deletion in adult mouse retina induces rapid RPE dystrophy and slow photoreceptor degeneration. PLoS ONE 2010, 5, e11673. [Google Scholar] [CrossRef]
- Sanyanusin, P.; Norrish, J.H.; Ward, T.A.; Nebel, A.; McNoe, L.A.; Eccles, M.R. Genomic structure of the human PAX2 gene. Genomics 1996, 35, 258–261. [Google Scholar] [CrossRef]
- Bower, M.; Salomon, R.; Allanson, J.; Antignac, C.; Benedicenti, F.; Benetti, E.; Binenbaum, G.; Jensen, U.B.; Cochat, P.; DeCramer, S.; et al. Update of PAX2 mutations in renal coloboma syndrome syndrome and establishment of a locus-specific database. Hum. Mutat. 2012, 33, 457–466. [Google Scholar] [CrossRef]
- Nornes, H.O.; Dressler, G.R.; Knapik, E.W.; Deutsch, U.; Gruss, P. Spatially and temporally restricted expression of Pax2 during murine neurogenesis. Development 1990, 109, 797–809. [Google Scholar]
- Otteson, D.C.; Shelden, E.; Jones, J.M.; Kameoka, J.; Hitchcock, P.F. Pax2 expression and retinal morphogenesis in the normal and Krd mouse. Dev. Biol. 1998, 193, 209–224. [Google Scholar] [CrossRef]
- Chu, Y.; Hughes, S.; Chan-Ling, T. Differentiation and migration of astrocyte precursor cells and astrocytes in human fetal retina: Relevance to optic nerve coloboma. FASEB J. 2001, 15, 2013–2015. [Google Scholar] [CrossRef]
- Chan-Ling, T.; Chu, Y.; Baxter, L.; Weible, M., II; Hughes, S. In vivo characterization of astrocyte precursor cells (APCs) and astrocytes in developing rat retinae: Differentiation, proliferation, and apoptosis. Glia 2009, 57, 39–53. [Google Scholar] [CrossRef]
- Tao, C.; Zhang, X. Development of astrocytes in the vertebrate eye. Dev. Dyn. 2014, 243, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Soukkarieh, C.; Agius, E.; Soula, C.; Cochard, P. Pax2 regulates neuronal-glial cell fate choice in the embryonic optic nerve. Dev. Biol. 2007, 303, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Boije, H.; Ring, H.; López-Gallardo, M.; Prada, C.; Hallböök, F. Pax2 is expressed in a subpopulation of Müller cells in the central chick retina. Dev. Dyn. 2010, 239, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Stanke, J.; Moose, H.E.; El-Hodiri, H.M.; Fischer, A.J. Comparative study of Pax2 expression in glial cells in the retina and optic nerve of birds and mammals. J. Comp. Neurol. 2010, 518, 2316–2333. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, R.; Karcavich, R.; Carlson, S.; Belecky-Adams, T.L. Ectopic Pax2 expression in chick ventral optic cup phenocopies loss of Pax2 expression. Dev. Biol. 2008, 319, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Bower, M.; Eccles, M.; Heidet, L.; Schimmenti, L.A. Clinical utility gene card for: Renal coloboma (Papillorenal) syndrome. Eur. J. Hum. Genet. 2011, 19, 1017. [Google Scholar] [CrossRef][Green Version]
- Bower, M.A.; Schimmenti, L.A.; Eccles, M.R. PAX2-Related Disorder. In Source Gene Reviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2007. [Google Scholar]
- Schimmenti, L.A.; Manligas, G.S.; Sieving, P.A. Optic nerve dysplasia and renal insufficiency in a family with a novel PAX2 mutation, Arg115X: Further ophthalmologic delineation of the renal-coloboma syndrome. Ophthalmic Genet. 2003, 24, 191–202. [Google Scholar] [CrossRef]
- Schimmenti, L.A. Renal coloboma syndrome. Eur. J. Hum. Genet. 2011, 19, 1207–1212. [Google Scholar] [CrossRef]
- Schimmenti, L.A. Genetic and developmental basis of renal coloboma (papillorenal) syndrome. Exp Rev. Ophthalmol. 2009, 4, 135–144. [Google Scholar] [CrossRef]
- Torres, M.; Gómez-Pardo, E.; Gruss, P. Pax2 contributes to inner ear patterning and optic nerve trajectory. Development 1996, 122, 3381–3391. [Google Scholar] [PubMed]
- Favor, J.; Sandulache, R.; Neuhäuser-Klaus, A.; Pretsch, W.; Chatterjee, B.; Senft, E.; Wurst, W.; Blanquet, V.; Grimes, P.; Spörle, R.; et al. The mouse Pax2(1Neu) mutation is identical to a human PAX2 mutation in a family with renal-coloboma syndrome and results in developmental defects of the brain, ear, eye, and kidney. Proc. Natl. Acad. Sci. USA 1996, 93, 13870–13875. [Google Scholar] [CrossRef] [PubMed]
- Alur, R.P.; Cox, T.A.; Crawford, M.A.; Gong, X.; Brooks, B.P. Optic nerve axon number in mouse is regulated by PAX2. J. AAPOS 2008, 12, 117–121. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cross, S.H.; McKie, L.; West, K.; Coghill, E.L.; Favor, J.; Bhattacharya, S.; Brown, S.D.; Jackson, I.J. The Opdc missense mutation of Pax2 has a milder than loss-of-function phenotype. Hum. Mol. Genet. 2011, 20, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Glaser, T.; Walton, D.S.; Maas, R.L. Genomic structure; evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat. Genet. 1992, 2, 232–239. [Google Scholar] [CrossRef]
- Mayran, A.; Pelletier, A.; Drouin, J. Pax factors in transcription and epigenetic remodelling. Semin. Cell. Dev. Biol. 2015, 44, 135–144. [Google Scholar] [CrossRef]
- Cvekl, A.; Callaerts, P. PAX6: 25th anniversary and more to learn. Exp. Eye Res. 2017, 156, 10–21. [Google Scholar] [CrossRef]
- Bäumer, N.; Marquardt, T.; Stoykova, A.; Ashery-Padan, R.; Chowdhury, K.; Gruss, P. Pax6 is required for establishing naso-temporal and dorsal characteristics of the optic vesicle. Development 2002, 129, 4535–4545. [Google Scholar]
- Bäumer, N.; Marquardt, T.; Stoykova, A.; Spieler, D.; Treichel, D.; Ashery-Padan, R.; Gruss, P. Retinal pigmented epithelium determination requires the redundant activities of Pax2 and Pax6. Development 2003, 130, 2903–2915. [Google Scholar] [CrossRef]
- Cavodeassi, F.; Bovolenta, P. New functions for old genes: Pax6 and Mitf in eye pigment biogenesis. Pigment Cell Melanoma Res. 2014, 27, 1005–1007. [Google Scholar] [CrossRef]
- Ashery-Padan, R.; Gruss, P. Pax6 lights-up the way for eye development. Curr. Opin. Cell. Biol. 2001, 13, 706–714. [Google Scholar] [CrossRef]
- Macdonald, R.; Wilson, S.W. Distribution of Pax6 protein during eye development suggests discrete roles in proliferative and differentiated visual cells. Dev. Genes Evol. 1997, 206, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Belecky-Adams, T.; Tomarev, S.; Li, H.S.; Ploder, L.; McInnes, R.R.; Sundin, O.; Adler, R. Pax-6, Prox 1, and Chx10 homeobox gene expression correlates with phenotypic fate of retinal precursor cells. Investig. Ophthalmol. Vis. Sci. 1997, 38, 1293–1303. [Google Scholar]
- Canto-Soler, M.V.; Huang, H.; Romero, M.S.; Adler, R. Transcription factors CTCF and Pax6 are segregated to different cell types during retinal cell differentiation. Dev. Dyn. 2008, 237, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Sunderland, M.E.; Coles, B.L.; Kam, A.; Holowacz, T.; Ashery-Padan, R.; Marquardt, T.; McInnes, R.R.; Van der Kooy, D. The proliferation and expansion of retinal stem cells require functional Pax6. Dev. Biol. 2007, 304, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, T.; Ashery-Padan, R.; Andrejewski, N.; Scardigli, R.; Guillemot, F.; Gruss, P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell 2001, 105, 43–55. [Google Scholar] [CrossRef]
- Philips, G.T.; Stair, C.N.; Young Lee, H.; Wroblewski, E.; Berberoglu, M.A.; Brown, N.L.; Mastick, G.S. Precocious retinal neurons: Pax6 controls timing of differentiation and determination of cell type. Dev. Biol. 2005, 279, 308–321. [Google Scholar] [CrossRef]
- Hitchcock, P.F.; Macdonald, R.E.; VanDeRyt, J.T.; Wilson, S.W. Antibodies against Pax6 immunostain amacrine and ganglion cells and neuronal progenitors, but not rod precursors, in the normal and regenerating retina of the goldfish. J. Neurobiol. 1996, 29, 399–413. [Google Scholar] [CrossRef]
- Rath, M.F.; Bailey, M.J.; Kim, J.S.; Coon, S.L.; Klein, D.C.; Møller, M. Developmental and daily expression of the Pax4 and Pax6 homeobox genes in the rat retina: Localization of Pax4 in photoreceptor cells. J. Neurochem. 2009, 108, 285–294. [Google Scholar] [CrossRef]
- Remez, L.A.; Onishi, A.; Menuchin-Lasowski, Y.; Biran, A.; Blackshaw, S.; Wahlin, K.J.; Zack, D.J.; Ashery-Padan, R. Pax6 is essential for the generation of late-born retinal neurons and for inhibition of photoreceptor-fate during late stages of retinogenesis. Dev. Biol. 2017, 432, 140–150. [Google Scholar] [CrossRef]
- Klimova, L.; Antosova, B.; Kuzelova, A.; Strnad, H.; Kozmik, Z. Onecut1 and Onecut2 transcription factors operate downstream of Pax6 to regulate horizontal cell development. Dev. Biol. 2015, 402, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Yasue, A.; Kono, H.; Habuta, M.; Bando, T.; Sato, K.; Inoue, J.; Oyadomari, S.; Noji, S.; Tanaka, E.; Ohuchi, H. Relationship between somatic mosaicism of Pax6 mutation and variable developmental eye abnormalities-an analysis of CRISPR genome-edited mouse embryos. Sci. Rep. 2017, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Azuma, N.; Yamaguchi, Y.; Handa, H.; Tadokoro, K.; Asaka, A.; Kawase, E.; Yamada, M. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am. J. Hum. Genet. 2003, 72, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, B.K.; Yang, Y.; Cveklová, K.; Cvekl, A. Functional properties of natural human PAX6 and PAX6(5a) mutants. Invest. Ophthalmol. Vis. Sci. 2004, 45, 385–392. [Google Scholar] [CrossRef]
- Vincent, M.C.; Gallai, R.; Olivier, D.; Speeg-Schatz, C.; Flament, J.; Calvas, P.; Dollfus, H. Variable phenotype related to a novel PAX 6 mutation (IVS4 + 5G > C) in a family presenting congenital nystagmus and foveal hypoplasia. Am. J. Ophthalmol. 2004, 138, 1016–1021. [Google Scholar] [CrossRef]
- Williamson, K.A.; FitzPatrick, D.R. The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur. J. Med. Genet. 2014, 57, 369–380. [Google Scholar] [CrossRef]
- Williamson, K.A.; Hall, H.N.; Owen, L.J.; Livesey, B.J.; Hanson, I.M.; Adams, G.G.W.; Bodek, S.; Calvas, P.; Castle, B.; Clarke, M.; et al. Recurrent heterozygous PAX6 missense variants cause severe bilateral microphthalmia via predictable effects on DNA-protein interaction. Genet. Med. 2019. published online 08 November 2019. [Google Scholar] [CrossRef]
- Verma, A.S.; Fitzpatrick, D.R. Anophthalmia and microphthalmia. Orphanet, J. Rare Dis. 2007, 2, 47. [Google Scholar] [CrossRef]
- Schmidt-Sidor, B.; Szymańska, K.; Williamson, K.; Van Heyningen, V.; Roszkowski, T.; Wierzba-Bobrowicz, T.; Zaremba, J. Malformations of the brain in two fetuses with a compound heterozygosity for two PAX6 mutations. Folia Neuropathol. 2009, 47, 372–382. [Google Scholar]
- Deml, B.; Reis, L.M.; Lemyre, E.; Clark, R.D.; Kariminejad, A.; Semina, E.V. Novel mutations in PAX6, OTX2 and NDP in anophthalmia, microphthalmia and coloboma. Eur. J. Hum. Genet. 2016, 24, 535–541. [Google Scholar] [CrossRef]
- Hogan, B.L.; Horsburgh, G.; Cohen, J.; Hetherington, C.M.; Fisher, G.; Lyon, M.F. Small eyes (Sey): A homozygous lethal mutation on chromosome 2 which affects the differentiation of both lens and nasal placodes in the mouse. J. Embryol. Exp. Morphol. 1986, 97, 95–110. [Google Scholar] [PubMed]
- Kaufman, M.H.; Chang, H.H.; Shaw, J.P. Craniofacial abnormalities in homozygous Small eye (Sey/Sey) embryos and newborn mice. J. Anat. 1995, 186, 607–617. [Google Scholar]
- Graw, J.; Löster, J.; Puk, O.; Münster, D.; Haubst, N.; Soewarto, D.; Fuchs, H.; Meyer, B.; Nürnberg, P.; Pretsch, W.; et al. Three novel Pax6 alleles in the mouse leading to the same small-eye phenotype caused by different consequences at target promoters. Invest. Ophthalmol. Vis. Sci. 2005, 46, 4671–4683. [Google Scholar] [CrossRef][Green Version]
- Nakayama, T.; Fisher, M.; Nakajima, K.; Odeleye, A.O.; Zimmerman, K.B.; Fish, M.B.; Yaoita, Y.; Chojnowski, J.L.; Lauderdale, J.D.; Netland, P.A.; et al. Xenopus pax6 mutants affect eye development and other organ systems; and have phenotypic similarities to human aniridia patients. Dev. Biol. 2015, 408, 328–344. [Google Scholar] [CrossRef]
- Favor, J.; Gloeckner, C.J.; Neuhäuser-Klaus, A.; Pretsch, W.; Sandulache, R.; Saule, S.; Zaus, I. Relationship of Pax6 activity levels to the extent of eye development in the mouse, Mus musculus. Genetics 2008, 179, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Manuel, M.; Pratt, T.; Liu, M.; Jeffery, G.; Price, D.J. Overexpression of Pax6 results in microphthalmia; retinal dysplasia and defective retinal ganglion cell axon guidance. BMC Dev. Biol. 2008, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Gregory-Evans, C.Y.; Wang, X.; Wasan, K.M.; Zhao, J.; Metcalfe, A.L.; Gregory-Evans, K. Postnatal manipulation of Pax6 dosage reverses congenital tissue malformation defects. J. Clin. Investig. 2014, 124, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Kozak, C.A.; Cepko, C.L. Rax, a novel paired-type homeobox gene, shows expression in the anterior neural fold and developing retina. Proc. Natl. Acad. Sci. USA 1997, 94, 3088–3093. [Google Scholar] [CrossRef]
- Orquera, D.P.; De Souza, F.S.J. Evolution of the Rax family of developmental transcription factors in vertebrates. Mech. Dev. 2017, 144, 163–170. [Google Scholar] [CrossRef]
- Chen, G.; Courey, A.J. Groucho/TLE family proteins and transcriptional repression. Gene 2000, 249, 1–16. [Google Scholar] [CrossRef]
- Muranishi, Y.; Terda, K.; Furukawa, T. An essential role for Rax in retina and neuroendocrine system development. Dev. Growth Differ. 2012, 54, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Giannaccini, M.; Giudetti, G.; Biasci, D.; Mariotti, S.; Martini, D.; Barsacchi, G.; Andreazzoli, M. Brief report: Rx1 defines retinal precursor identity by repressing alternative fates through the activation of TLE2 and Hes4. Stem Cells 2013, 31, 2842–2847. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Mukherjee, S.; Bao, Z.Z.; Morrow, E.M.; Cepko, C.L. rax, Hes1, and notch1 promote the formation of Müller glia by postnatal retinal progenitor cells. Neuron 2000, 26, 383–394. [Google Scholar] [CrossRef]
- Pan, Y.; Martinez-De Luna, R.I.; Lou, C.H.; Nekkalapudi, S.; Kelly, L.E.; Sater, A.K.; El-Hodiri, H.M. Regulation of photoreceptor gene expression by the retinal homeobox (Rx) gene product. Dev. Biol. 2010, 339, 494–506. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dixit, R.; Tachibana, N.; Touahri, Y.; Zinyk, D.; Logan, C.; Schuurmans, C. Gene expression is dynamically regulated in retinal progenitor cells prior to and during overt cellular differentiation. Gene Expr. Patterns 2014, 14, 42–54. [Google Scholar] [CrossRef]
- Harris, W.A.; Perron, M. Molecular recapitulation: The growth of the vertebrate retina. Int. J. Dev. Biol. 1998, 42, 299–304. [Google Scholar] [PubMed]
- Lequeux, L.; Rio, M.; Vigouroux, A.; Titeux, M.; Etchevers, H.; Malecaze, F.; Chassaing, N.; Calvas, P. Confirmation of RAX gene involvement in human anophthalmia. Clin. Genet. 2008, 74, 392–395. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, J.; Pelcastre, E.L.; Tovilla-Canales, J.L.; Garcia-Ortiz, J.E.; Amato-Almanza, M.; Villanueva-Mendoza, C.; Espinosa-Mattar, Z.; Zenteno, J.C. Mutational screening of CHX10, GDF6, OTX2, RA.X and SOX2 genes in 50 unrelated microphthalmia-anophthalmia-coloboma (MAC) spectrum cases. Br. J. Ophthalmol. 2010, 94, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- Abouzeid, H.; Youssef, M.A.; Bayoumi, N.; ElShakankiri, N.; Marzouk, I.; Hauser, P.; Schorderet, D.F. RAX and anophthalmia in humans: Evidence of brain anomalies. Mol. Vis. 2012, 18, 1449–1456. [Google Scholar]
- Voronina, V.A.; Kozlov, S.; Mathers, P.H.; Lewandoski, M. Conditional alleles for activation and inactivation of the mouse Rx homeobox gene. Genesis 2005, 41, 160–164. [Google Scholar] [CrossRef]
- Bailey, T.J.; El-Hodiri, H.; Zhang, L.; Shah, R.; Mathers, P.H.; Jamrich, M. Regulation of vertebrate eye development by Rx genes. Int. J. Dev. Biol. 2004, 48, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.M.; Park, L.; Stenkamp, D.L. Retinal homeobox 1 is required for retinal neurogenesis and photoreceptor differentiation in embryonic zebrafish. Dev. Biol. 2009, 328, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Andreazzoli, M.; Gestri, G.; Angeloni, D.; Menna, E.; Barsacchi, G. Role of Xrx1 in Xenopus eye and anterior brain development. Development 1999, 126, 2451–2460. [Google Scholar] [PubMed]
- Chuang, J.C.; Raymond, P.A. Zebrafish genes rx1 and rx2 help define the region of forebrain that gives rise to retina. Dev. Biol. 2001, 231, 13–30. [Google Scholar] [CrossRef][Green Version]
- Winkler, S.; Loosli, F.; Henrich, T.; Wakamatsu, Y.; Wittbrodt, J. The conditional medaka mutation eyeless uncouples patterning and morphogenesis of the eye. Development 2000, 127, 1911–1919. [Google Scholar]
- Pan, Y.; Kelly, L.E.; El-Hodiri, H.M. Identification of retinal homeobox (rax) gene-dependent genes by a microarray approach: The DNA endoglycosylase neil3 is a major downstream component of the rax genetic pathway. Dev. Dyn. 2018, 247, 1199–1210. [Google Scholar] [CrossRef]
- Wang, Q.L.; Chen, S.; Esumi, N.; Swain, P.K.; Haines, H.S.; Peng, G.; Melia, B.M.; McIntosh, I.; Heckenlively, J.R.; Jacobson, S.G.; et al. QRX, a novel homeobox gene, modulates photoreceptor gene expression. Hum. Mol. Genet. 2004, 13, 1025–1040. [Google Scholar] [CrossRef]
- Chen, C.M.; Cepko, C.L. The chicken RaxL gene plays a role in the initiation of photoreceptor differentiation. Development 2002, 129, 5363–5375. [Google Scholar] [CrossRef]
- Zhong, Y.F.; Holland, P.W. The dynamics of vertebrate homeobox gene evolution: Gain and loss of genes in mouse and human lineages. BMC Evol. Biol. 2011, 11, 169. [Google Scholar]
- Sánchez-Arrones, L.; Ferán, J.L.; Rodríguez-Gallardo, L.; Puelles, L. Incipient forebrain boundaries traced by differential gene expression and fate mapping in the chick neural plate. Dev. Biol. 2009, 335, 43–65. [Google Scholar] [CrossRef]
- Pinelli, M.; Carissimo, A.; Cutillo, L.; Lai, C.H.; Mutarelli, M.; Moretti, M.N.; Singh, M.V.; Karali, M.; Carrella, D.; Pizzo, M.; et al. An atlas of gene expression and gene co-regulation in the human retina. Nucleic Acids Res. 2016, 44, 5773–5784. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.C.; Mathers, P.H.; Raymond, P.A. Expression of three Rx homeobox genes in embryonic and adult zebrafish. Mech. Dev. 1999, 84, 195–198. [Google Scholar] [CrossRef]
- Yang, P.; Chiang, P.W.; Weleber, R.G.; Pennesi, M.E. Autosomal Dominant Retinal Dystrophy with Electronegative Waveform Associated with a Novel RAX2 Mutation. JAMA Ophthalmol. 2015, 133, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Van de Sompele, S.; Smith, C.; Karali, M.; Corton, M.; Van Schil, K.; Peelman, F.; Cherry, T.; Rosseel, T.; Verdin, H.; Derolez, J.; et al. Biallelic sequence and structural variants in RAX2 are a novel cause for autosomal recessive inherited retinal disease. Genet. Med. 2019, 21, 1319–1329. [Google Scholar] [CrossRef]
- Wu, H.Y.; Perron, M.; Hollemann, T. The role of Xenopus Rx-L in photoreceptor cell determination. Dev. Biol. 2009, 327, 352–365. [Google Scholar] [CrossRef]
- Bertuzzi, S.; Hindges, R.; Mui, S.H.; O’Leary, D.D.; Lemke, G. The homeodomain protein vax1 is required for axon guidance and major tract formation in the developing forebrain. Genes Dev. 1999, 13, 3092–3105. [Google Scholar] [CrossRef]
- Kim, N.; Min, K.W.; Kang, K.H.; Lee, E.J.; Kim, H.T.; Moon, K.; Choi, J.; Le, D.; Lee, S.H.; Kim, J.W. Regulation of retinal axon growth by secreted Vax1 homeodomain protein. Elife 2014, 3, e02671. [Google Scholar] [CrossRef]
- Hallonet, M.; Hollemann, T.; Wehr, R.; Jenkins, N.A.; Copeland, N.G.; Pieler, T.; Gruss, P. Vax1 is a novel homeobox-containing gene expressed in the developing anterior ventral forebrain. Development 1998, 125, 2599–2610. [Google Scholar]
- Hallonet, M.; Hollemann, T.; Pieler, T.; Gruss, P. Vax1, a novel homeobox-containing gene, directs development of the basal forebrain and visual system. Genes Dev. 1999, 13, 3106–3114. [Google Scholar] [CrossRef]
- Mui, S.H.; Kim, J.W.; Lemke, G.; Bertuzzi, S. Vax genes ventralize the embryonic eye. Genes Dev. 2005, 19, 1249–1259. [Google Scholar] [CrossRef]
- Take-uchi, M.; Clarke, J.D.; Wilson, S.W. Hedgehog signalling maintains the optic stalk-retinal interface through the regulation of Vax gene activity. Development 2003, 130, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Slavotinek, A.M.; Chao, R.; Vacik, T.; Yahyavi, M.; Abouzeid, H.; Bardakjian, T.; Schneider, A.; Shaw, G.; Sherr, E.H.; Lemke, G.; et al. VAX1 mutation associated with microphthalmia, corpus callosum agenesis, and orofacial clefting: The first description of a VAX1 phenotype in humans. Hum. Mutat. 2012, 33, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Butali, A.; Suzuki, S.; Cooper, M.E.; Mansilla, A.M.; Cuenco, K.; Leslie, E.J.; Suzuki, Y.; Niimi, T.; Yamamoto, M.; Ayanga, G.; et al. Replication of genome wide association identified candidate genes confirm the role of common and rare variants in PAX7 and VAX1 in the etiology of nonsyndromic CL(P). Am. J. Med. Genet. A. 2013, 161A, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Setó-Salvia, N.; Stanier, P. Genetics of cleft lip and/or cleft palate: Association with other common anomalies. Eur. J. Med. Genet. 2014, 57, 381–393. [Google Scholar] [CrossRef]
- Schulte, D.; Furukawa, T.; Peters, M.A.; Kozak, C.A.; Cepko, C.L. Misexpression of the Emx-related homeobox genes cVax and mVax2 ventralizes the retina and perturbs the retinotectal map. Neuron 1999, 24, 541–553. [Google Scholar] [CrossRef]
- Ohsaki, K.; Morimitsu, T.; Ishida, Y.; Kominami, R.; Takahashi, N. Expression of the Vax family homeobox genes suggests multiple roles in eye development. Genes Cells 1999, 4, 267–276. [Google Scholar] [CrossRef]
- Barbieri, A.M.; Lupo, G.; Bulfone, A.; Andreazzoli, M.; Mariani, M.; Fougerousse, F.; Consalez, G.G.; Borsani, G.; Beckmann, J.S.; Barsacchi, G.; et al. A homeobox gene, vax2, controls the patterning of the eye dorsoventral axis. Proc. Natl. Acad. Sci. USA 1999, 96, 10729–10734. [Google Scholar] [CrossRef]
- Alfano, G.; Conte, I.; Caramico, T.; Avellino, R.; Arnò, B.; Pizzo, M.T.; Tanimoto, N.; Beck, S.C.; Huber, G.; Dollé, P.; et al. Vax2 regulates retinoic acid distribution and cone opsin expression in the vertebrate eye. Development 2011, 138, 261–271. [Google Scholar] [CrossRef]
- Kim, J.W.; Lemke, G. Hedgehog-regulated localization of Vax2 controls eye development. Genes Dev. 2006, 20, 2833–2847. [Google Scholar] [CrossRef]
- Alfano, G.; Shah, A.Z.; Jeffery, G.; Bhattacharya, S.S. First insights into the expression of VAX2 in humans and its localization in the adult primate retina. Exp. Eye Res. 2016, 148, 24–29. [Google Scholar] [CrossRef]
- Alfano, G.; Waseem, N.H.; Webster, A.R.; Bhattacharya, S.S. Identification and characterization of the VAX2 p.Leu139Arg variant: Possible involvement of VAX2 in cone dystrophy. Ophthalmic Genet. 2018, 39, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Norgett, E.E.; Yii, A.; Blake-Palmer, K.G.; Sharifian, M.; Allen, L.E.; Najafi, A.; Kariminejad, A.; Karet Frankl, F.E. A role for VAX2 in correct retinal function revealed by a novel genomic deletion at 2p13.3 causing distal Renal Tubular Acidosis: Case report. BMC Med. Genet. 2015, 16, 38. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barbieri, A.M.; Broccoli, V.; Bovolenta, P.; Alfano, G.; Marchitiello, A.; Mocchetti, C.; Crippa, L.; Bulfone, A.; Marigo, V.; Ballabio, A.; et al. Vax2 inactivation in mouse determines alteration of the eye dorsal-ventral axis; misrouting of the optic fibres and eye coloboma. Development 2002, 129, 805–813. [Google Scholar] [PubMed]
- Mui, S.H.; Hindges, R.; O’Leary, D.D.; Lemke, G.; Bertuzzi, S. The homeodomain protein Vax2 patterns the dorsoventral and nasotemporal axes of the eye. Development 2002, 129, 797–804. [Google Scholar] [PubMed]
- Levine, E.M.; Passini, M.; Hitchcock, P.F.; Glasgow, E.; Schechter, N. Vsx-1 and Vsx-2: Two Chx10-like homeobox genes expressed in overlapping domains in the adult goldfish retina. J. Comp. Neurol. 1997, 387, 439–448. [Google Scholar] [CrossRef]
- Hayashi, T.; Huang, J.; Deeb, S.S. RINX(VSX1), a novel homeobox gene expressed in the inner nuclear layer of the adult retina. Genomics 2000, 67, 128–139. [Google Scholar] [CrossRef]
- Semina, E.V.; Mintz-Hittner, H.A.; Murray, J.C. Isolation and characterization of a novel human paired-like homeodomain-containing transcription factor gene, VSX1, expressed in ocular tissues. Genomics 2000, 63, 289–293. [Google Scholar] [CrossRef]
- Dorval, K.M.; Bobechko, B.P.; Ahmad, K.F.; Bremner, R. Transcriptional activity of the paired-like homeodomain proteins CHX10 and VSX1. J. Biol. Chem. 2005, 280, 10100–10108. [Google Scholar] [CrossRef]
- Hayashi, T.; Huang, J.; Deeb, S.S. Expression of rinx/vsx1 during postnatal eye development in cone-bipolar, differentiating ganglion, and lens fiber cells. Jpn. J. Ophthalmol. 2005, 49, 93–105. [Google Scholar] [CrossRef]
- Levine, E.M.; Hitchcock, P.F.; Glasgow, E.; Schechter, N. Restricted expression of a new paired-class homeobox gene in normal and regenerating adult goldfish retina. J. Comp. Neurol. 1994, 348, 596–606. [Google Scholar] [CrossRef]
- Passini, M.A.; Levine, E.M.; Canger, A.K.; Raymond, P.A.; Schechter, N. Vsx-1 and Vsx-2: Differential expression of two paired-like homeobox genes during zebrafish and goldfish retinogenesis. J. Comp. Neurol. 1997, 388, 495–505. [Google Scholar] [CrossRef]
- Ohtoshi, A.; Wang, S.W.; Maeda, H.; Saszik, S.M.; Frishman, L.J.; Klein, W.H.; Behringer, R.R. Regulation of retinal cone bipolar cell differentiation and photopic vision by the CVC homeobox gene Vsx1. Curr. Biol. 2004, 14, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Chow, R.L.; Snow, B.; Novak, J.; Looser, J.; Freund, C.; Vidgen, D.; Ploder, L.; McInnes, R.R. Vsx1, a rapidly evolving paired-like homeobox gene expressed in cone bipolar cells. Mech. Dev. 2001, 109, 315–322. [Google Scholar] [CrossRef]
- Chow, R.L.; Volgyi, B.; Szilard, R.K.; Ng, D.; McKerlie, C.; Bloomfield, S.A.; Birch, D.G.; McInnes, R.R. Control of late off-center cone bipolar cell differentiation and visual signaling by the homeobox gene Vsx1. Proc. Natl. Acad. Sci. USA 2004, 101, 1754–1759. [Google Scholar] [CrossRef]
- D’Autilia, S.; Decembrini, S.; Casarosa, S.; He, R.Q.; Barsacchi, G.; Cremisi, F.; Andreazzoli, M. Cloning and developmental expression of the Xenopus homeobox gene Xvsx1. Dev. Genes Evol. 2006, 216, 829–834. [Google Scholar] [CrossRef]
- Decembrini, S.; Andreazzoli, M.; Vignali, R.; Barsacchi, G.; Cremisi, F. Timing the generation of distinct retinal cells by homeobox proteins. PLoS Biol. 2006, 4, e272. [Google Scholar] [CrossRef]
- Héon, E.; Greenberg, A.; Kopp, K.K.; Rootman, D.; Vincent, A.L.; Billingsley, G.; Priston, M.; Dorval, K.M.; Chow, R.L.; McInnes, R.R.; et al. VSX1: A gene for posterior polymorphous dystrophy and keratoconus. Hum. Mol. Genet. 2002, 11, 1029–1036. [Google Scholar] [CrossRef]
- Mintz-Hittner, H.A.; Semina, E.V.; Frishman, L.J.; Prager, T.C.; Murray, J.C. VSX1 (RINX) mutation with craniofacial anomalies, empty sella, corneal endothelial changes, and abnormal retinal and auditory bipolar cells. Ophthalmology 2004, 111, 828–836. [Google Scholar] [CrossRef]
- Litke, A.M.; Samuelson, S.; Delaney, K.R.; Sauvé, Y.; Chow, R.L. Investigating the Pathogenicity of VSX1 Missense Mutations and Their Association with Corneal Disease. Invest. Ophthalmol. Vis. Sci. 2018, 59, 5824–5835. [Google Scholar] [CrossRef]
- Aldave, A.J.; Yellore, V.S.; Salem, A.K.; Yoo, G.L.; Rayner, S.A.; Yang, H.; Tang, G.Y.; Piconell, Y.; Rabinowitz, Y.S. No VSX1 gene mutations associated with keratoconus. Invest. Ophthalmol. Vis. Sci. 2006, 47, 2820–2822. [Google Scholar] [CrossRef]
- Tanwar, M.; Kumar, M.; Nayak, B.; Pathak, D.; Sharma, N.; Titiyal, J.S.; Dada, R. VSX1 gene analysis in keratoconus. Mol. Vis. 2010, 16, 2395–2401. [Google Scholar] [PubMed]
- Valleix, S.; Nedelec, B.; Rigaudiere, F.; Dighiero, P.; Pouliquen, Y.; Renard, G.; Le Gargasson, J.F.; Delpech, M. H244R VSX1 is associated with selective cone ON bipolar cell dysfunction and macular degeneration in a PPCD family. Invest. Ophthalmol. Vis. Sci. 2006, 47, 48–54. [Google Scholar] [CrossRef]
- Liang, L.; Sandell, J.H. Focus on molecules: Homeobox protein Chx10. Exp Eye Res. 2008, 86, 541–542. [Google Scholar] [CrossRef]
- Dorval, K.M.; Bobechko, B.P.; Fujieda, H.; Chen, S.; Zack, D.J.; Bremner, R. CHX10 targets a subset of photoreceptor genes. J. Biol. Chem. 2006, 281, 744–751. [Google Scholar] [CrossRef]
- Zou, C.; Levine, E.M. Vsx2 controls eye organogenesis and retinal progenitor identity via homeodomain and non-homeodomain residues required for high affinity DNA binding. PLoS Genet. 2012, 8, e1002924. [Google Scholar] [CrossRef] [PubMed]
- Liu, I.S.; Chen, J.D.; Ploder, L.; Vidgen, D.; Van der Kooy, D.; Kalnins, V.I.; McInnes, R.R. Developmental expression of a novel murine homeobox gene (Chx10): Evidence for roles in determination of the neuroretina and inner nuclear layer. Neuron 1994, 13, 377–393. [Google Scholar] [CrossRef]
- Rowan, S.; Cepko, C.L. A POU factor binding site upstream of the Chx10 homeobox gene is required for Chx10 expression in subsets of retinal progenitor cells and bipolar cells. Dev. Biol. 2005, 281, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Comiskey, D.F.; Kelly, L.E.; Chandler, D.S.; El-Hodiri, H.M. Regulation of photoreceptor gene transcription via a highly conserved transcriptional regulatory element by vsx gene products. Mol. Vis. 2016, 22, 1421–1428. [Google Scholar] [PubMed]
- Rowan, S.; Chen, C.M.; Young, T.L.; Fisher, D.E.; Cepko, C.L. Transdifferentiation of the retina into pigmented cells in ocular retardation mice defines a new function of the homeodomain gene Chx10. Development 2004, 131, 5139–5152. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, B.; Singhal, S.; Lawrence, J.M.; Khaw, P.T.; Limb, G.A. Distribution of Müller stem cells within the neural retina: Evidence for the existence of a ciliary margin-like zone in the adult human eye. Exp. Eye Res. 2009, 89, 373–382. [Google Scholar] [CrossRef]
- Green, E.S.; Stubbs, J.L.; Levine, E.M. Genetic rescue of cell number in a mouse model of microphthalmia: Interactions between Chx10 and G1-phase cell cycle regulators. Development 2003, 130, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Horsford, D.J.; Nguyen, M.T.; Sellar, G.C.; Kothary, R.; Arnheiter, H.; McInnes, R.R. Chx10 repression of Mitf is required for the maintenance of mammalian neuroretinal identity. Development 2005, 132, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Faiyaz-Ul-Haque, M.; Zaidi, S.H.; Al-Mureikhi, M.S.; Peltekova, I.; Tsui, L.C.; Teebi, A.S. Mutations in the CHX10 gene in non-syndromic microphthalmia/anophthalmia patients from Qatar. Clin. Genet. 2007, 72, 164–166. [Google Scholar] [CrossRef]
- Iseri, S.U.; Wyatt, A.W.; Nürnberg, G.; Kluck, C.; Nürnberg, P.; Holder, G.E.; Blair, E.; Salt, A.; Ragge, N.K. Use of genome-wide SNP homozygosity mapping in small pedigrees to identify new mutations in VSX2 causing recessive microphthalmia and a semidominant inner retinal dystrophy. Hum. Genet. 2010, 128, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Burkitt Wright, E.M.; Perveen, R.; Bowers, N.; Ramsden, S.; McCann, E.; O’Driscoll, M.; Lloyd, I.C.; Clayton-Smith, J.; Black, G.C. VSX2 in microphthalmia: A novel splice site mutation producing a severe microphthalmia phenotype. Br. J. Ophthalmol. 2010, 94, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, M.; Novak, J.; Liang, M.Y.; Basu, S.; Ploder, L.; Hawes, N.L.; Vidgen, D.; Hoover, F.; Goldman, D.; Kalnins, V.I.; et al. Ocular retardation mouse caused by Chx10 homeobox null allele: Impaired retinal progenitor proliferation and bipolar cell differentiation. Nat. Genet. 1996, 12, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.M.; Yun, S.; Veien, E.S.; Wu, Y.Y.; Chow, R.L.; Dorsky, R.I.; Levine, E.M. Negative regulation of Vsx1 by its paralog Chx10/Vsx2 is conserved in the vertebrate retina. Brain Res. 2008, 1192, 99–113. [Google Scholar] [CrossRef]
- Toy, J.; Norton, J.S.; Jibodh, S.R.; Adler, R. Effects of homeobox genes on the differentiation of photoreceptor and nonphotoreceptor neurons. Invest. Ophthalmol. Vis. Sci. 2002, 43, 3522–3529. [Google Scholar]
- Rutherford, A.D.; Dhomen, N.; Smith, H.K.; Sowden, J.C. Delayed expression of the Crx gene and photoreceptor development in the Chx10-deficient retina. Invest. Ophthalmol. Vis. Sci. 2004, 45, 375–384. [Google Scholar] [CrossRef][Green Version]
- Phillips, M.J.; Jiang, P.; Howden, S.; Barney, P.; Min, J.; York, N.W.; Chu, L.F.; Capowski, E.E.; Cash, A.; Jain, S.; et al. A Novel Approach to Single Cell RNA-Sequence Analysis Facilitates In Silico Gene Reporting of Human Pluripotent Stem Cell-Derived Retinal Cell Types. Stem Cells 2018, 36, 313–324. [Google Scholar] [CrossRef]
- Bryant, L.; Lozynska, O.; Maguire, A.M.; Aleman, T.S.; Bennett, J. Prescreening whole exome sequencing results from patients with retinal degeneration for variants in genes associated with retinal degeneration. Clin. Ophthalmol. 2017, 12, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, S.; Sun, W.; Xiao, X.; Jia, X.; Liu, M.; Xu, L.; Long, Y.; Zhang, Q. An Ophthalmic Targeted Exome Sequencing Panel as a Powerful Tool to Identify Causative Mutations in Patients Suspected of Hereditary Eye Diseases. Transl. Vis. Sci. Technol. 2019, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Hazen, J.L.; Faust, G.G.; Rodriguez, A.R.; Ferguson, W.C.; Shumilina, S.; Clark, R.A.; Boland, M.J.; Martin, G.; Chubukov, P.; Tsunemoto, R.K.; et al. The Complete Genome Sequences, Unique Mutational Spectra, and Developmental Potency of Adult Neurons Revealed by Cloning. Neuron 2016, 89, 1223–1236. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Calton, M.A.; Abell, N.S.; Benchorin, G.; Gloudemans, M.J.; Chen, M.; Hu, J.; Li, X.; Balliu, B.; Bok, D.; et al. Genetic analyses of human fetal retinal pigment epithelium gene expression suggest ocular disease mechanisms. Commun. Biol. 2019, 2, 186. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, X.; Hu, B.; Mao, Y.; Chen, Y.; Yan, L.; Yong, J.; Dong, J.; Wei, Y.; Wang, W.; et al. Dissecting the transcriptome landscape of the human fetal neural retina and retinal pigment epithelium by single-cell RNA-seq analysis. PLoS Biol. 2019, 17, e3000365. [Google Scholar] [CrossRef]
- Voigt, A.P.; Mulfaul, K.; Mullin, N.K.; Flamme-Wiese, M.J.; Giacalone, J.C.; Stone, E.M.; Tucker, B.A.; Scheetz, T.E.; Mullins, R.F. Single-cell transcriptomics of the human retinal pigment epithelium and choroid in health and macular degeneration. Proc. Natl. Acad. Sci. USA 2019, 116, 24100–24107. [Google Scholar] [CrossRef]
- Liu, X.; Chen, J.; Liu, Z.; Li, J.; Yao, K.; Wu, Y. Potential Therapeutic Agents Against Retinal Diseases Caused by Aberrant Metabolism of Retinoids. Invest. Ophthalmol. Vis. Sci. 2016, 57, 1017–1030. [Google Scholar] [CrossRef]
- Lodato, M.A.; Woodworth, M.B.; Lee, S.; Evrony, G.D.; Mehta, B.K.; Karger, A.; Lee, S.; Chittenden, T.W.; D’Gama, A.M.; Cai, X.; et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 2015, 350, 94–98. [Google Scholar] [CrossRef]
- Lukowski, S.W.; Lo, C.Y.; Sharov, A.A.; Nguyen, Q.; Fang, L.; Hung, S.S.; Zhu, L.; Zhang, T.; Grünert, U.; Nguyen, T.; et al. A single-cell transcriptome atlas of the adult human retina. EMBO J. 2019, 38, e100811. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Aleman, T.S.; Boye, S.L.; Schwartz, S.B.; Kaushal, S.; Roman, A.J.; Pang, J.J.; Sumaroka, A.; Windsor, E.A.; Wilson, J.M.; et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc. Natl. Acad. Sci. USA 2008, 105, 15112–15117. [Google Scholar] [CrossRef]
- Bacchi, N.; Casarosa, S.; Denti, M.A. Splicing-correcting therapeutic approaches for retinal dystrophies: Where endogenous gene regulation and specificity matter. Invest. Ophthalmol. Vis. Sci. 2014, 55, 3285–3294. [Google Scholar] [CrossRef] [PubMed]
- Zelinger, L.; Swaroop, A. RNA Biology in Retinal Development and Disease. Trends Genet. 2018, 34, 341–351. [Google Scholar] [CrossRef] [PubMed]
- DiStefano, T.; Chen, H.Y.; Panebianco, C.; Kaya, K.D.; Brooks, M.J.; Gieser, L.; Morgan, N.Y.; Pohida, T.; Swaroop, A. Accelerated and Improved Differentiation of Retinal Organoids from Pluripotent Stem Cells in Rotating-Wall Vessel Bioreactors. Stem Cell Rep. 2018, 10, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Sanjurjo-Soriano, C.; Kalatzis, V. Guiding Lights in Genome Editing Inherited Retinal Disorders: Implications for Gene and Cell Therapy. Neural Plast. 2018, 2018, 5056279. [Google Scholar] [CrossRef] [PubMed]
- Bennicelli, J.; Wright, J.F.; Komaromy, A.; Jacobs, J.B.; Hauck, B.; Zelenaia, O.; Mingozzi, F.; Hui, D.; Chung, D.; Rex, T.S.; et al. Reversal of blindness in animal models of leber congenital amaurosis using optimized AAV2-mediated gene transfer. Mol. Ther. 2008, 16, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Wu, Y.; Gembrling, M.P.; Oliver, M.L.; Waller, M.A.; Bohning, J.D.; Robinson-Hamm, J.N.; Bulaklak, K.; Castellanos Rivera, R.M.; Collier, J.H.; et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019, 25, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 29, 490–495. [Google Scholar] [CrossRef]
- Kulcsár, P.I.; Tálas, A.; Huszár, K.; Ligeti, Z.; Tóth, E.; Weinhardt, N.; Fodor, E.; Welker, E. Crossing enhanced and high fidelity SpCas9 nucleases to optimize specificity and cleavage. Genome Biol. 2017, 18, 190. [Google Scholar] [CrossRef]
- Fagerlund, R.D.; Staals, R.H.; Fineran, P.C. The Cpf1 CRISPR-Cas protein expands genome-editing tools. Genome Biol. 2015, 16, 251. [Google Scholar] [CrossRef]
- Stern, J.H.; Tian, Y.; Funderburgh, J.; Pellegrini, G.; Zhang, K.; Goldberg, J.L.; Ali, R.R.; Young, M.; Xie, Y.; Temple, S. Regenerating Eye Tissues to Preserve and Restore Vision. Cell Stem Cell 2018, 22, 834–849. [Google Scholar] [CrossRef]
- Llonch, S.; Carido, M.; Ader, M. Organoid technology for retinal repair. Dev. Biol. 2018, 433, 132–143. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Qi, S.; Su, G. Retinal organotypic culture—A candidate for research on retinas. Tissue Cell 2018, 51, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuzzani, O.; Binette, F.; Sternberg, H.; West, M.D.; Nasonkin, I.O. Pluripotent Stem Cells for Retinal Tissue Engineering: Current Status and Future Prospects. Stem Cell Rev. Rep. 2018, 14, 463–483. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Kaya, K.D.; Dong, L.; Swaroop, A. Three-dimensional retinal organoids from mouse pluripotent stem cells mimic in vivo development with enhanced stratification and rod photoreceptor differentiation. Mol. Vis. 2016, 22, 1077–1094. [Google Scholar] [PubMed]
- Kuriyan, A.E.; Albini, T.A.; Townsend, J.H.; Rodriguez, M.; Pandya, H.K.; Leonard, R.E.; Parrott, M.B.; Rosenfeld, P.J.; Flynn, H.W.; Goldberg, J.L. Vision Loss after Intravitreal Injection of Autologous "Stem Cells" for AMD. N. Engl. J. Med. 2017, 376, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Okamoto, S.; Takahashi, M. Induction of retinal pigment epithelial cells from monkey iPS cells. Invest. Ophthalmol. Vis. Sci. 2011, 52, 8785–8790. [Google Scholar] [CrossRef]
- Maeda, T.; Lee, M.J.; Palczewska, G.; Marsili, S.; Tesar, P.J.; Palczewskim, K.; Takahashi, M.; Maeda, A. Retinal pigmented epithelial cells obtained from human induced pluripotent stem cells possess functional visual cycle enzymes in vitro and in vivo. J. Biol. Chem. 2013, 288, 34484–34493. [Google Scholar] [CrossRef]
- Welby, E.; Lakowski, J.; Di Foggia, V.; Budinger, D.; Gonzalez-Cordero, A.; Lun, A.T.L.; Epstein, M.; Patel, A.; Cuevas, E.; Kruczek, K.; et al. Isolation and Comparative Transcriptome Analysis of Human Fetal and iPSC-Derived Cone Photoreceptor Cells. Stem Cell Rep. 2017, 9, 1898–1915. [Google Scholar] [CrossRef]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef]
- Roy, S.; Amin, S.; Roy, S. Retinal fibrosis in diabetic retinopathy. Exp. Eye. Res. 2016, 142, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Rusciano, D.; Pezzino, S.; Mutolo, M.G.; Giannotti, R.; Librando, A.; Pescosolido, N. Neuroprotection in Glaucoma: Old and New Promising Treatments. Adv. Pharmacol. Sci. 2017, 2017, 4320408. [Google Scholar] [CrossRef] [PubMed]
- Pardue, M.T.; Allen, R.S. Neuroprotective strategies for retinal disease. Prog. Retin. Eye Res. 2018, 65, 50–76. [Google Scholar] [CrossRef] [PubMed]
- Wubben, T.J.; Besirli, C.G.; Johnson, M.W.; Zacks, D.N. Retinal Neuroprotection: Overcoming the Translational Roadblocks. Am. J. Ophthalmol. 2018, 192, xv–xxii. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, E.; Novikova, Y.; Kilina, O.; Philippov, P. New antioxidant SkQ1 is an effective protector of rat neural retina under conditions of long-term organotypic cultivation. Adv. Aging Res. 2013, 2, 65–71. [Google Scholar] [CrossRef]
- Tokarz, P.; Kaarniranta, K.; Blasiak, J. Role of antioxidant enzymes and small molecular weight antioxidants in the pathogenesis of age-related macular degeneration (AMD). Biogerontology 2013, 14, 461–482. [Google Scholar] [CrossRef] [PubMed]
- Bennis, A.; Gorgels, T.G.; Ten Brink, J.B.; Van der Spek, P.J.; Bossers, K.; Heine, V.M.; Bergen, A.A. Comparison of Mouse and Human Retinal Pigment Epithelium Gene Expression Profiles: Potential Implications for Age-Related Macular Degeneration. PLoS ONE 2015, 10, e0141597. [Google Scholar] [CrossRef]
- Powell, C.; Grant, A.R.; Cornblath, E.; Goldman, D. Analysis of DNA methylation reveals a partial reprogramming of the Müller glia genome during retina regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 19814–19819. [Google Scholar] [CrossRef]
- Jorstad, N.L.; Wilken, M.S.; Grimes, W.N.; Wohl, S.G.; VandenBosch, L.S.; Yoshimatsu, T.; Wong, R.O.; Rieke, F.; Reh, T.A. Stimulation of functional neuronal regeneration from Müller glia in adult mice. Nature 2017, 548, 103–107. [Google Scholar] [CrossRef]
- Bhatia, B.; Singhal, S.; Jayaram, H.; Khaw, P.T.; Limb, G.A. Adult retinal stem cells revisited. Open Ophthalmol. J. 2010, 4, 30–38. [Google Scholar] [CrossRef]
- Hamon, A.; Roger, J.E.; Yang, X.J.; Perron, M. Müller glial cell-dependent regeneration of the neural retina: An overview across vertebrate model systems. Dev. Dyn. 2016, 245, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Boda, E.; Nato, G.; Buffo, A. Emerging pharmacological approaches to promote neurogenesis from endogenous glial cells. Biochem. Pharmacol. 2017, 141, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.J.; Hyde, D.R. Opportunities for CRISPR/Cas9 Gene Editing in Retinal Regeneration Research. Front. Cell Dev. Biol. 2017, 5, 99. [Google Scholar] [CrossRef] [PubMed]
- Hodgetts, S.I.; Harvey, A.R. Neurotrophic Factors Used to Treat Spinal Cord Injury. Vitam. Horm. 2017, 104, 405–457. [Google Scholar]
- Önger, M.E.; Delibaş, B.; Türkmen, A.P.; Erener, E.; Altunkaynak, B.Z.; Kaplan, S. The role of growth factors in nerve regeneration. Drug Discov. Ther. 2017, 10, 285–291. [Google Scholar] [CrossRef][Green Version]
- Leaver, S.G.; Cui, Q.; Plant, G.W.; Arulpragasam, A.; Hisheh, S.; Verhaagen, J.; Harvey, A.R. AAV-mediated expression of CNTF promotes long-term survival and regeneration of adult rat retinal ganglion cells. Gene Ther. 2006, 13, 1328–1341. [Google Scholar] [CrossRef]
- Logan, A.; Ahmed, Z.; Baird, A.; Gonzalez, A.M.; Berry, M. Neurotrophic factor synergy is required for neuronal survival and disinhibited axon regeneration after CNS injury. Brain 2006, 129, 490–502. [Google Scholar] [CrossRef]
- Vigneswara, V.; Esmaeilim, M.; Deer, L.; Berry, M.; Logan, A.; Ahmed, Z. Eye drop delivery of pigment epithelium-derived factor-34 promotes retinal ganglion cell neuroprotection and axon regeneration. Mol. Cell. Neurosci. 2015, 68, 212–221. [Google Scholar] [CrossRef]
- Tönges, L.; Ostendorf, T.; Lamballe, F.; Genestine, M.; Dono, R.; Koch, J.C.; Bähr, M.; Maina, F.; Lingor, P. Hepatocyte growth factor protects retinal ganglion cells by increasing neuronal survival and axonal regeneration in vitro and in vivo. J. Neurochem. 2011, 117, 892–903. [Google Scholar] [CrossRef]
- Leal, G.; Comprido, D.; Duarte, C.B. BDNF-induced local protein synthesis and synaptic plasticity. Neuropharmacology 2014, 76, 639–656. [Google Scholar] [CrossRef]
- Sasi, M.; Vignoli, B.; Canossa, M.; Blum, R. Neurobiology of local and intercellular BDNF signaling. Pflugers Arch. 2017, 469, 593–610. [Google Scholar] [CrossRef] [PubMed]
- Katsman, D.; Stackpole, E.J.; Domin, D.R.; Farber, D.B. Embryonic stem cell-derived microvesicles induce gene expression changes in Müller cells of the retina. PLoS ONE 2012, 7, e50417. [Google Scholar] [CrossRef] [PubMed]
- Thorne, R.G.; Frey, W.H. Delivery of neurotrophic factors to the central nervous system: Pharmacokinetic considerations. Clin. Pharmacokinet. 2001, 40, 907–946. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Dexter, D.T. Neuroprotective and symptomatic effects of targeting group III mGlu receptors in neurodegenerative disease. J. Neurochem. 2014, 129, 4–20. [Google Scholar] [CrossRef]
- Blackiston, D.J.; Vien, K.; Levin, M. Serotonergic stimulation induces nerve growth and promotes visual learning via posterior eye grafts in a vertebrate model of induced sensory plasticity. NPJ Regen. Med. 2017, 2, 8. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, M.; Laties, A.M.; Mitchell, C.H. Balance of purines may determine life or death of retinal ganglion cells as A3 adenosine receptors prevent loss following P2X7 receptor stimulation. J. Neurochem. 2006, 98, 566–575. [Google Scholar] [CrossRef]
- Webster, M.K.; Barnett, B.J.; Stanchfield, M.L.; Paris, J.R.; Webster, S.E.; Cooley-Themm, C.A.; Levine, E.M.; Otteson, D.C.; Linn, C.L. Stimulation of Retinal Pigment Epithelium With an α7 nAChR Agonist Leads to Müller Glia Dependent Neurogenesis in the Adult Mammalian Retina. Invest. Ophthalmol. Vis. Sci. 2019, 60, 570–579. [Google Scholar] [CrossRef]
- Duncan, J.L.; Pierce, E.A.; Laster, A.M.; Daiger, S.P.; Birch, D.G.; Ash, J.D.; Iannaccone, A.; Flannery, J.G.; Sahel, J.A.; Zack, D.J.; et al. Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Transl. Vis. Sci. Technol. 2018, 7, 6. [Google Scholar] [CrossRef]
- Grassmann, F.; Kiel, C.; Zimmermann, M.E.; Gorski, M.; Grassmann, V.; Stark, K.; International AMD Genomics Consortium (IAMDGC); Heid, I.M.; Weber, B.H. Genetic pleiotropy between age-related macular degeneration and 16 complex diseases and traits. Genome Med. 2017, 9, 29. [Google Scholar] [CrossRef]
- Werdich, X.Q.; Place, E.M.; Pierce, E.A. Systemic diseases associated with retinal dystrophies. Semin. Ophthalmol. 2014, 29, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Domínguez, I.; Garanto, A.; Collin, R.W.J. Molecular Therapies for Inherited Retinal Diseases-Current Standing, Opportunities and Challenges. Genes 2019, 10, 654. [Google Scholar] [CrossRef]
- Faber, S.; Roepman, R. Balancing the Photoreceptor Proteome: Proteostasis Network Therapeutics for Inherited Retinal Disease. Genes 2019, 10, 557. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P.M.; Buck, T.M.; Mulder, A.A.; Ohonin, C.; Alves, C.H.; Vos, R.M.; Bialecka, M.; Van Herwaarden, T.; Van Dijk, E.H.C.; Talib, M.; et al. Human iPSC-Derived Retinas Recapitulate the Fetal CRB1 CRB2 Complex Formation and Demonstrate that Photoreceptors and Müller Glia Are Targets of AAV5. Stem Cell Rep. 2019, 12, 906–919. [Google Scholar] [CrossRef] [PubMed]
- Cacheiro, P.; Haendel, M.A.; Smedley, D.; International Mouse Phenotyping Consortium and the Monarch Initiative. New models for human disease from the International Mouse Phenotyping Consortium. Mamm. Genome 2019, 30, 143–150. [Google Scholar] [CrossRef]
- Sridhar, A.; Hoshino, A.; Finkbeiner, C.R.; Chitsazan, A.; Dai, L.; Haugan, A.K.; Eschenbacher, K.M.; Jackson, D.L.; Trapnell, C.; Bermingham-McDonogh, O.; et al. Single-Cell Transcriptomic Comparison of Human Fetal Retina, hPSC-Derived Retinal Organoids, and Long-Term Retinal Cultures. Cell Rep. 2020, 30, 1644–1659. [Google Scholar] [CrossRef]
- Chen, F.K.; McLenachan, S.; Edel, M.; Da Cruz, L.; Coffey, P.J.; Mackey, D.A. iPS Cells for Modelling and Treatment of Retinal Diseases. J. Clin Med. 2014, 3, 1511–1541. [Google Scholar] [CrossRef]
- Delyfer, M.N.; Raffelsberger, W.; Mercier, D.; Korobelnik, J.F.; Gaudric, A.; Charteris, D.G.; Tadayoni, R.; Metge, F.; Caputo, G.; Barale, P.O.; et al. Transcriptomic analysis of human retinal detachment reveals both inflammatory response and photoreceptor death. PLoS ONE 2011, 6, e28791. [Google Scholar] [CrossRef]
- Gasparini, S.J.; Llonch, S.; Borsch, O.; Ader, M. Transplantation of photoreceptors into the degenerative retina: Current state and future perspectives. Prog. Retin. Eye Res. 2019, 69, 1–37. [Google Scholar] [CrossRef]
- Karl, M.O.; Reh, T.A. Regenerative medicine for retinal diseases: Activating endogenous repair mechanisms. Trends Mol. Med. 2010, 16, 193–202. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, C.; Su, G. Cellular Signaling in Müller Glia: Progenitor Cells for Regenerative and Neuroprotective Responses in Pharmacological Models of Retinal Degeneration. J. Ophthalmol. 2019, 2019, 5743109. [Google Scholar] [CrossRef] [PubMed]
- Markitantova, Y.V.; Simirskii, V.N. The Role of the Redox System in Initiation of Neural Eye Tissues Regenerative Response in Vertebrates. Russian J. Dev. Biol. 2020, 51, 16–30. [Google Scholar]


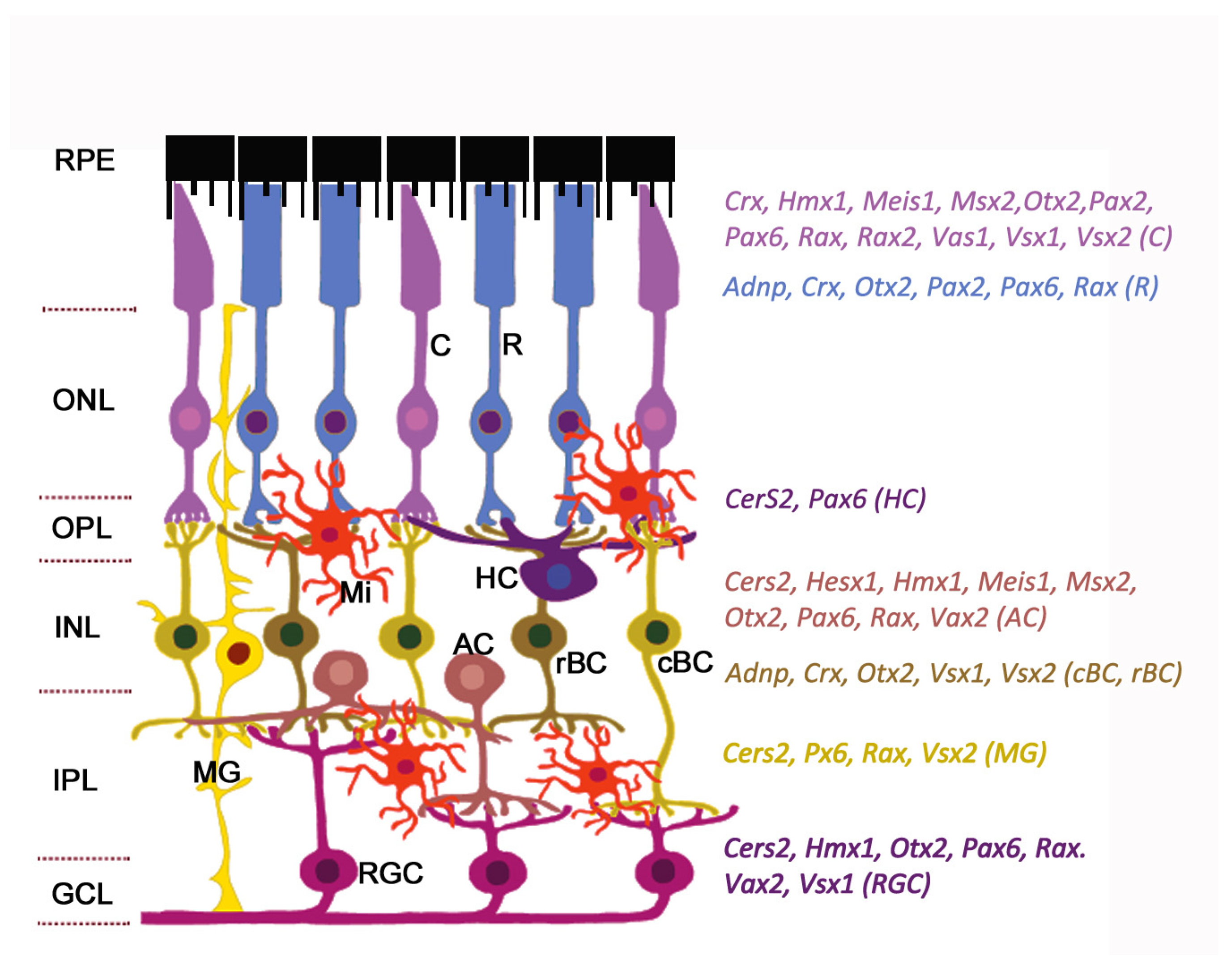{kind=link}
| Gene | Full Name | Synonyms | Ensembl ID | Location | Transcripts (Total/Protein-Coding) (Ensembl) | Homeobox Class | Expression in the Retina | |
|---|---|---|---|---|---|---|---|---|
| RNA (Microarray) (BioGPS) | Protein (ProteomicsDB) | |||||||
| ADNP | Activity-dependent neuroprotector homeobox | ADNP1, KIAA0784 | ENSG00000101126 | 20q13.13 | 9/8 | ZF | 20 | 4 |
| ALX1 | Aristaless-like homeobox 1 | CART1, HEL23 | ENSG00000180318 | 12q21.31 | 1/1 | PRD | 3 | 0 |
| CERS2 | Ceramide synthase 2 | LASS2, TRH3, TMSG1 | ENSG00000143418 | 1q21.3 | 14/9 | CERS | 40 | 3 |
| CRX | Cone-rod homeobox | CORD2, OTX3, LCA7 | ENSG00000105392 | 19q13.33 | 7/4 | PRD | 43 | 18 |
| HESX1 | Homeobox gene expressed in embryonic stem cells 1 | ANF, CPHD5, RPX | ENSG00000163666 | 3p14.3 | 4/4 | PRD | 4 | – |
| HMX1 | H6 family homeobox 1 | Nkx5-3, H6 | ENSG00000215612 | 4p16.1 | 2/2 | ANTP | 13 | 0 |
| LMX1B | LIM homeobox transcription factor 1 beta | NPS1, LMX-1.2 | ENSG00000136944 | 9q33.3 | 4/4 | LIM | 3 | 0 |
| MEIS1 | Myeloid ecotropic insertion site homeobox 1 | – | ENSG00000143995 | 2p14 | 17/6 | TALE | 6 | 2 |
| MSX2 | Muscle segment homeobox 2 | MSH, HOX8 | ENSG00000120149 | 5q35.2 | 2/2 | ANTP | 4 | 0 |
| OTX2 | Orthodenticle homeobox | – | ENSG00000165588 | 14q22.3 | 11/11 | PRD | 4 | 12 |
| PAX2 | Paired box 2 | – | ENSG00000075891 | 10q24.31 | 9/6 | PRD | 3 | 0 |
| PAX6 | Paired box 6 | – | ENSG00000007372 | 11p13 | 82/57 | PRD | 75 | 5 |
| RAX | Retina and anterior neural fold homeobox | RX | ENSG00000134438 | 18q21.32 | 4/3 | PRD | 4 | 0.52 |
| RAX2 | Retina and anterior neural fold homeobox 2 | RAXL1, QRX | ENSG00000173976 | 19p13.3 | 2/2 | PRD | 13 | 5 |
| VAX1 | Ventral anterior homeobox 1 | – | ENSG00000148704 | 10q25.3 | 2/2 | ANTP | – | – |
| VAX2 | Ventral anterior homeobox 2 | – | ENSG00000116035 | 2p13.3 | 3/3 | ANTP | 3 | – |
| VSX1 | Visual system homeobox 1 | RINX, KTCN | ENSG00000100987 | 20p11.21 | 7/6 | PRD | 3 | – |
| VSX2 | Visual system homeobox 2 | CHX10, HOX10, RET1 | ENSG00000119614 | 14q24.3 | 1/1 | PRD | 4 | 4 |
| Gene | Full Name | Expression in the Retina/RPE | Cell Functions | Disease or Syndrome | ||
|---|---|---|---|---|---|---|
| Single-Cell RNAseq (mouse) | RT-PCR, IHC | Disease/Syndrome (OMIM#) | Ocular Manifestations in Humans | |||
| ADNP (611386) | Activity-dependent neuroprotector homeobox | BC, rods | INP, OPL | Neuroprotection, promotion of neuronal growth and differentiation, autophagy | Helsmoortel–van der Aa syndrome (615873) | Macular laminations, foveal hypoplasia, cone degeneration |
| ALX1 (601527) | Aristaless-like homeobox 1 | Early RPC (E14) | Retinal margin | Cranial neural crest migration and differentiation, retinogenesis | Frontonasal dysplasia, type 3 (613456) | Anophthalmia, microphthalmia, optic nerve hypoplasia, coloboma |
| CERS2 (606920) | Ceramide synthase 2 | AC | All neurons, MG | Apoptosis, inflammation, signal transduction, lipid metabolism | Rhegmatogenous retinal detachment (Stickler syndrome) (PS108300) AMD | Retinal detachment, photoreceptor degradation, lipofuscin granules, RPE atrophy |
| CRX (602225) | Cone-rod homeobox | BC, cones, rods | RPC | Development and maintenance of Phr, renewal of Phr disks | Cone-rod retinal dystrophy type 2 (120970) Leber congenital amaurosis type 7 (613829) Dominant retinitis pigmentosa (268000) | Cone-rod retinal dystrophy, widespread retinal pigmentation, chorioretinal atrophy, attenuated retinal vessels, cystoid macular edema |
| HESX1 (601802) | Homeobox gene expressed in embryonic stem cells 1 | – | Early RPC, AC | Maintenance of stemness, neural cell determination | Septo-optic dysplasia (182230) Ocular coloboma (120200) | Optic nerve hypoplasia, hypoplastic optic discs, microphthalmia |
| HMX1 (142992) | H6 family homeobox 1 | AC, cones, RGC | Optic vesicle, early RPC, nasal RGC | Neurogenesis, nasotemporal patterning of retina | Oculoauricular syndrome (612109) | Microphthalmia, ocular coloboma, RPE anomalies, rod-cone dystrophy, macular hypoplasia |
| LMX1B (602575) | LIM homeobox transcription factor 1 beta | – | Periocular mesenchyme | Optic cup morphogenesis, nasotemporal patterning of retina | Nail-patella syndrome (161200) Primary open-angle glaucoma (137760) | Isolated optic disk excavation, ocular hypertension |
| MEIS1 (601739) | Myeloid ecotropic insertion site homeobox 1 | RPC, AC, cones, PhR precursors | – | Control of cell cycle in RPE, dorsoventral and nasotemporal patterning of retina | Microphthalmia (?) | Microphthalmia |
| MSX2 (123101) | Muscle segment homeobox 2 | AC, cones | – | EMT, suppression of transcription, apoptosis, RGC commitment and differentiation | Craniosynostosis type 2 (Boston-type) (604757) | Chorioretinal coloboma |
| OTX2 (600037) | Orthodenticle homeobox 2 | Late RPC, AC, rods, cones, BC, RGC (E14-P0) | RPC, RPE, RGC, PhR, BC | RPE specification, differentiation of photoreceptors and bipolar cells | Microphthalmia, syndromic type 5 (610125) Early-onset retinal dystrophy (610125) | Microphthalmia or anophthalmia, ocular coloboma, retinal dystrophy, optic nerve dysplasia |
| PAX2 (167409) | Paired box 2 | Rods, cones (E18) | Ventral optic vesicle, optic fissure, optic stalk, astrocytes | Inhibition of neurogenesis, induction of gliogenesis, axon guidance | Papillorenal syndrome (120330) | Retinal and optic nerve colobomas, optic disc dysplasia or hyperplasia, microphthalmia, gliosis of optic nerve, abnormal retinal vessels, chorioretinal degeneration |
| PAX6 (607108) | Paired box 6 | Early and late RPC, AC, cones, rods, RGC, MG | RPC, AC, RGC, HC | Maintenance of RPC multipotency, proliferation and differentiation of RGC and RPE, differentiation of HC | Foveal hypoplasia 1 (136520) Optic nerve hypoplasia (165550) Coloboma of optic nerve (120430) | Optic nerve hypoplasia, coloboma of optic nerve, microphthalmia |
| RAX (601881) | Retina and anterior neural fold homeobox | Early and late RPC, AC, rods, cones, RGC | RPC, rods, cones, MG | Proliferation of RPC, differentiation of PhR | Microphthalmia, isolated, type 3 (611038) | Microphthalmia (retinal size reduction) |
| RAX2 | Retina and anterior neural fold homeobox 2 | – | RPC, cones | Differentiation of cones | Cone-rod dystrophy type 11 (610381); AMD type 6 (613757) | Progressive macular atrophy, pigment granularity of peripheral retina, mixed rod and cone dysfunction on electroretinography, atrophy of macular RPE, progressive attenuation of retinal vessels, macular degeneration |
| VAX1 (604294) | Ventral anterior homeobox 1 | Late RPC | Glia of the optic nerve, ventral optic stalk | Repression of retinogenesis, RGC axon growth and guidance, dorsoventral patterning of retina | Microphthalmia, syndromic type 11 (614402) | Bilateral severe microphthalmia, small optic nerve |
| VAX2 (604295) | Ventral anterior homeobox 2 | AC | Cones, RGC | Repression of retinogenesis, dorsoventral patterning of retina, differentiation of cones, organization of nerve fiber layer within retina | Cone dystrophy, coloboma | Macular and cone degeneration |
| VSX1 (605020) | Visual system homeobox 1 | BC, cones | Cone BC (cBC), RGC | cBC differentiation | Craniofacial anomalies and anterior segment dysgenesis syndrome (614195) | Macular degeneration, BC dysfunction |
| VSX2 (142993) | Visual system homeobox 2 | Early and late RPC, BC, cones | RPC, all BC, MG | RPC proliferation, neuroretina specification | Microphthalmia, isolated type 2 (610093) Microphthalmia with coloboma type 3 (610092) | Microphthalmia, coloboma |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markitantova, Y.; Simirskii, V. Inherited Eye Diseases with Retinal Manifestations through the Eyes of Homeobox Genes. Int. J. Mol. Sci. 2020, 21, 1602. https://doi.org/10.3390/ijms21051602
Markitantova Y, Simirskii V. Inherited Eye Diseases with Retinal Manifestations through the Eyes of Homeobox Genes. International Journal of Molecular Sciences. 2020; 21(5):1602. https://doi.org/10.3390/ijms21051602
Chicago/Turabian StyleMarkitantova, Yuliya, and Vladimir Simirskii. 2020. "Inherited Eye Diseases with Retinal Manifestations through the Eyes of Homeobox Genes" International Journal of Molecular Sciences 21, no. 5: 1602. https://doi.org/10.3390/ijms21051602
APA StyleMarkitantova, Y., & Simirskii, V. (2020). Inherited Eye Diseases with Retinal Manifestations through the Eyes of Homeobox Genes. International Journal of Molecular Sciences, 21(5), 1602. https://doi.org/10.3390/ijms21051602