VLA-4 Expression and Activation in B Cell Malignancies: Functional and Clinical Aspects



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Integrins in the Hematopoietic System
2. VLA-4 Functions during B Cell Development in Bone Marrow
3. Marginal Zone B Cells and Other Mature B Cell Subsets
4. The Peculiar Connection of B Cell Activation and VLA-4 in Chronic Lymphocytic Leukemia
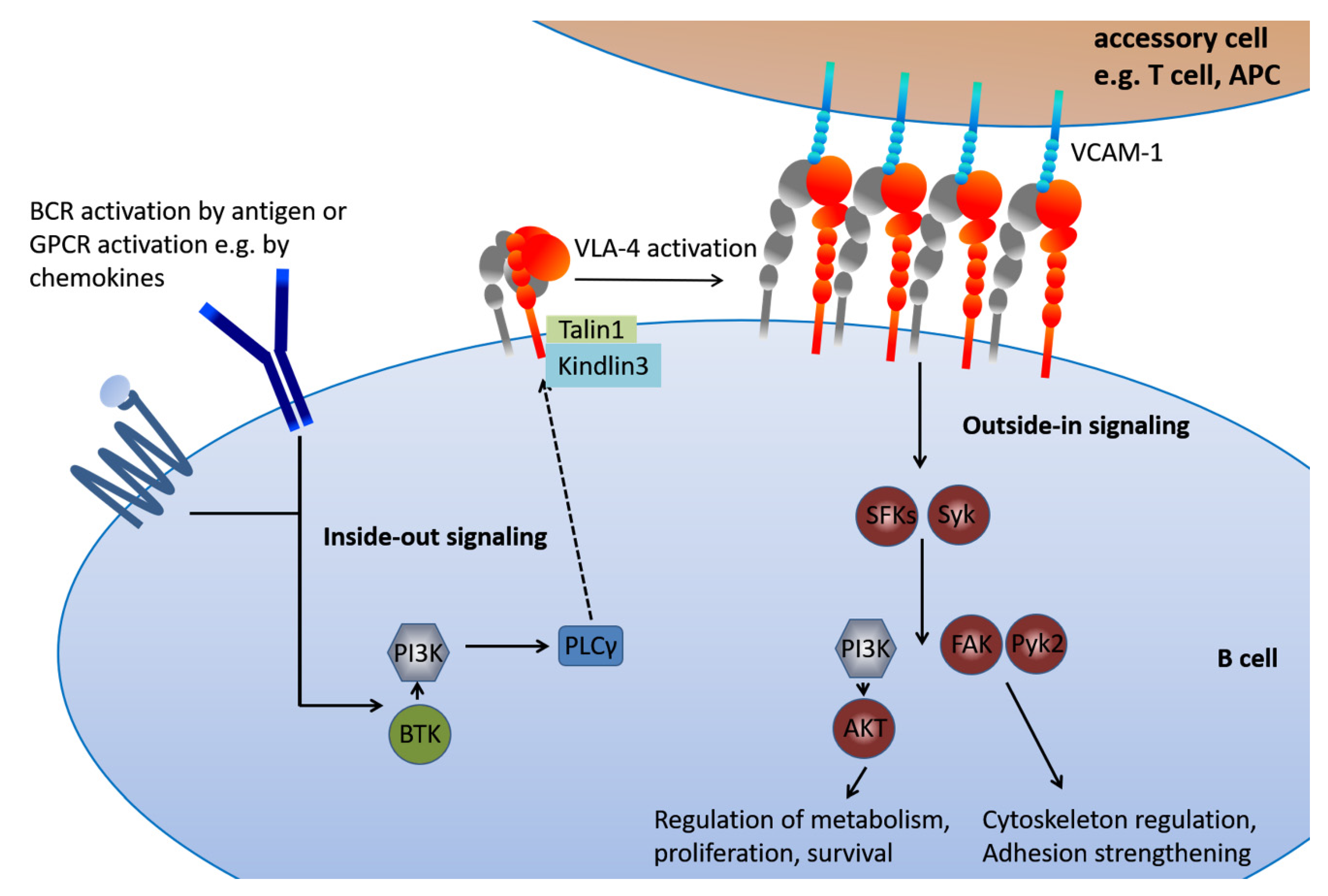
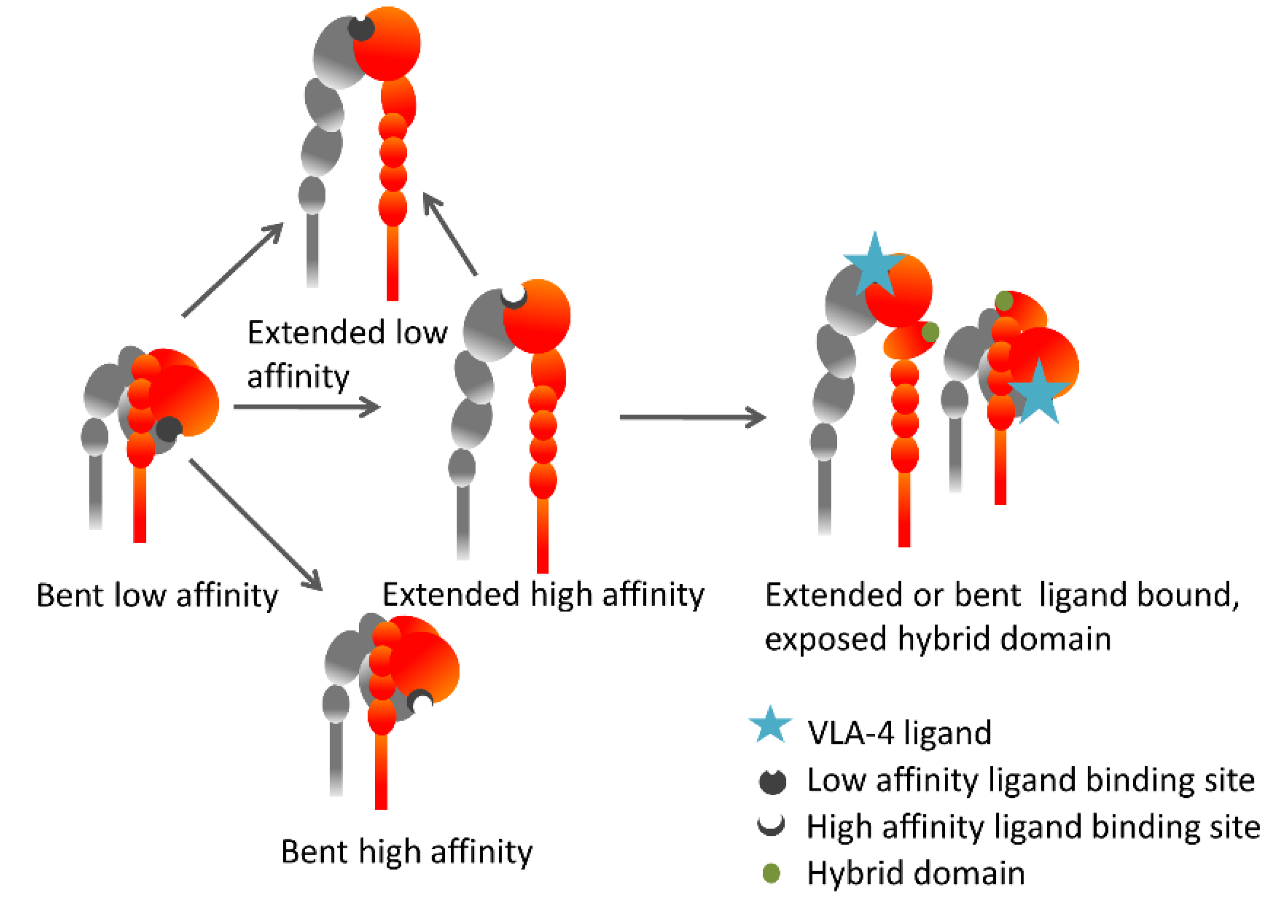
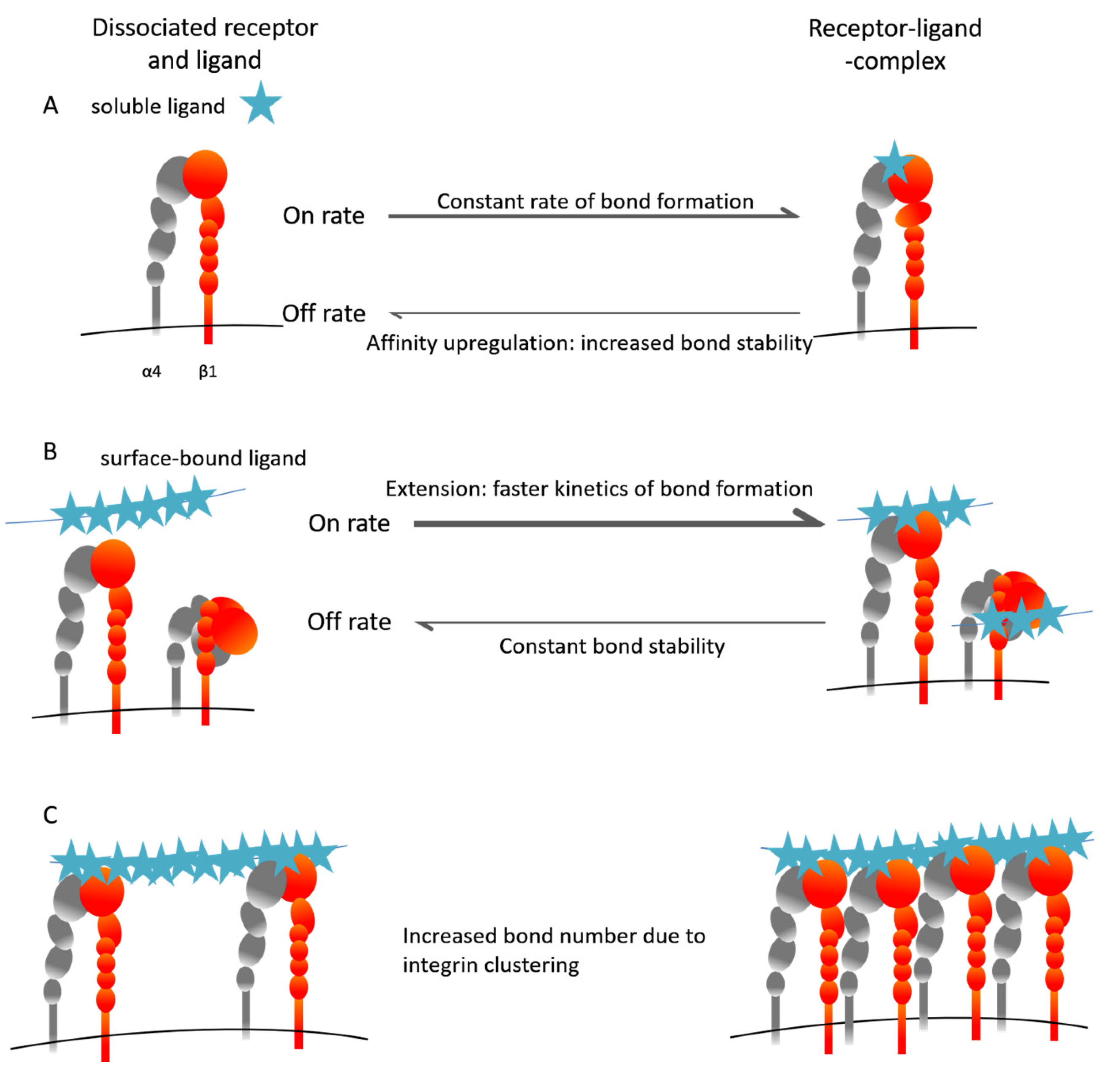
5. The Inside-Out VLA-4 Activation Cascade in Detail
6. Lessons Learned from Natalizumab
7. Conclusion and Future Perspectives of VLA-4 as a Therapeutic Target
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALL | Acute Lymphoblastic Leukemia |
| APC | Antigen Presenting Cell |
| BCR | B Cell Receptor |
| BTK | Bruton’s Tyrosine Kinase |
| CLL | Chronic Lymphocytic Leukemia |
| CNS | Central Nervous System |
| EAE | Experimental Autoimmune Encephalitis |
| FAK | Focal Adhesion Kinase |
| GPCR | G-Protein–Coupled Receptor |
| ICAP-1 | Integrin Cytoplasmic Domain-Associated Protein-1 |
| JCV | John Cunningham Virus |
| LFA-1 | Lymphocyte Function-associated Antigen 1 |
| MS | Multiple Sclerosis |
| PI3K | Phosphatidylinositide 3-Kinase |
| PKC | Protein Kinase C |
| PLCγ | Phospholipase Cγ |
| PML | Progressive Multifocal Leukoencephalopathy |
| Pyk2 | Proline-rich Tyrosine Kinase 2 |
| SFK | Src Family Kinase |
| Syk | Spleen Tyosine Kinase |
| VLA-4 | Very Late Antigen-4 |
| WM | Waldenström Macroglobulinemia |
References
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin Ligands at a Glance. J. Cell Sci. 2006, 119 Pt 19, 3901–3903. [Google Scholar] [CrossRef]
- Bertoni, A.; Alabiso, O.; Galetto, A.S.; Baldanzi, G. Integrins in T Cell Physiology. Int. J. Mol. Sci. 2018, 19, 485. [Google Scholar] [CrossRef]
- Chigaev, A.; Sklar, L.A. Aspects of Vla-4 and Lfa-1 Regulation That May Contribute to Rolling and Firm Adhesion. Front. Immunol. 2012, 3, 242. [Google Scholar] [CrossRef] [PubMed]
- Huhtala, M.; Heino, J.; Casciari, D.; de Luise, A.; Johnson, M.S. Integrin Evolution: Insights from Ascidian and Teleost Fish Genomes. Matrix Biol. 2005, 24, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, Y.R.; Batista, F.D. B-Cell Activation by Membrane-Bound Antigens Is Facilitated by the Interaction of Vla-4 with Vcam-1. EMBO J. 2006, 25, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, Y.R.; Fleire, S.J.; Cameron, T.; Dustin, M.L.; Batista, F.D. Lfa-1/Icam-1 Interaction Lowers the Threshold of B Cell Activation by Facilitating B Cell Adhesion and Synapse Formation. Immunity 2004, 20, 589–599. [Google Scholar] [CrossRef]
- Chen, C.C.; Manning, A.M. Transcriptional Regulation of Endothelial Cell Adhesion Molecules: A Dominant Role for Nf-Kappa B. Agents Actions Suppl. 1995, 47, 135–141. [Google Scholar] [PubMed]
- Hui, T.; Sorensen, E.S.; Rittling, S.R. Osteopontin Binding to the Alpha 4 Integrin Requires Highest Affinity Integrin Conformation, but Is Independent of Post-Translational Modifications of Osteopontin. Matrix Biol. 2015, 41, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Spessotto, P.; Bulla, R.; Danussi, C.; Radillo, O.; Cervi, M.; Monami, G.; Bossi, F.; Tedesco, F.; Doliana, R.; Colombatti, A. Emilin1 Represents a Major Stromal Element Determining Human Trophoblast Invasion of the Uterine Wall. J. Cell Sci. 2006, 119 Pt 21, 4574–4584. [Google Scholar] [CrossRef]
- Verdone, G.; Doliana, R.; Corazza, A.; Colebrooke, S.A.; Spessotto, P.; Bot, S.; Bucciotti, F.; Capuano, A.; Silvestri, A.; Viglino, P.; et al. The Solution Structure of Emilin1 Globular C1q Domain Reveals a Disordered Insertion Necessary for Interaction with the Alpha4beta1 Integrin. J. Biol. Chem. 2008, 283, 18947–18956. [Google Scholar] [CrossRef]
- Imhof, B.A.; Aurrand-Lions, M. Adhesion Mechanisms Regulating the Migration of Monocytes. Nat. Rev. Immunol. 2004, 4, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, A.G.; Yang, J.T.; Rayburn, H.; Hynes, R.O. Differential Requirements for Alpha4 Integrins During Fetal and Adult Hematopoiesis. Cell 1996, 85, 997–1008. [Google Scholar] [CrossRef]
- Arroyo, A.G.; Yang, J.T.; Rayburn, H.; Hynes, R.O. Alpha4 Integrins Regulate the Proliferation/Differentiation Balance of Multilineage Hematopoietic Progenitors in Vivo. Immunity 1999, 11, 555–566. [Google Scholar] [CrossRef]
- Ryan, D.H.; Nuccie, B.L.; Abboud, C.N.; Winslow, J.M. Vascular Cell Adhesion Molecule-1 and the Integrin Vla-4 Mediate Adhesion of Human B Cell Precursors to Cultured Bone Marrow Adherent Cells. J. Clin. Investig. 1991, 88, 995–1004. [Google Scholar] [CrossRef]
- Postigo, A.A.; Pulido, R.; Campanero, M.R.; Acevedo, A.; Garcia-Pardo, A.; Corbi, A.L.; Sanchez-Madrid, F.; de Landazuri, M.O. Differential Expression of Vla-4 Integrin by Resident and Peripheral Blood B Lymphocytes. Acquisition of Functionally Active Alpha 4 Beta 1-Fibronectin Receptors Upon B Cell Activation. Eur. J. Immunol. 1991, 21, 2437–2445. [Google Scholar] [CrossRef]
- Rossi, B.; Espeli, M.; Schiff, C.; Gauthier, L. Clustering of Pre-B Cell Integrins Induces Galectin-1-Dependent Pre-B Cell Receptor Relocalization and Activation. J. Immunol. 2006, 177, 796–803. [Google Scholar] [CrossRef]
- Patrick, C.W.; Juneja, H.S., Jr.; Lee, S.; Schmalstieg, F.C.; McIntire, L.V. Heterotypic Adherence between Human B-Lymphoblastic and Pre-B-Lymphoblastic Cells and Marrow Stromal Cells Is a Biphasic Event: Integrin Very Late Antigen-4 Alpha Mediates Only the Early Phase of the Heterotypic Adhesion. Blood 1995, 85, 168–178. [Google Scholar] [CrossRef]
- Spiegel, A.; Kollet, O.; Peled, A.; Abel, L.; Nagler, A.; Bielorai, B.; Rechavi, G.; Vormoor, J.; Lapidot, T. Unique Sdf-1-Induced Activation of Human Precursor-B All Cells as a Result of Altered Cxcr4 Expression and Signaling. Blood 2004, 103, 2900–2907. [Google Scholar] [CrossRef]
- Harima, A.; Nakaseko, C.; Yokota, A.; Kitagawa, M.; Morimoto, C.; Harigaya, K.; Saito, Y. Fibronectin Promotes Cell Proliferation of Human Pre-B Cell Line Via Its Interactions with Vla-4 and Vla-5. Hematology 2008, 13, 236–243. [Google Scholar] [CrossRef]
- Shalapour, S.; Hof, J.; Kirschner-Schwabe, R.; Bastian, L.; Eckert, C.; Prada, J.; Henze, G.; von Stackelberg, A.; Seeger, K. High Vla-4 Expression Is Associated with Adverse Outcome and Distinct Gene Expression Changes in Childhood B-Cell Precursor Acute Lymphoblastic Leukemia at First Relapse. Haematologica 2011, 96, 1627–1635. [Google Scholar] [CrossRef]
- Hinterseer, E.; Stiefel, O.; Neureiter, D.; Kandler, G.; Vogt, S.; Hutter, J.; Strasser, G.; Greil, R.; Hartmann, T.N.; Hopfinger, G. Vla-4 and Cxcr4 Overexpression in Bone Marrow of an Aleukemic B-Cell Acute Lymphoblastic Leukemia Presenting with Osteolytic Bone Lesions. Leuk. Lymphoma 2015, 56, 2465–2467. [Google Scholar] [CrossRef] [PubMed]
- Beck, T.C.; Gomes, A.C.; Cyster, J.G.; Pereira, J.P. Cxcr4 and a Cell-Extrinsic Mechanism Control Immature B Lymphocyte Egress from Bone Marrow. J. Exp. Med. 2014, 211, 2567–2581. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.P.; An, J.; Xu, Y.; Huang, Y.; Cyster, J.G. Cannabinoid Receptor 2 Mediates the Retention of Immature B Cells in Bone Marrow Sinusoids. Nat. Immunol. 2009, 10, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Tedford, K.; Steiner, M.; Koshutin, S.; Richter, K.; Tech, L.; Eggers, Y.; Jansing, I.; Schilling, K.; Hauser, A.E.; Korthals, M.; et al. The Opposing Forces of Shear Flow and Sphingosine-1-Phosphate Control Marginal Zone B Cell Shuttling. Nat. Commun. 2017, 8, 2261. [Google Scholar] [CrossRef]
- Camponeschi, A.; Gerasimcik, N.; Wang, Y.; Fredriksson, T.; Chen, D.; Farroni, C.; Thorarinsdottir, K.; Ottsjo, L.S.; Aranburu, A.; Cardell, S.; et al. Dissecting Integrin Expression and Function on Memory B Cells in Mice and Humans in Autoimmunity. Front. Immunol. 2019, 10, 534. [Google Scholar] [CrossRef] [PubMed]
- Heinig, K.; Gatjen, M.; Grau, M.; Stache, V.; Anagnostopoulos, I.; Gerlach, K.; Niesner, R.A.; Cseresnyes, Z.; Hauser, A.E.; Lenz, P.; et al. Access to Follicular Dendritic Cells Is a Pivotal Step in Murine Chronic Lymphocytic Leukemia B-Cell Activation and Proliferation. Cancer Discov. 2014, 4, 1448–1465. [Google Scholar] [CrossRef]
- Stache, V.; Verlaat, L.; Gatjen, M.; Heinig, K.; Westermann, J.; Rehm, A.; Hopken, U.E. The Splenic Marginal Zone Shapes the Phenotype of Leukemia B Cells and Facilitates Their Niche-Specific Retention and Survival. Oncoimmunology 2017, 6, e1323155. [Google Scholar] [CrossRef]
- Tian, Y.F.; Ahn, H.; Schneider, R.S.; Yang, S.N.; Roman-Gonzalez, L.; Melnick, A.M.; Cerchietti, L.; Singh, A. Integrin-Specific Hydrogels as Adaptable Tumor Organoids for Malignant B and T Cells. Biomaterials 2015, 73, 110–119. [Google Scholar] [CrossRef]
- Robles, E.F.; Mena-Varas, M.; Barrio, L.; Merino-Cortes, S.V.; Balogh, P.; Du, M.Q.; Akasaka, T.; Parker, A.; Roa, S.; Panizo, C.; et al. Homeobox Nkx2-3 Promotes Marginal-Zone Lymphomagenesis by Activating B-Cell Receptor Signalling and Shaping Lymphocyte Dynamics. Nat. Commun. 2016, 7, 11889. [Google Scholar] [CrossRef]
- Chopin, M.; Quemeneur, L.; Ripich, T.; Jessberger, R. Swap-70 Controls Formation of the Splenic Marginal Zone through Regulating T1b-Cell Differentiation. Eur. J. Immunol. 2010, 40, 3544–3556. [Google Scholar] [CrossRef]
- Manevich-Mendelson, E.; Grabovsky, V.; Feigelson, S.W.; Cinamon, G.; Gore, Y.; Goverse, G.; Monkley, S.J.; Margalit, R.; Melamed, D.; Mebius, R.E.; et al. Talin1 Is Required for Integrin-Dependent B Lymphocyte Homing to Lymph Nodes and the Bone Marrow but Not for Follicular B-Cell Maturation in the Spleen. Blood 2010, 116, 5907–5918. [Google Scholar] [CrossRef] [PubMed]
- Ousman, S.S.; Kubes, P. Immune Surveillance in the Central Nervous System. Nat. Neurosci. 2012, 15, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Stuve, O.; Marra, C.M.; Jerome, K.R.; Cook, L.; Cravens, P.D.; Cepok, S.; Frohman, E.M.; Phillips, J.T.; Arendt, G.; Hemmer, B.; et al. Immune Surveillance in Multiple Sclerosis Patients Treated with Natalizumab. Ann. Neurol. 2006, 59, 743–747. [Google Scholar] [CrossRef]
- Parker Harp, C.R.; Archambault, A.S.; Cheung, M.; Williams, J.W.; Czepielewski, R.S.; Duncker, P.C.; Kilgore, A.J.; Miller, A.T.; Segal, B.M.; Kim, A.H.J.; et al. Neutrophils Promote Vla-4-Dependent B Cell Antigen Presentation and Accumulation within the Meninges during Neuroinflammation. Proc. Natl. Acad. Sci. USA 2019, 116, 24221–24230. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Horn, K.; Sagan, S.A.; Bernard, C.C.; Sobel, R.A.; Zamvil, S.S. B-Cell Very Late Antigen-4 Deficiency Reduces Leukocyte Recruitment and Susceptibility to Central Nervous System Autoimmunity. Ann. Neurol. 2015, 77, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Horn, K.; Sagan, S.A.; Winger, R.C.; Spencer, C.M.; Bernard, C.C.; Sobel, R.A.; Zamvil, S.S. Cns Accumulation of Regulatory B Cells Is Vla-4-Dependent. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e212. [Google Scholar] [CrossRef]
- Spaargaren, M.; Beuling, E.A.; Rurup, M.L.; Meijer, H.P.; Klok, M.D.; Middendorp, S.; Hendriks, R.W.; Pals, S.T. The B Cell Antigen Receptor Controls Integrin Activity through Btk and Plcgamma2. J. Exp. Med. 2003, 198, 1539–1550. [Google Scholar] [CrossRef]
- Zucchetto, A.; Caldana, C.; Benedetti, D.; Tissino, E.; Rossi, F.M.; Hutterer, E.; Pozzo, F.; Bomben, R.; Bo, M.D.; D’Arena, G.; et al. Cd49d Is Overexpressed by Trisomy 12 Chronic Lymphocytic Leukemia Cells: Evidence for a Methylation-Dependent Regulation Mechanism. Blood 2013, 122, 3317–3321. [Google Scholar] [CrossRef]
- Benedetti, D.; Tissino, E.; Pozzo, F.; Bittolo, T.; Caldana, C.; Perini, C.; Martorelli, D.; Bravin, V.; D’Agaro, T.; Rossi, F.M.; et al. Notch1 Mutations Are Associated with High Cd49d Expression in Chronic Lymphocytic Leukemia: Link between the Notch1 and the Nf-Kappab Pathways. Leukemia 2018, 32, 654–662. [Google Scholar] [CrossRef]
- Bulian, P.; Shanafelt, T.D.; Fegan, C.; Zucchetto, A.; Cro, L.; Nuckel, H.; Baldini, L.; Kurtova, A.V.; Ferrajoli, A.; Burger, J.A.; et al. Cd49d Is the Strongest Flow Cytometry-Based Predictor of Overall Survival in Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2014, 32, 897–904. [Google Scholar] [CrossRef]
- Gattei, V.; Bulian, P.; del Principe, M.I.; Zucchetto, A.; Maurillo, L.; Buccisano, F.; Bomben, R.; Dal-Bo, M.; Luciano, F.; Rossi, F.M.; et al. Relevance of Cd49d Protein Expression as Overall Survival and Progressive Disease Prognosticator in Chronic Lymphocytic Leukemia. Blood 2008, 111, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Shanafelt, T.D.; Geyer, S.M.; Bone, N.D.; Tschumper, R.C.; Witzig, T.E.; Nowakowski, G.S.; Zent, C.S.; Call, T.G.; Laplant, B.; Dewald, G.W.; et al. Cd49d Expression Is an Independent Predictor of Overall Survival in Patients with Chronic Lymphocytic Leukaemia: A Prognostic Parameter with Therapeutic Potential. Br. J. Haematol. 2008, 140, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Tissino, E.; Pozzo, F.; Benedetti, D.; Caldana, C.; Bittolo, T.; Rossi, F.M.; Bomben, R.; Nanni, P.; Chivilo, H.; Cattarossi, I.; et al. Cd49d Promotes Disease Progression in Chronic Lymphocytic Leukemia: New Insights from Cd49d Bimodal Expression. Blood 2020. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Malavasi, F. Chronic Lymphocytic Leukemia Microenvironment: Shifting the Balance from Apoptosis to Proliferation. Haematologica 2009, 94, 752–756. [Google Scholar] [CrossRef][Green Version]
- Lucas, F.; Woyach, J.A. Inhibiting Bruton’s Tyrosine Kinase in Cll and Other B-Cell Malignancies. Target Oncol. 2019, 14, 125–138. [Google Scholar] [CrossRef]
- Tissino, E.; Benedetti, D.; Herman, S.E.M.; Hacken, E.T.; Ahn, I.E.; Chaffee, K.G.; Rossi, F.M.; Bo, M.D.; Bulian, P.; Bomben, R.; et al. Functional and Clinical Relevance of Vla-4 (Cd49d/Cd29) in Ibrutinib-Treated Chronic Lymphocytic Leukemia. J. Exp. Med. 2018, 215, 681–697. [Google Scholar] [CrossRef]
- Arana, E.; Vehlow, A.; Harwood, N.E.; Vigorito, E.; Henderson, R.; Turner, M.; Tybulewicz, V.L.; Batista, F.D. Activation of the Small Gtpase Rac2 Via the B Cell Receptor Regulates B Cell Adhesion and Immunological-Synapse Formation. Immunity 2008, 28, 88–99. [Google Scholar] [CrossRef]
- Szenes, E.; Harzschel, A.; Decker, S.; Tissino, E.; Pischeli, J.; Gutjahr, J.C.; Kissel, S.; Pennisi, S.; Hopner, J.P.; Egle, A.; et al. Tcl1 Transgenic Mice as a Model for Cd49d-High Chronic Lymphocytic Leukemia. Leukemia 2020, 1–5. [Google Scholar] [CrossRef]
- Moreno, O.; Butler, T.; Zann, V.; Willson, A.; Leung, P.; Connor, A. Safety, Pharmacokinetics, and Pharmacodynamics of Me-401, an Oral, Potent, and Selective Inhibitor of Phosphatidylinositol 3-Kinase P110delta, Following Single Ascending Dose Administration to Healthy Volunteers. Clin. Ther. 2018, 40, 1855–1867. [Google Scholar] [CrossRef]
- Alon, R.; Kassner, P.D.; Carr, M.W.; Finger, E.B.; Hemler, M.E.; Springer, T.A. The Integrin Vla-4 Supports Tethering and Rolling in Flow on Vcam-1. J. Cell Biol. 1995, 128, 1243–1253. [Google Scholar] [CrossRef]
- Chigaev, A.; Waller, A.; Amit, O.; Halip, L.; Bologa, C.G.; Sklar, L.A. Real-Time Analysis of Conformation-Sensitive Antibody Binding Provides New Insights into Integrin Conformational Regulation. J. Biol. Chem. 2009, 284, 14337–14346. [Google Scholar] [CrossRef] [PubMed]
- Askari, J.A.; Buckley, P.A.; Mould, A.P.; Humphries, M.J. Linking Integrin Conformation to Function. J. Cell Sci. 2009, 122 Pt 2, 165–170. [Google Scholar] [CrossRef]
- Chigaev, A.; Waller, A.; Zwartz, G.J.; Buranda, T.; Sklar, L.A. Regulation of Cell Adhesion by Affinity and Conformational Unbending of Alpha4beta1 Integrin. J. Immunol. 2007, 178, 6828–6839. [Google Scholar] [CrossRef] [PubMed]
- Chigaev, A.; Blenc, A.M.; Braaten, J.V.; Kumaraswamy, N.; Kepley, C.L.; Andrews, R.P.; Oliver, J.M.; Edwards, B.S.; Prossnitz, E.R.; Larson, R.S.; et al. Real Time Analysis of the Affinity Regulation of Alpha 4-Integrin. The Physiologically Activated Receptor Is Intermediate in Affinity between Resting and Mn(2+) or Antibody Activation. J. Biol. Chem. 2001, 276, 48670–48678. [Google Scholar] [CrossRef] [PubMed]
- Luque, A.; Gomez, M.; Puzon, W.; Takada, Y.; Sanchez-Madrid, F.; Cabanas, C. Activated Conformations of Very Late Activation Integrins Detected by a Group of Antibodies (Huts) Specific for a Novel Regulatory Region (355–425) of the Common Beta 1 Chain. J. Biol. Chem. 1996, 271, 11067–11075. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Mobley, J.L.; Dwir, O.; Shimron, F.; Grabovsky, V.; Lobb, R.R.; Shimizu, Y.; Alon, R. High Affinity Very Late Antigen-4 Subsets Expressed on T Cells Are Mandatory for Spontaneous Adhesion Strengthening but Not for Rolling on Vcam-1 in Shear Flow. J. Immunol. 1999, 162, 1084–1095. [Google Scholar]
- Grabovsky, V.; Feigelson, S.; Chen, C.; Bleijs, D.A.; Peled, A.; Cinamon, G.; Baleux, F.; Arenzana-Seisdedos, F.; Lapidot, T.; van Kooyk, Y.; et al. Subsecond Induction of Alpha4 Integrin Clustering by Immobilized Chemokines Stimulates Leukocyte Tethering and Rolling on Endothelial Vascular Cell Adhesion Molecule 1 under Flow Conditions. J. Exp. Med. 2000, 192, 495–506. [Google Scholar] [CrossRef]
- Laudanna, C.; Kim, J.Y.; Constantin, G.; Butcher, E. Rapid Leukocyte Integrin Activation by Chemokines. Immunol. Rev. 2002, 186, 37–46. [Google Scholar] [CrossRef]
- De Rooij, M.F.; Kuil, A.; Kraan, W.; Kersten, M.J.; Treon, S.P.; Pals, S.T.; Spaargaren, M. Ibrutinib and Idelalisib Target B Cell Receptor- but Not Cxcl12/Cxcr4-Controlled Integrin-Mediated Adhesion in Waldenstrom Macroglobulinemia. Haematologica 2016, 101, e111–e115. [Google Scholar] [CrossRef]
- Montresor, A.; Toffali, L.; Rigo, A.; Ferrarini, I.; Vinante, F.; Laudanna, C. Cxcr4- and Bcr-Triggered Integrin Activation in B-Cell Chronic Lymphocytic Leukemia Cells Depends on Jak2-Activated Bruton’s Tyrosine Kinase. Oncotarget 2018, 9, 35123–35140. [Google Scholar] [CrossRef]
- Laufer, J.M.; Lyck, R.; Legler, D.F. Zap70 Expression Enhances Chemokine-Driven Chronic Lymphocytic Leukemia Cell Migration and Arrest by Valency Regulation of Integrins. FASEB J. 2018, 32, 4824–4835. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Moreno, M.; Leiva, M.; Aguilera-Montilla, N.; Sevilla-Movilla, S.; de Val, S.I.; Arellano-Sanchez, N.; Gutierrez, N.C.; Maldonado, R.; Martinez-Lopez, J.; Buno, I.; et al. In Vivo Adhesion of Malignant B Cells to Bone Marrow Microvasculature is Regulated by Alpha4beta1 Cytoplasmic-Binding Proteins. Leukemia 2016, 30, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Perez, D.R.; Smagley, Y.; Garcia, M.; Carter, M.B.; Evangelisti, A.; Matlawska-Wasowska, K.; Winter, S.S.; Sklar, L.A.; Chigaev, A. Cyclic Amp Efflux Inhibitors as Potential Therapeutic Agents for Leukemia. Oncotarget 2016, 7, 33960–33982. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brandstadter, R.; Sand, I.K. The Use of Natalizumab for Multiple Sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1691–1702. [Google Scholar] [CrossRef]
- Armuzzi, A.; Felice, C. Natalizumab in Crohn’s Disease: Past and Future Areas of Applicability. Ann. Gastroenterol. 2013, 26, 189–190. [Google Scholar]
- Hoepner, R.; Faissner, S.; Salmen, A.; Gold, R.; Chan, A. Efficacy and Side Effects of Natalizumab Therapy in Patients with Multiple Sclerosis. J. Cent. Nerv. Syst. Dis. 2014, 6, 41–49. [Google Scholar] [CrossRef]
- Ley, K.; Rivera-Nieves, J.; Sandborn, W.J.; Shattil, S. Integrin-Based Therapeutics: Biological Basis, Clinical Use and New Drugs. Nat. Rev. Drug Discov. 2016, 15, 173–183. [Google Scholar] [CrossRef]
- Bonig, H.; Wundes, A.; Chang, K.H.; Lucas, S.; Papayannopoulou, T. Increased Numbers of Circulating Hematopoietic Stem/Progenitor Cells Are Chronically Maintained in Patients Treated with the Cd49d Blocking Antibody Natalizumab. Blood 2008, 111, 3439–3441. [Google Scholar] [CrossRef]
- Kaufmann, M.; Haase, R.; Proschmann, U.; Ziemssen, T.; Akgun, K. Real-World Lab Data in Natalizumab Treated Multiple Sclerosis Patients up to 6 Years Long-Term Follow up. Front. Neurol. 2018, 9, 1071. [Google Scholar] [CrossRef]
- Sheremata, W.A.; Vollmer, T.L.; Stone, L.A.; Willmer-Hulme, A.J.; Koller, M. A Safety and Pharmacokinetic Study of Intravenous Natalizumab in Patients with Ms. Neurology 1999, 52, 1072–1074. [Google Scholar] [CrossRef]
- Yoshimura, N.; Watanabe, M.; Motoya, S.; Tominaga, K.; Matsuoka, K.; Iwakiri, R.; Watanabe, K.; Hibi, T.; Group, A.J.M.S. Safety and Efficacy of Ajm300, an Oral Antagonist of Alpha4 Integrin, in Induction Therapy for Patients with Active Ulcerative Colitis. Gastroenterology 2015, 149, 1775–1783.e2. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Härzschel, A.; Zucchetto, A.; Gattei, V.; Hartmann, T.N. VLA-4 Expression and Activation in B Cell Malignancies: Functional and Clinical Aspects. Int. J. Mol. Sci. 2020, 21, 2206. https://doi.org/10.3390/ijms21062206
Härzschel A, Zucchetto A, Gattei V, Hartmann TN. VLA-4 Expression and Activation in B Cell Malignancies: Functional and Clinical Aspects. International Journal of Molecular Sciences. 2020; 21(6):2206. https://doi.org/10.3390/ijms21062206
Chicago/Turabian StyleHärzschel, Andrea, Antonella Zucchetto, Valter Gattei, and Tanja Nicole Hartmann. 2020. "VLA-4 Expression and Activation in B Cell Malignancies: Functional and Clinical Aspects" International Journal of Molecular Sciences 21, no. 6: 2206. https://doi.org/10.3390/ijms21062206
APA StyleHärzschel, A., Zucchetto, A., Gattei, V., & Hartmann, T. N. (2020). VLA-4 Expression and Activation in B Cell Malignancies: Functional and Clinical Aspects. International Journal of Molecular Sciences, 21(6), 2206. https://doi.org/10.3390/ijms21062206