Norepinephrine Regulation of Ventromedial Hypothalamic Nucleus Metabolic-Sensory Neuron 5′-AMP-Activated Protein Kinase Activity: Impact of Estradiol

{kind=link}
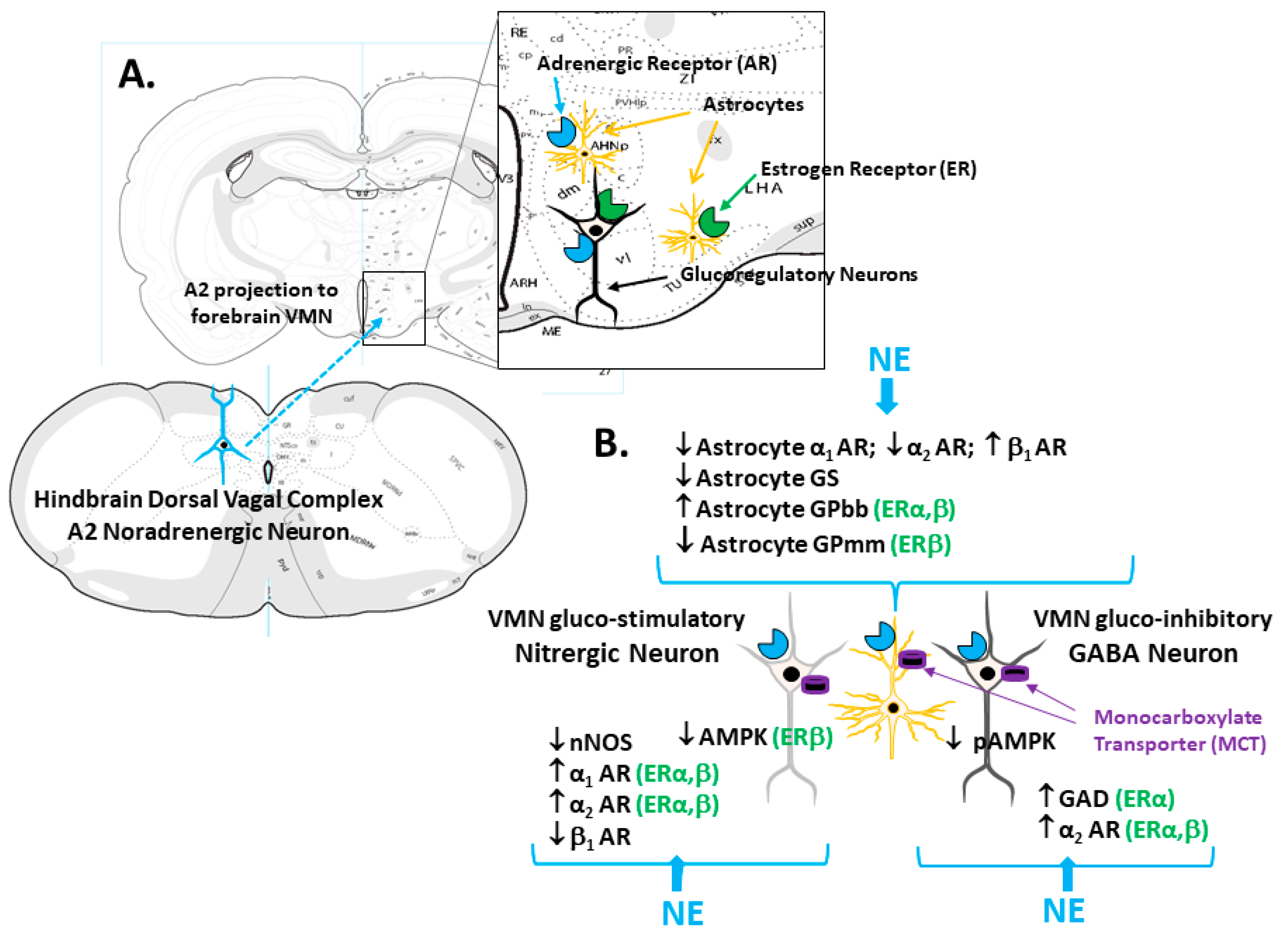
Abstract
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-hydroxydopamine |
| α1-AR | alpha1-adrenergic receptor |
| α2-AR | alpha2-adrenergic receptor |
| β1-AR | beta1-adrenergic receptor |
| AMPK | 5′-AMP-activated protein kinase |
| CA | catecholamine |
| Cc | Compound C |
| ERα | estrogen receptor-alpha |
| ERβ | estrogen receptor-beta |
| GABA | γ-aminobutyric acid |
| GAD | glutamate decarboxylase65/67 |
| GP | glycogen phosphorylase |
| GS | glycogen synthase |
| IIH | insulin-induced hypoglycemia |
| MBH | mediobasal hypothalamus |
| MPP | 1,3-Bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole dihydrochloride |
| NE | norepinephrine |
| NO | nitric oxide |
| nNOS | neuronal nitric oxide synthase |
| OVX | ovariectomy |
| pAMKP | phospho-AMPK |
| PHTPP | 4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol |
| RIIH | recurrent insulin-induced hypoglycemia |
| VMN | ventromedial hypothalamic nucleus |
References
- Watts, A.G.; Donovan, C.M. Sweet talk in the brain: glucosensing, neural networks, and hypoglycemic counterregulation. Front. Neuroendocrinol. 2010, 31, 32–43. [Google Scholar] [CrossRef]
- Donovan, C.M.; Watts, A.G. Peripheral and central glucose sensing in hypoglycemic detection. Physiology 2014, 29, 314–324. [Google Scholar] [CrossRef]
- Borg, M.A.; Tamborlane, W.V.; Shulman, G.I.; Sherwin, R.S. Local lactate perfusion of the ventromedial hypothalamus suppresses hypoglycemic counterregulation. Diabetes 2003, 52, 663–666. [Google Scholar] [CrossRef]
- Borg, M.A.; Sherwin, R.S.; Borg, W.P.; Tamborlane, W.V.; Shulman, G.I. Local ventromedial hypothalamus glucose perfusion blocks counterregulation during systemic hypoglycemia in awake rats. J. Clin. Invest. 1997, 99, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Oomura, Y.; Ono, T.; Ooyama, H.; Wayner, M.J. Glucose and osmosensitive neurones of the rat hypothalamus. Nature 1969, 222, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Ashford, M.L.J.; Boden, P.R.; Treherne, J.M. Glucose-induced excitation of hypothalamic neurons is mediated by ATP-sensitive K+ channels. Pfugers Arch. 1990, 415, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Silver, I.A.; Erecińska, M. Glucose-induced intracellular ion changes in sugar-sensitive hypothalamic neurons. J. Neurophysiol. 1998, 79, 1733–1745. [Google Scholar] [CrossRef]
- Chan, O.; Zhu, W.; Ding, Y.; McCrimmon, R.J.; Sherwin, R.S. Blockade of GABA(A) receptors in the ventromedial hypothalamus further stimulates glucagon and sympathoadrenal but not the hypothalamo-pituitary-adrenal response to hypoglycemia. Diabetes 2006, 55, 1080–1087. [Google Scholar] [CrossRef]
- Fioramonti, X.; Marsollier, N.; Song, Z.; Fakira, K.A.; Patel, R.M.; Brown, S.; Duparc, T.; Pica-Mendez, A.; Sanders, N.M.; Knauf, C.; et al. Ventromedial hypothalamic nitric oxide production is necessary for hypoglycemia detection and counterregulation. Diabetes 2010, 59, 519–528. [Google Scholar] [CrossRef]
- Routh, V.H.; Hao, L.; Santiago, A.M.; Sheng, Z.; Zhou, C. Hypothalamic glucose sensing: making ends meet. Front. Syst. Neurosci. 2014, 8, 236. [Google Scholar] [CrossRef]
- Fioramonti, X.; Song, Z.; Vazirani, R.P.; Beuve, A.; Routh, V.H. Hypothalamic nitric oxide in hypoglycemia detection and counterregulation: a two-edged sword. Antioxid. Redox Signal. 2011, 14, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Han, S.M.; Namkoong, C.; Jang, P.G.; Park, I.S.; Hong, S.W.; Katakami, H.; Chun, S.; Kim, S.W.; Park, J.Y.; Lee, K.U.; et al. Hypothalamic AMP-activated protein kinase mediates counter-regulatory responses to hypoglycaemia in rats. Diabetologia 2005, 48, 2170–2178. [Google Scholar] [CrossRef] [PubMed]
- McCrimmon, R.J.; Shaw, M.; Fan, X.; Cheng, H.; Ding, Y.; Vella, M.C.; Zhou, L.; McNay, E.C.; Sherwin, R.S. Key role for AMP-activated protein kinase in the ventromedial hypothalamus in regulating counterregulatory hormone responses to acute hypoglycemia. Diabetes 2008, 57, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Lόpez, M. Hypothalamic AMPK and energy balance. Eur. J. Clin. Invest. 2018, 48, e12996. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Cheng, K.K. Hypothalamic AMPK as a mediator of hormonal regulation of energy balance. Int. J. Mol. Sci. 2018, 19, 3552. [Google Scholar] [CrossRef] [PubMed]
- Patil, G.D.; Briski, K.P. Lactate is a critical ‘sensed’ variable in caudal hindbrain monitoring of CNS metabolic stasis. Amer. J. Physiol. 2005, 289, R1777–R1786. [Google Scholar] [CrossRef][Green Version]
- Alenazi, F.S.H.; Ibrahim, B.A.; Briski, K.P. Estradiol regulates effects of hindbrain activator 5-aminoimidazole-4-carboxamide-riboside administration on hypothalamic adenosine 5′-monophosphate-activated protein kinase activity and metabolic neurotransmitter mRNA and protein expression. J. Neurosci. Res. 2014, 93, 651–659. [Google Scholar] [CrossRef]
- Alenazi, F.S.H.; Ibrahim, B.A.; Alhamami, H.; Shakya, M.; Briski, K.P. Role of estradiol in intrinsic hindbrain AMPK regulation of hypothalamic AMPK, metabolic neuropeptide, and norepinephrine activity and food intake in the female rat. Neuroscience 2016, 314, 35–46. [Google Scholar] [CrossRef][Green Version]
- Mandal, S.K.; Briski, K.P. Hindbrain dorsal vagal complex AMPK controls hypothalamic AMPK activation and metabolic neurotransmitter protein expression and counter-regulatory hormone secretion in the hypoglycemic male rat. Brain Res. Bull. 2018, 144, 171–179. [Google Scholar] [CrossRef]
- Gujar, A.D.; Ibrahim, B.A.; Tamrakar, P.; Koshy Cherian, A.; Briski, K.P. Hindbrain lactostasis regulates hypothalamic AMPK activity and hypothalamic metabolic neurotransmitter mRNA and protein responses to hypoglycemia. Amer. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R457–R469. [Google Scholar] [CrossRef]
- Briski, K.P.; Koshy Cherian, A.; Genabai, N.K.; Vavaiya, K.V. In situ coexpression of glucose and monocarboxylate transporter mRNAs in metabolic-sensitive dorsal vagal complex catecholaminergic neurons: transcriptional reactivity to insulin-induced hypoglycemia and caudal hindbrain glucose or lactate repletion during insulin-induced hypoglycemia. Neuroscience 2009, 164, 1152–1160. [Google Scholar] [PubMed]
- Cherian, A.; Briski, K.P. Quantitative RT PCR and immunoblot analyses reveal acclimated A2 noradrenergic neuron substrate fuel transporter, glucokinase, phospho-AMPK, and dopamine-beta-hydroxylase responses to hypoglycemia. J. Neurosci. Res. 2011, 89, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, P.K.; Tamarkar, P.; Ibrahim, B.A.; Briski, K.P. Hindbrain medulla catecholamine cell group involvement in lactate-sensitive hypoglycemia-associated patterns of hypothalamic norepinephrine and epinephrine activity. Neuroscience 2014, 278, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M.H.; Alhamami, H.N.; Briski, K.P. Norepinephrine regulation of ventromedial hypothalamic nucleus metabolic transmitter biomarker and astrocyte enzyme and receptor expression: role of 5′-AMP-activated protein kinase. Brain Res. 2019, 1711, 48–57. [Google Scholar] [CrossRef]
- Wade, G.N.; Schneider, J.E. Metabolic fuels and reproduction in female mammals. Neurosci. Biobehav. Res. 1992, 16, 235–272. [Google Scholar] [CrossRef]
- Nedungadi, T.P.; Goleman, W.L.; Paranjape, S.A.; Kale, A.Y.; Briski, K.P. Effects of estradiol on glycemic and CNS neuronal activational responses to recurrent insulin-induced hypoglycemia in the ovariectomized female rat. Neuroendocrinology 2006, 84, 235–243. [Google Scholar] [CrossRef]
- Briski, K.P.; Nedungadi, T.P. Adaptation of feeding and counter-regulatory hormone responses to intermediate insulin-induced hypoglycaemia in the ovariectomised female rat: effects of oestradiol. J. Neuroendocrinol. 2009, 21, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, B.A.; Tamrakar, P.; Gujar, A.D.; Koshy Cherian, A.; Briski, K.P. Caudal fourth ventricular administration of the AMPK activator 5-aminoimiazole-4-carboxamide-riboside regulates glucose and counterregulatory hormone profiles, dorsal vagal complex metabolosensory neuron function, and hypothalamic Fos expression. J. Neurosci. Res. 2013, 91, 1226–1238. [Google Scholar] [CrossRef]
- Nedungadi, T.P.; Briski, K.P. Site-specific effects of intracranial estradiol administration on recurrent insulin-induced hypoglycemia in ovariectomized female rats. Neuroendocrinology 2012, 96, 311–323. [Google Scholar] [CrossRef]
- Mahmood, A.S.M.H.; Uddin, M.M.; Mandal, S.K.; Ibrahim, M.M.H.; Alhamami, H.N.; Briski, K.P. Sex differences in forebrain estrogen receptor regulation of hypoglycemic patterns of counter-regulatory hormone secretion and ventromedial hypothalamic nucleus gluco-regulatory neurotransmitter and astrocyte glycogen metabolic enzyme expression. Neuropeptides 2018, 72, 65–74. [Google Scholar] [CrossRef]
- Uddin, M.M.; Mahmood, A.S.M.H.; IBRAHIM, M.M.H.; Briski, K.P. Sex dimorphic estrogen receptor regulation of ventromedial hypothalamic nucleus glucoregulatory neuron adrenergic receptor expression in hypoglycemic male and female rats. Brain Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Butcher, R.L.; Collins, W.E.; Fugo, N.W. Plasma concentrations of LH, FSH, progesterone, and estradiol-17beta throughout the 4-day estrous cycle of the rat. Endocrinology 1974, 94, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R.L. A quantitative analysis of the physiological role of estradiol and progesterone in the control of tonic and surge secretion of luteinizing hormone in the rat. Endocrinology 1978, 102, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Briski, K.P.; Marshall, E.S.; Sylvester, P.W. Effects of estradiol on glucoprivic transactivation of catecholaminergic neurons in the female rat caudal brainstem. Neuroendocrinology 2001, 73, 369–377. [Google Scholar] [CrossRef]
- Mahmood, A.S.M.H.; Napit, P.R.; Ali, M.H.; Briski, K.P. Estrogen receptor involvement in noradrenergic regulation of ventromedial hypothalamic nucleus glucoregulatory neurotransmitter and stimulus-specific glycogen phosphorylase enzyme isoform expression. ASN Neuro 2020, in press. [Google Scholar]
- Martínez de Morentin, P.B.; González-García, I.; Martins, L.; Lage, R.; Fernández-Mallo, D.; Martínez-Sánchez, N.; Ruíz-Pino, F.; Liu, J.; Morgan, D.A.; Pinilla, L.; et al. Estradiol regulates brown adipose tissue thermogenesis via hypothalamic AMPK. Cell Metab. 2014, 20, 41–53. [Google Scholar] [CrossRef]
- Stobart, J.L.; Anderson, C.M. Role of astrocytes as gatekeepers of neuronal energy supply. Front. Cell. Neurosci. 2013, 7, 1–21. [Google Scholar] [CrossRef]
- Laming, P.R.; Kimelberg, H.; Robinson, S.; Salm, A.; Hawrylak, N.; Muller, C.; Roots, B.; Ng, K. Neuronal-glial interactions and behavior. Neurosci. Biobehav. Rev. 2000, 24, 295–340. [Google Scholar] [CrossRef]
- Broer, S.; Rahman, B.; Pellegri, G.; Pellerin, L.; Martin, J.L.; Verleysdonk, S.; Hamprecht, B.; Magistretti, P.J. Comparison of lactate transport in astroglial cells and monocarboxylate transporter (MCT 1) expressing Xenopus laevis oocytes. Expression of two different monocarboxylate transporters in astroglial cells and neurons. J. Biol. Chem. 1997, 272, 30096–30102. [Google Scholar] [CrossRef]
- Alhamami, H.N.; UDDIN, M.M.; MAHMOOD, A.S.M.H.; Briski, K.P. Lateral but not medial hypothalamic AMPK activation occurs at the hypoglycemic nadir in insulin-injected male rats: Impact of caudal dorsomedial hindbrain catecholamine signaling. Neuroscience 2018, 379, 103–114. [Google Scholar] [CrossRef]
- Fillenz, M.; Lowry, J.P.; Boutelle, M.G.; Fray, A.E. The role of astrocytes and noradrenaline in neuronal glucose metabolism. Acta Physiol. Scand. 1999, 167, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.H.; Chen, X.; Cui, M.; Yu, X.; Pang, Q.; Sun, J.P. Β2-adrenergic receptor and astrocyte glucose metabolism. J. Mol. Neurosci. 2012, 48, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Tamrakar, P.; Briski, K.P. Estradiol regulates hypothalamic astrocyte adenosine 5′-monophosphate-activated protein kinase activation by hypoglycemia: role of hindbrain catecholamine signaling. Brain Res. Bull. 2015, 110, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Cryer, P.E. Hypoglycemia in type 1 diabetes mellitus. Endocrinol. Metab. Clin. North Am. 2010, 39, 641–654. [Google Scholar] [CrossRef]
- Cryer, P.E.; Davis, S.N.; Shamoon, H. Hypoglycemia in diabetes. Diabetes Care 2003, 26, 1902–1912. [Google Scholar] [CrossRef]
- Kale, A.Y.; Paranjape, S.A.; Briski, K.P. I.c.v. administration of the nonsteroidal glucocorticoid receptor antagonist, CP4-72555, prevents exacerbated hypoglycemia during repeated insulin administration. Neuroscience 2006, 140, 555–565. [Google Scholar] [CrossRef]
- Paranjape, S.A.; Briski, K.P. Recurrent insulin-induced hypoglycemia causes site-specific patterns of habituation or amplification of CNS neuronal genomic activation. Neuroscience 2005, 130, 957–970. [Google Scholar] [CrossRef]
- Smith, D.; Amiel, S.A. Hypoglycaemia unawareness and the brain. Diabetologia 2002, 45, 949–958. [Google Scholar] [CrossRef]
- Briski, K.P.; Mandal, S.K.; Bheemanapally, K.; Ibrahim, M.M.H. Effects of acute versus recurrent insulin-induced hypoglycemia on ventromedial hypothalamic nucleus metabolic-sensory neuron AMPK activity: impact of alpha1-adrenergic receptor signaling. Brain Res. Bull. 2020, 157, 41–50. [Google Scholar] [CrossRef]
- Chen, J.Q.; Cammarata, P.R.; Baines, C.P.; Yager, J.D. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim. Biophys. Acta 2009, 1793, 1540–1570. [Google Scholar] [CrossRef]
- Tamrakar, P.; Briski, K.P. Impact of recurrent hypoglycemic stress on metabolic signaling in dorsal vagal complex neurons: modulation by estradiol. Acta Neurobiol. Exper. 2017, 77, 31–44. [Google Scholar]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmood, A.S.M.H.; Uddin, M.M.; Ibrahim, M.M.H.; Briski, K.P. Norepinephrine Regulation of Ventromedial Hypothalamic Nucleus Metabolic-Sensory Neuron 5′-AMP-Activated Protein Kinase Activity: Impact of Estradiol. Int. J. Mol. Sci. 2020, 21, 2013. https://doi.org/10.3390/ijms21062013
Mahmood ASMH, Uddin MM, Ibrahim MMH, Briski KP. Norepinephrine Regulation of Ventromedial Hypothalamic Nucleus Metabolic-Sensory Neuron 5′-AMP-Activated Protein Kinase Activity: Impact of Estradiol. International Journal of Molecular Sciences. 2020; 21(6):2013. https://doi.org/10.3390/ijms21062013
Chicago/Turabian StyleMahmood, A. S. M. Hasan, Md. Main Uddin, Mostafa M. H. Ibrahim, and Karen P. Briski. 2020. "Norepinephrine Regulation of Ventromedial Hypothalamic Nucleus Metabolic-Sensory Neuron 5′-AMP-Activated Protein Kinase Activity: Impact of Estradiol" International Journal of Molecular Sciences 21, no. 6: 2013. https://doi.org/10.3390/ijms21062013
APA StyleMahmood, A. S. M. H., Uddin, M. M., Ibrahim, M. M. H., & Briski, K. P. (2020). Norepinephrine Regulation of Ventromedial Hypothalamic Nucleus Metabolic-Sensory Neuron 5′-AMP-Activated Protein Kinase Activity: Impact of Estradiol. International Journal of Molecular Sciences, 21(6), 2013. https://doi.org/10.3390/ijms21062013