Overexpression of NRF1-742 or NRF1-772 Reduces Arsenic-Induced Cytotoxicity and Apoptosis in Human HaCaT Keratinocytes


Abstract
:1. Introduction
2. Results
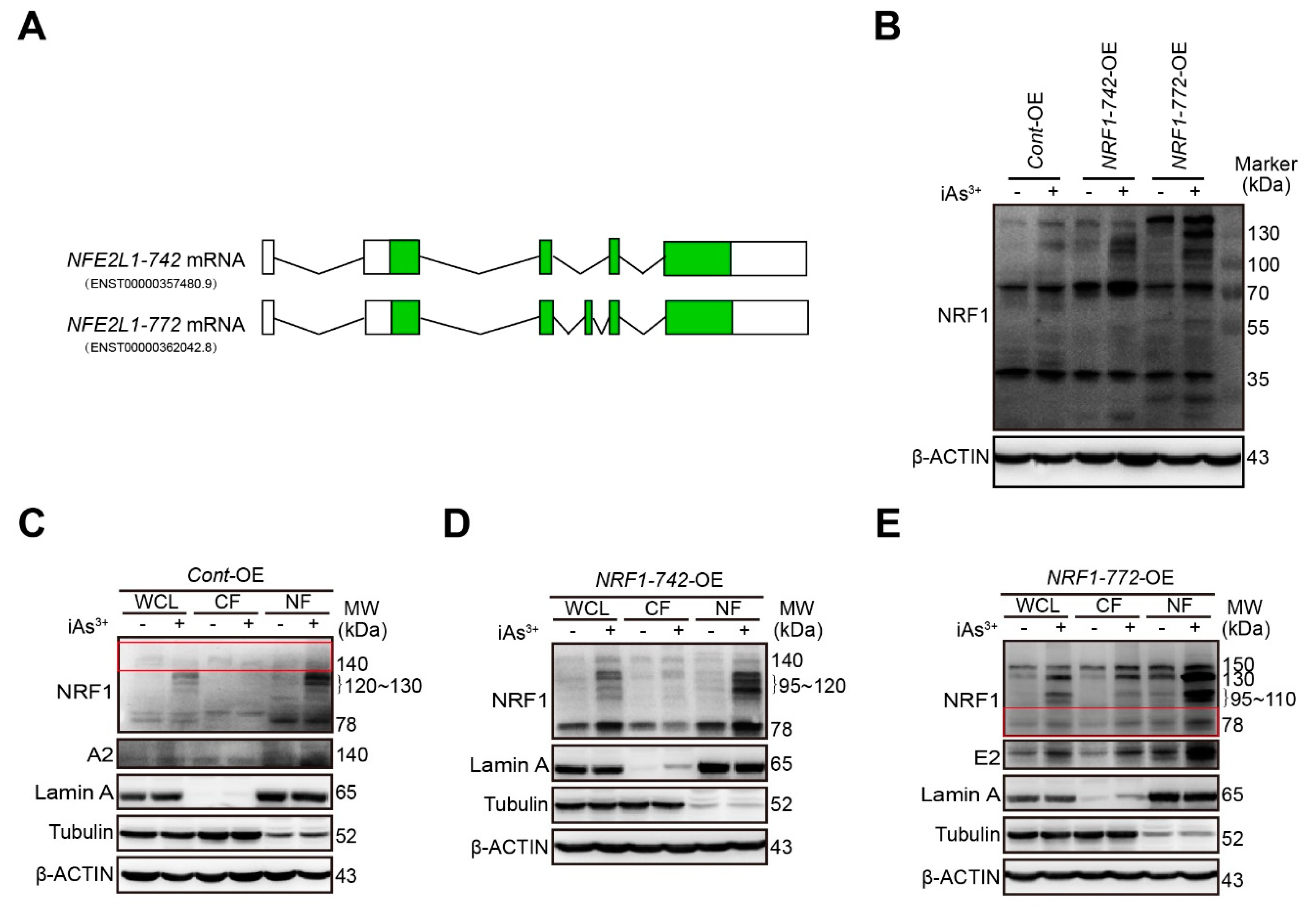
2.1. Characterization of Human Endogenous NRF1-742 and NRF1-772 Proteins and Their Derivative Isoforms
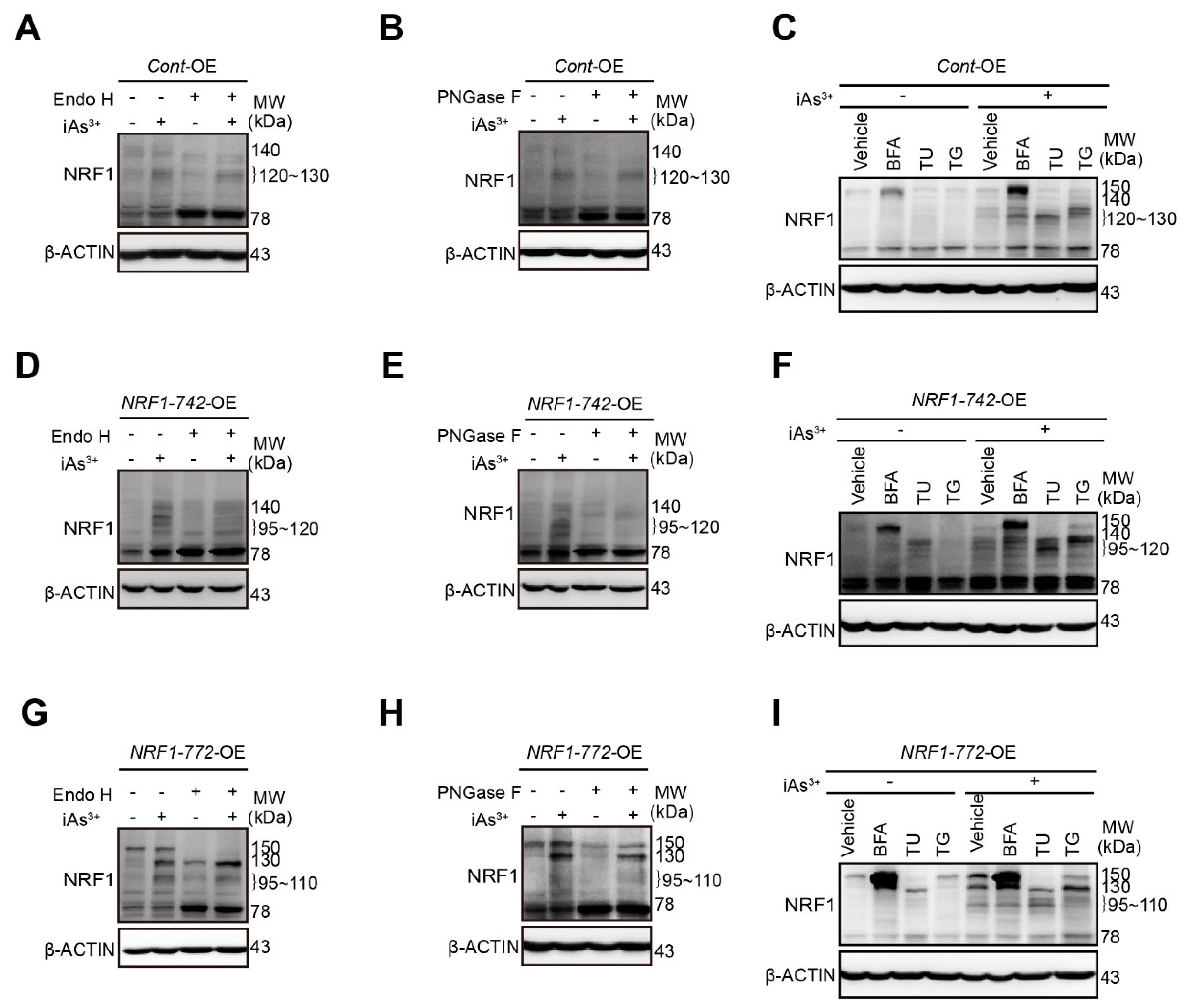
2.2. Modification of NRF1 Protein Isoforms by Glycosylation and Deglycosylation
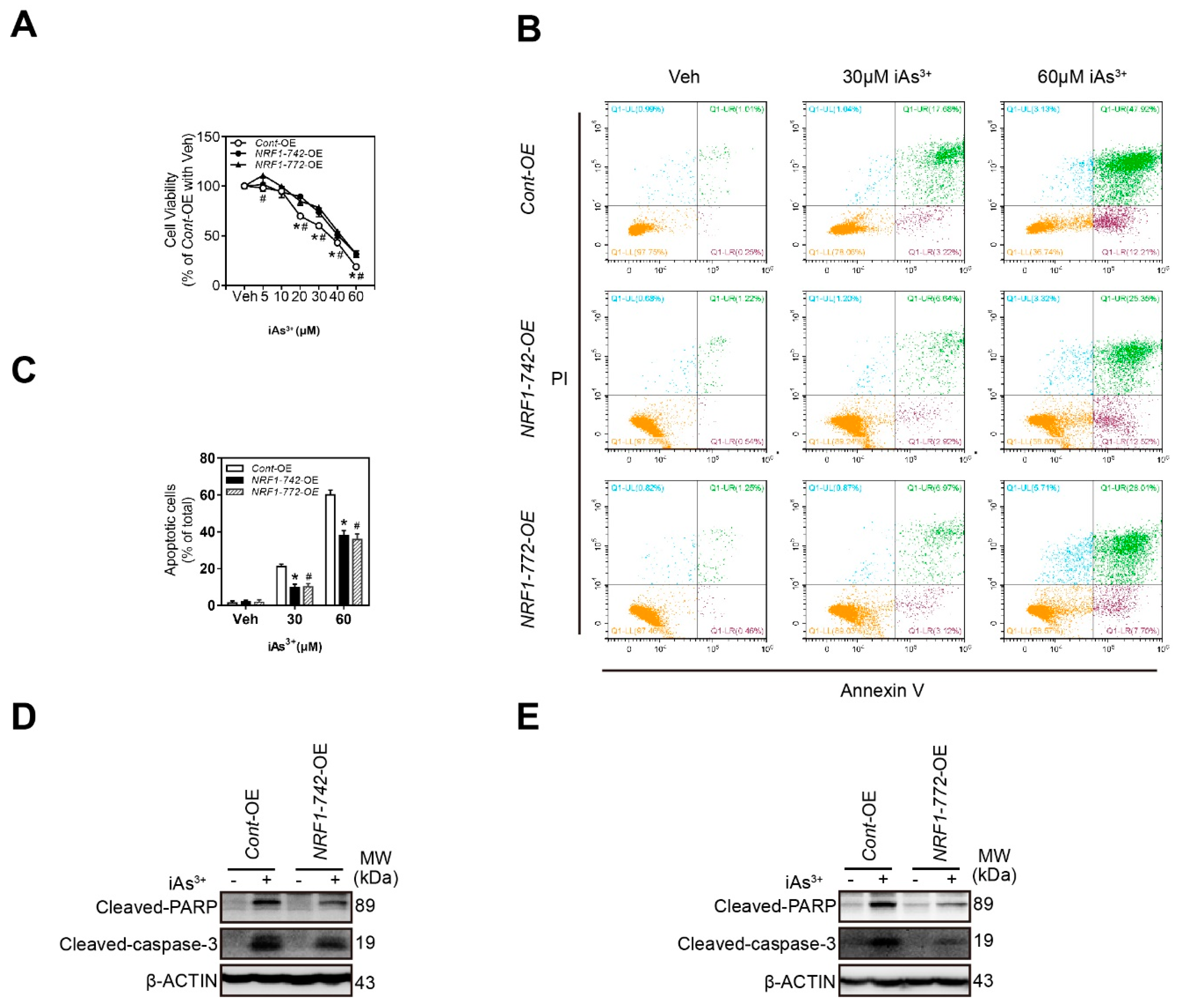
2.3. NRF1-742-OE and NRF1-772-OE Cells Are Resistant to Acute iAs3+-Induced Cell Damage
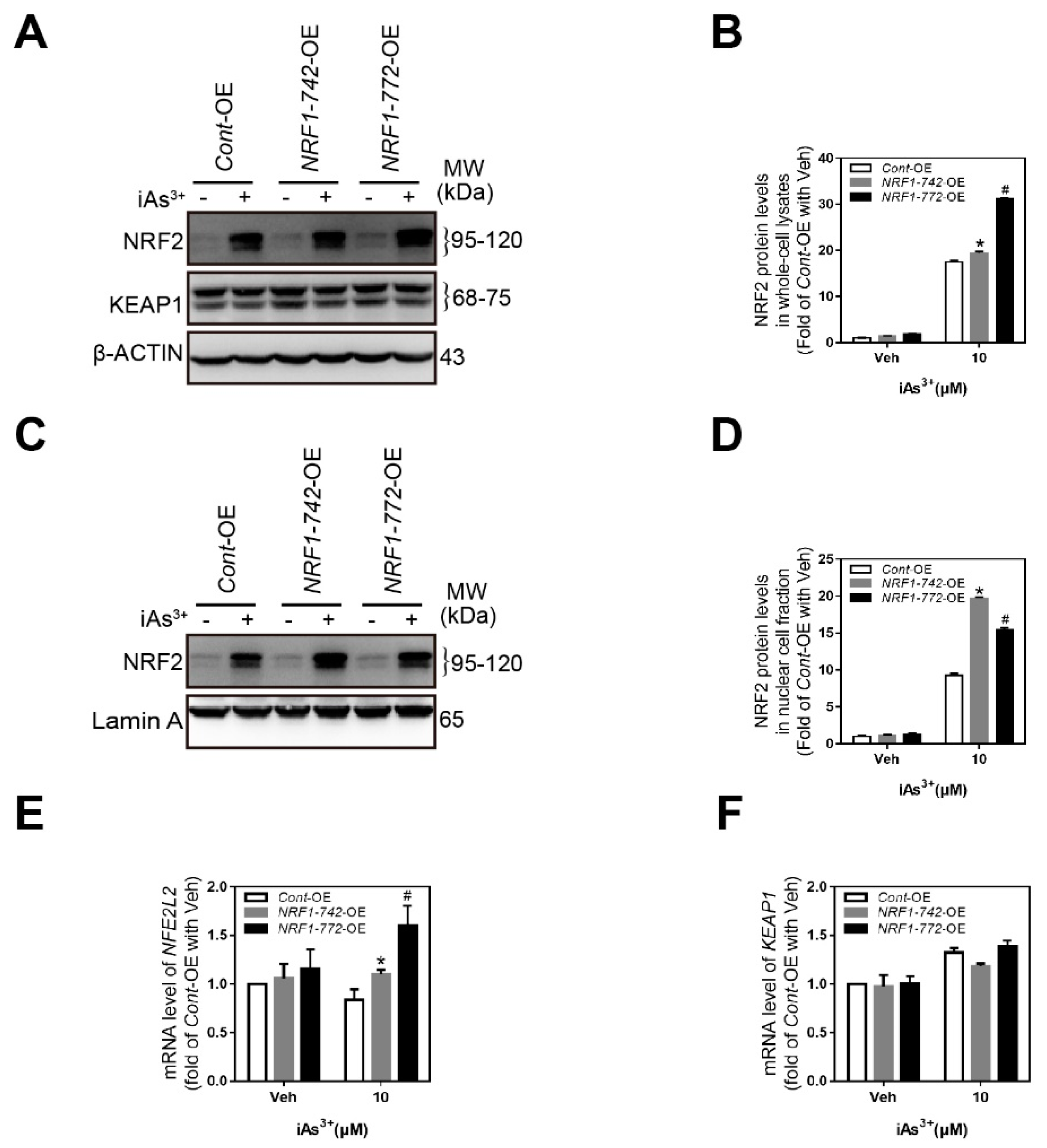
2.4. Crosstalk among NRF1, NRF2, and KEAP1
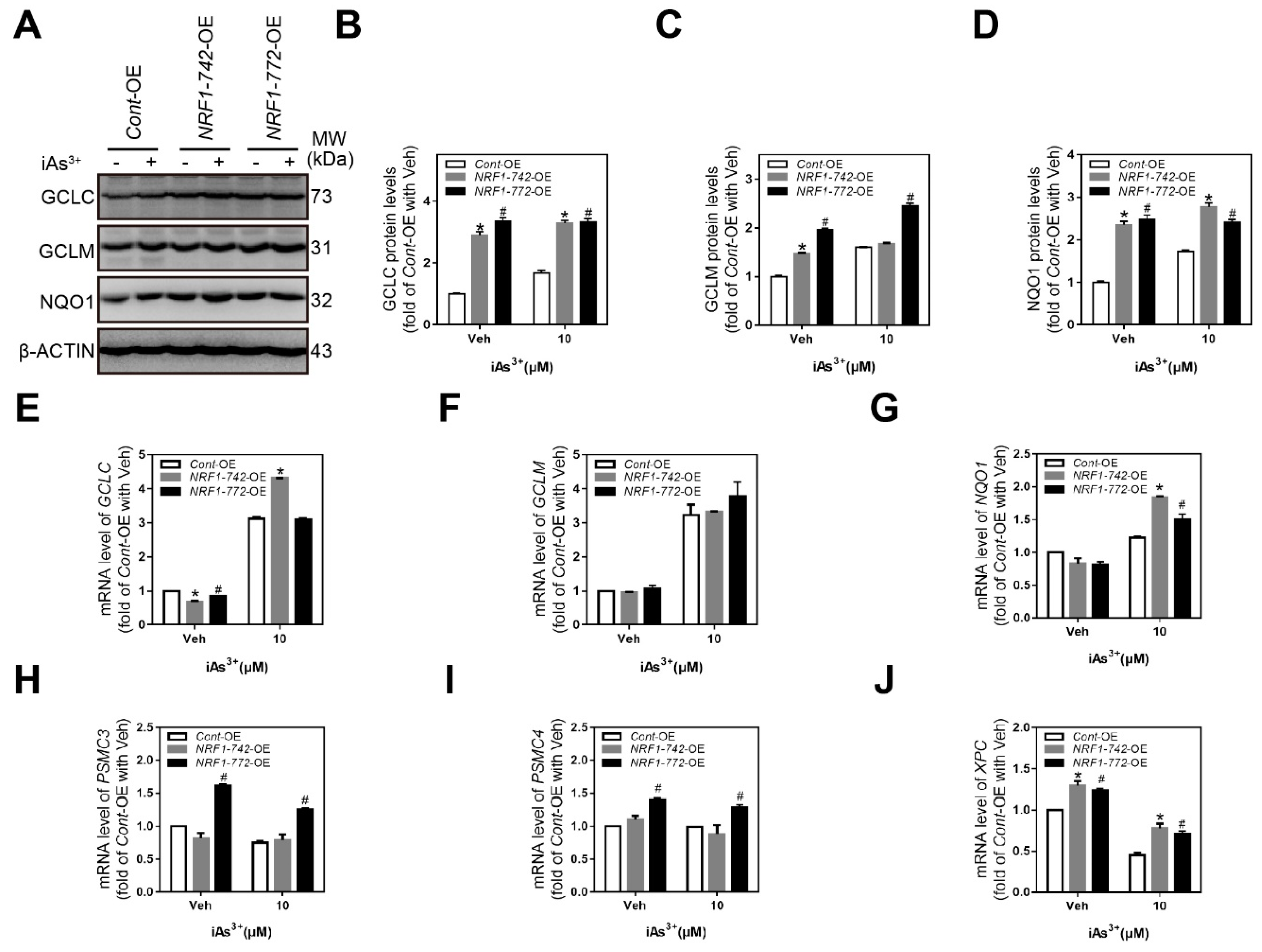
2.5. Expression of NRF1 Target Genes Involved in the Antioxidant Response, Proteasome Function, and Nucleotide Excision Repair
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Lentiviral-Based NRF1 Overexpression
4.3. In Vitro Deglycosylation
4.4. Western Blot Analysis
4.5. Cell Viability
4.6. RT-qPCR
4.7. Apoptosis Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NRF1 | Nuclear factor erythroid 2-like 1 |
| ARE | Antioxidant response element |
| iAs | Inorganic arsenic |
| NRF2 | Nuclear factor erythroid 2-like 2 |
| EpRE | Electrophile response element |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| TU | Tunicamycin |
| TG | Thapsigargin |
| BFA | Brefeldin |
| qPCR | Quantitative polymerase chain reaction |
| WCL | Whole-cell lysates |
| NF | Nuclear fraction |
| CF | Cytosolic fraction |
| GCLC | Glutamate-cysteine ligase catalytic |
| GCLM | Glutamate-cysteine ligase modifier |
| NQO1 | NAD(P)H quinone oxidoreductase 1 |
| XPC | Xeroderma pigmentosum C |
| UPS | Ubiquitin-proteasome system |
| PSMC3 | Proteasome 26S subunit, ATPase 3 |
| PSMC4 | Proteasome 26S subunit, ATPase 4 |
References
- Mohammed Abdul, K.S.; Jayasinghe, S.S.; Chandana, E.P.S.; Jayasumana, C.; De Silva, P.M.C.S. Arsenic and human health effects: A review. Environ. Toxicol. Pharmacol. 2015, 40, 828–846. [Google Scholar] [CrossRef]
- Rahman, M.M.; Ng, J.C.; Naidu, R. Chronic exposure of arsenic via drinking water and its adverse health impacts on humans. Environ. Geochem. Health 2009, 31 (Suppl. S1), 189–200. [Google Scholar] [CrossRef]
- Sinha, D.; Prasad, P. Health effects inflicted by chronic low-level arsenic contamination in groundwater: A global public health challenge. J. Appl. Toxicol. 2020, 40, 87–131. [Google Scholar] [CrossRef]
- Chen, C. Health hazards and mitigation of chronic poisoning from arsenic in drinking water: Taiwan experiences. Rev. Environ. Health 2014, 29, 13–19. [Google Scholar] [CrossRef]
- Smith, A.H.; Marshall, G.; Roh, T.; Ferreccio, C.; Liaw, J.; Steinmaus, C. Lung, Bladder, and Kidney Cancer Mortality 40 Years After Arsenic Exposure Reduction. JNCI: J. Natl. Cancer Inst. 2018, 110, 241–249. [Google Scholar] [CrossRef]
- Bryant, S.E.O.; Edwards, M.; Menon, C.; Gong, G.; Barber, R. Long-Term Low-Level Arsenic Exposure Is Associated with Poorer Neuropsychological Functioning: A Project FRONTIER Study. Int. J. Environ. Res. Public Health 2011, 8, 861–874. [Google Scholar]
- Robles-Osorio, M.L.; Sabath-Silva, E.; Sabath, E. Arsenic-mediated nephrotoxicity. Ren. Fail. 2015, 37, 542–547. [Google Scholar] [CrossRef]
- Guha Mazumder, D.N. Effect of chronic intake of arsenic-contaminated water on liver. Toxicol. Appl. Pharmacol. 2005, 206, 169–175. [Google Scholar] [CrossRef]
- Tokar, E.J.; Person, R.J.; Sun, Y.; Perantoni, A.O.; Waalkes, M.P. Chronic Exposure of Renal Stem Cells to Inorganic Arsenic Induces a Cancer Phenotype. Chem. Res. Toxicol. 2013, 26, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Kadono, T.; Inaoka, T.; Murayama, N.; Ushijima, K.; Nagano, M.; Nakamura, S.; Watanabe, C.; Tamaki, K.; Ohtsuka, R. Skin manifestations of arsenicosis in two villages in Bangladesh. Int. J. Dermatol. 2002, 41, 841–846. [Google Scholar] [CrossRef]
- Hsu, L.; Chen, G.; Lee, C.; Yang, T.; Chen, Y.; Wang, Y.; Hsueh, Y.; Chiou, H.; Wu, M.; Chen, C. Use of Arsenic-Induced Palmoplantar Hyperkeratosis and Skin Cancers to Predict Risk of Subsequent Internal Malignancy. Am. J. Epidemiol. 2013, 177, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Yajima, I.; Ahsan, N.; Akhand, A.A.; AI Hossain, M.A.; Yoshinaga, M.; Ohgami, N.; Iida, M.; Oshino, R.; Naito, M.; Wakai, K.; et al. Arsenic levels in cutaneous appendicular organs are correlated with digitally evaluated hyperpigmented skin of the forehead but not the sole in Bangladesh residents. J. Expo. Sci. Environ. Epidemiol. 2018, 28, 64–68. [Google Scholar] [CrossRef]
- Saleem, M.D.; Oussedik, E.; Picardo, M.; Schoch, J.J. Acquired disorders with hypopigmentation: A clinical approach to diagnosis and treatment. J. Am. Acad. Dermatol. 2019, 80, 1233–1250. [Google Scholar] [CrossRef]
- Ghosh, P.; Banerjee, M.; De Chaudhuri, S.; Das, J.K.; Sarma, N.; Basu, A.; Giri, A.K. Increased chromosome aberration frequencies in the Bowen’s patients compared to non-cancerous skin lesions individuals exposed to arsenic. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2007, 632, 104–110. [Google Scholar] [CrossRef]
- Namdar, N.D.; Arikan, I.; Kahraman, C.; Kocaturk, E.; Dagci, M.; Ece, E. Evaluation of the quality of life in patients with arsenic keratosis. Cutan Ocul Toxicol. 2018, 37, 167–171. [Google Scholar] [CrossRef]
- Lee, C.; Liao, W.; Yu, H. Aberrant immune responses in arsenical skin cancers. Kaohsiung J. Med Sci. 2011, 27, 396–401. [Google Scholar] [CrossRef] [Green Version]
- Pi, J.; Yamauchi, H.; Kumagai, Y.; Sun, G.; Yoshida, T.; Aikawa, H.; Hopenhayn-Rich, C.; Shimojo, N. Evidence for induction of oxidative stress caused by chronic exposure of Chinese residents to arsenic contained in drinking water. Environ. Health Perspect. 2002, 110, 331–336. [Google Scholar] [CrossRef] [Green Version]
- Pi, J.; Qu, W.; Reece, J.M.; Kumagai, Y.; Waalkes, M.P. Transcription factor Nrf2 activation by inorganic arsenic in cultured keratinocytes: Involvement of hydrogen peroxide. Exp. Cell Res. 2003, 290, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuji, M.; Katsuoka, F.; Kobayashi, A.; Aburatani, H.; Hayes, J.D.; Yamamoto, M. Nrf1 and Nrf2 Play Distinct Roles in Activation of Antioxidant Response Element-dependent Genes. J. Biol. Chem. 2008, 283, 33554–33562. [Google Scholar] [CrossRef] [Green Version]
- Biswas, M.; Chan, J.Y. Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol. Appl. Pharmacol. 2010, 244, 16–20. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lucocq, J.M.; Hayes, J.D. The Nrf1 CNC/bZIP protein is a nuclear envelope-bound transcription factor that is activated by t-butyl hydroquinone but not by endoplasmic reticulum stressors. Biochem. J. 2009, 418, 293–310. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, S.K.; den Besten, W.; Deshaies, R.J. p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. eLife 2014, 3, e01856. [Google Scholar] [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef]
- Berg, D.T.; Gupta, A.; Richardson, M.A.; O’Brien, L.A.; Calnek, D.; Grinnell, B.W. Negative Regulation of Inducible Nitric-oxide Synthase Expression Mediated through Transforming Growth Factor-β-dependent Modulation of Transcription Factor TCF11. J. Biol. Chem. 2007, 282, 36837–36844. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.S.; Lee, C.; Hu, T.; Nguyen, J.M.; Zhang, J.; Martin, M.V.; Vawter, M.P.; Huang, E.J.; Chan, J.Y. Loss of nuclear factor E2-related factor 1 in the brain leads to dysregulation of proteasome gene expression and neurodegeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 8408–8413. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.S.; Ho, D.V.; Chan, J.Y. Nuclear factor-erythroid 2-related factor 1 regulates expression of proteasome genes in hepatocytes and protects against endoplasmic reticulum stress and steatosis in mice. FEBS J. 2013, 280, 3609–3620. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Fu, J.; Xue, P.; Zhao, R.; Dong, J.; Liu, D.; Yamamoto, M.; Tong, Q.; Teng, W.; Qu, W.; et al. CNC-bZIP Protein Nrf1-Dependent Regulation of Glucose-Stimulated Insulin Secretion. Antioxid. Redox Signal. 2015, 22, 819–831. [Google Scholar] [CrossRef] [Green Version]
- Jacob, A.; Zhang, Y.; George, A. Transcriptional regulation of dentin matrix protein 1 (DMP1) in odontoblasts and osteoblasts. Connect. Tissue Res. 2014, 55 (Suppl. S1), 107–112. [Google Scholar] [CrossRef]
- Zhao, R.; Hou, Y.; Xue, P.; Woods, C.G.; Fu, J.; Feng, B.; Guan, D.; Sun, G.; Chan, J.; Waalkes, M.; et al. Long isoforms of NRF1 contribute to arsenic-induced antioxidant response in human keratinocytes. Environ. Health Perspect. 2011, 119, 56–62. [Google Scholar] [CrossRef]
- Zhao, R.; Hou, Y.; Zhang, Q.; Woods, C.; Xue, P.; Fu, J.; Yarborough, K.; Guan, D.; Andersen, M.; Pi, J. Cross-regulations among NRFs and KEAP1 and effects of their silencing on arsenic-induced antioxidant response and cytotoxicity in human keratinocytes. Environ. Health Perspect. 2012, 120, 583–589. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Fu, J.; Hu, Y.; Li, Y.; Yang, B.; Li, L.; Sun, J.; Chen, C.; Sun, G.; Xu, Y.; et al. Deficiency of long isoforms of Nfe2l1 sensitizes MIN6 pancreatic beta cells to arsenite-induced cytotoxicity. Toxicol. Appl. Pharmacol. 2017, 329, 67–74. [Google Scholar] [CrossRef]
- Zhang, Y.; Lucocq, J.M.; Yamamoto, M.; Hayes, J.D. The NHB1 (N-terminal homology box 1) sequence in transcription factor Nrf1 is required to anchor it to the endoplasmic reticulum and also to enable its asparagine-glycosylation. Biochem. J. 2007, 408, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Wang, M.; Hu, S.; Qiu, L.; Yang, F.; Zhang, Z.; Yu, S.; Pi, J.; Zhang, Y. Mechanisms controlling the multistage post-translational processing of endogenous Nrf1α/TCF11 proteins to yield distinct isoforms within the coupled positive and negative feedback circuits. Toxicol. Appl. Pharmacol. 2018, 360, 212–235. [Google Scholar] [CrossRef]
- Xie, Y. Feedback regulation of proteasome gene expression and its implications in cancer therapy. Cancer Metastasis Rev. 2010, 29, 687–693. [Google Scholar] [CrossRef]
- Han, W.; Ming, M.; Zhao, R.; Pi, J.; Wu, C.; He, Y. Nrf1 CNC-bZIP Protein Promotes Cell Survival and Nucleotide Excision Repair through Maintaining Glutathione Homeostasis. J. Biol. Chem. 2012, 287, 18788–18795. [Google Scholar] [CrossRef] [Green Version]
- Holcomb, N.; Goswami, M.; Han, S.G.; Scott, T.D.; Orazio, J.; Orren, D.K.; Gairola, C.G.; Mellon, I. Inorganic arsenic inhibits the nucleotide excision repair pathway and reduces the expression of XPC. DNA Repair 2017, 52, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Pi, J.; Kumagai, Y.; Sun, G.; Yamauchi, H.; Yoshida, T.; Iso, H.; Endo, A.; Yu, L.; Yuki, K.; Miyauchi, T.; et al. Decreased serum concentrations of nitric oxide metabolites among Chinese in an endemic area of chronic arsenic poisoning in inner Mongolia. Free Radic. Biol. Med. 2000, 28, 1137–1142. [Google Scholar] [CrossRef]
- YOSHIDA, T. Chronic health effects in people exposed to arsenic via the drinking water: Dose? response relationships in review. Toxicol. Appl. Pharmacol. 2004, 198, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Qiu, L.; Li, S.; Xiang, Y.; Chen, J.; Ren, Y. The C-terminal domain of Nrf1 negatively regulates the full-length CNC-bZIP factor and its shorter isoform LCR-F1/Nrf1beta; both are also inhibited by the small dominant-negative Nrf1gamma/delta isoforms that down-regulate ARE-battery gene expression. PLoS ONE 2014, 9, e109159. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, S.; Xiang, Y.; Qiu, L.; Zhao, H.; Hayes, J.D. The selective post-translational processing of transcription factor Nrf1 yields distinct isoforms that dictate its ability to differentially regulate gene expression. Sci. Rep. 2015, 5, 12983. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Chan, J.Y. Nrf1 Is Targeted to the Endoplasmic Reticulum Membrane by an N-terminal Transmembrane Domain. J. Biol. Chem. 2006, 281, 19676–19687. [Google Scholar] [CrossRef] [Green Version]
- Chepelev, N.L.; Zhang, H.; Liu, H.; McBride, S.; Seal, A.J.; Morgan, T.E.; Finch, C.E.; Willmore, W.G.; Davies, K.J.A.; Forman, H.J. Competition of nuclear factor-erythroid 2 factors related transcription factor isoforms, Nrf1 and Nrf2, in antioxidant enzyme induction. Redox Biol. 2013, 1, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Jomova, K.; Jenisova, Z.; Feszterova, M.; Baros, S.; Liska, J.; Hudecova, D.; Rhodes, C.J.; Valko, M. Arsenic: Toxicity, oxidative stress and human disease. J. Appl. Toxicol. 2011, 31, 95–107. [Google Scholar] [CrossRef]
- Sattar, A.; Xie, S.; Hafeez, M.A.; Wang, X.; Hussain, H.I.; Iqbal, Z.; Pan, Y.; Iqbal, M.; Shabbir, M.A.; Yuan, Z. Metabolism and toxicity of arsenicals in mammals. Environ. Toxicol. Pharmacol. 2016, 48, 214–224. [Google Scholar] [CrossRef]
- Pan, J.; Sinclair, E.; Xuan, Z.; Dyba, M.; Fu, Y.; Sen, S.; Berry, D.; Creswell, K.; Hu, J.; Roy, R.; et al. Nucleotide excision repair deficiency increases levels of acrolein-derived cyclic DNA adduct and sensitizes cells to apoptosis induced by docosahexaenoic acid and acrolein. Mutat. Res./Fundam. Mol. Mech. Mutagenesis 2016, 789, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Clément, V.; Dunand-Sauthier, I.; Clarkson, S.G. Suppression of UV-induced apoptosis by the human DNA repair protein XPG. Cell Death Differ. 2006, 13, 478–488. [Google Scholar] [CrossRef] [Green Version]
- Ho, D.V.; Chan, J.Y. Induction of Herpud1 expression by ER stress is regulated by Nrf1. FEBS Lett. 2015, 589, 615–620. [Google Scholar] [CrossRef]
- Sha, Z.; Goldberg, A.L. Proteasome-Mediated Processing of Nrf1 Is Essential for Coordinate Induction of All Proteasome Subunits and p97. Curr. Biol. 2014, 24, 1573–1583. [Google Scholar] [CrossRef] [Green Version]
- Xue, P.; Hou, Y.; Zuo, Z.; Wang, Z.; Wang, H.; Ren, S.; Dong, J.; Fu, J.; Andersen, M.E.; Zhang, Q.; et al. Long isoforms of NRF1 negatively regulate adipogenesis via suppression of PPARγ expression. Redox Biol. 2019, 30, 101414. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, M.; Wang, L.; Yu, T.; Jiang, S.; Jiang, P.; Sun, Y.; Pi, J.; Zhao, R.; Guan, D. Activation of cannabinoid type 2 receptor protects skeletal muscle from ischemia-reperfusion injury partly via Nrf2 signaling. Life Sci. 2019, 230, 55–67. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Veh | iAs3+ | ||
|---|---|---|---|---|
| WCL | NF | CF | ||
| Cont-OE | 78, 140 | 78, 120–130,140 | 78,120–130,140 | 78, 140 |
| NRF1-742-OE | 78, 110–120, 140 | 78, 95–120, 140 | 78, 95–120,140 | 78, 110–120, 140 |
| NRF1-772-OE | 30, 60, 78,150 | 30, 60, 78,95–110, 130, 150 | 78, 95–110, 130, 150 | 78, 95–110, 130, 150 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Cheng, H.; Wang, L.; Zhao, R.; Guan, D. Overexpression of NRF1-742 or NRF1-772 Reduces Arsenic-Induced Cytotoxicity and Apoptosis in Human HaCaT Keratinocytes. Int. J. Mol. Sci. 2020, 21, 2014. https://doi.org/10.3390/ijms21062014
Wang S, Cheng H, Wang L, Zhao R, Guan D. Overexpression of NRF1-742 or NRF1-772 Reduces Arsenic-Induced Cytotoxicity and Apoptosis in Human HaCaT Keratinocytes. International Journal of Molecular Sciences. 2020; 21(6):2014. https://doi.org/10.3390/ijms21062014
Chicago/Turabian StyleWang, Shuai, Hao Cheng, Linlin Wang, Rui Zhao, and Dawei Guan. 2020. "Overexpression of NRF1-742 or NRF1-772 Reduces Arsenic-Induced Cytotoxicity and Apoptosis in Human HaCaT Keratinocytes" International Journal of Molecular Sciences 21, no. 6: 2014. https://doi.org/10.3390/ijms21062014