Novel Mutations in the TMPRSS3 Gene May Contribute to Taiwanese Patients with Nonsyndromic Hearing Loss


Abstract
:1. Introduction
2. Results
2.1. TMPRSS3 Gene Screen in 230 Patients with NSHL
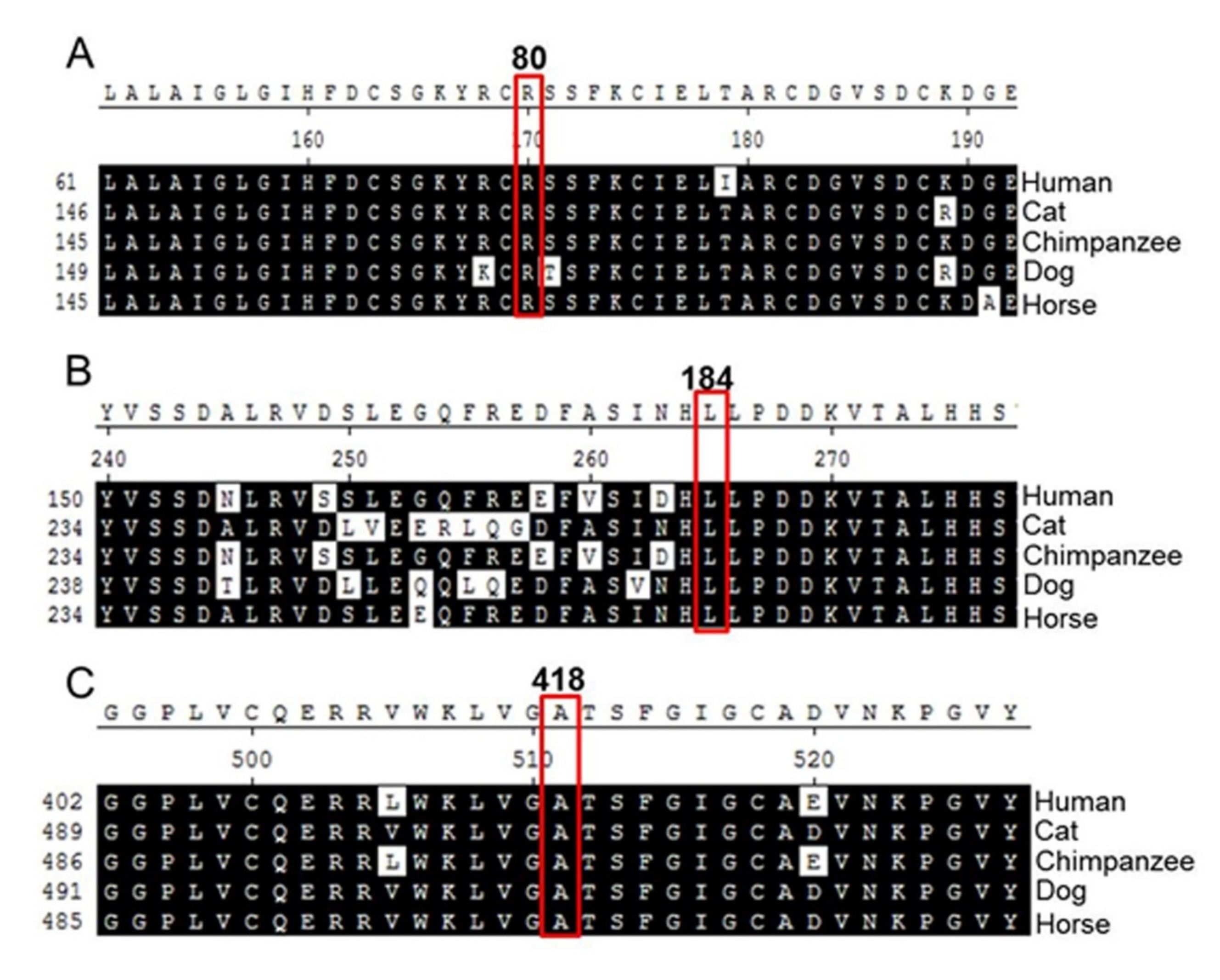
2.2. Analysis of Amino Acid Sequence Homology
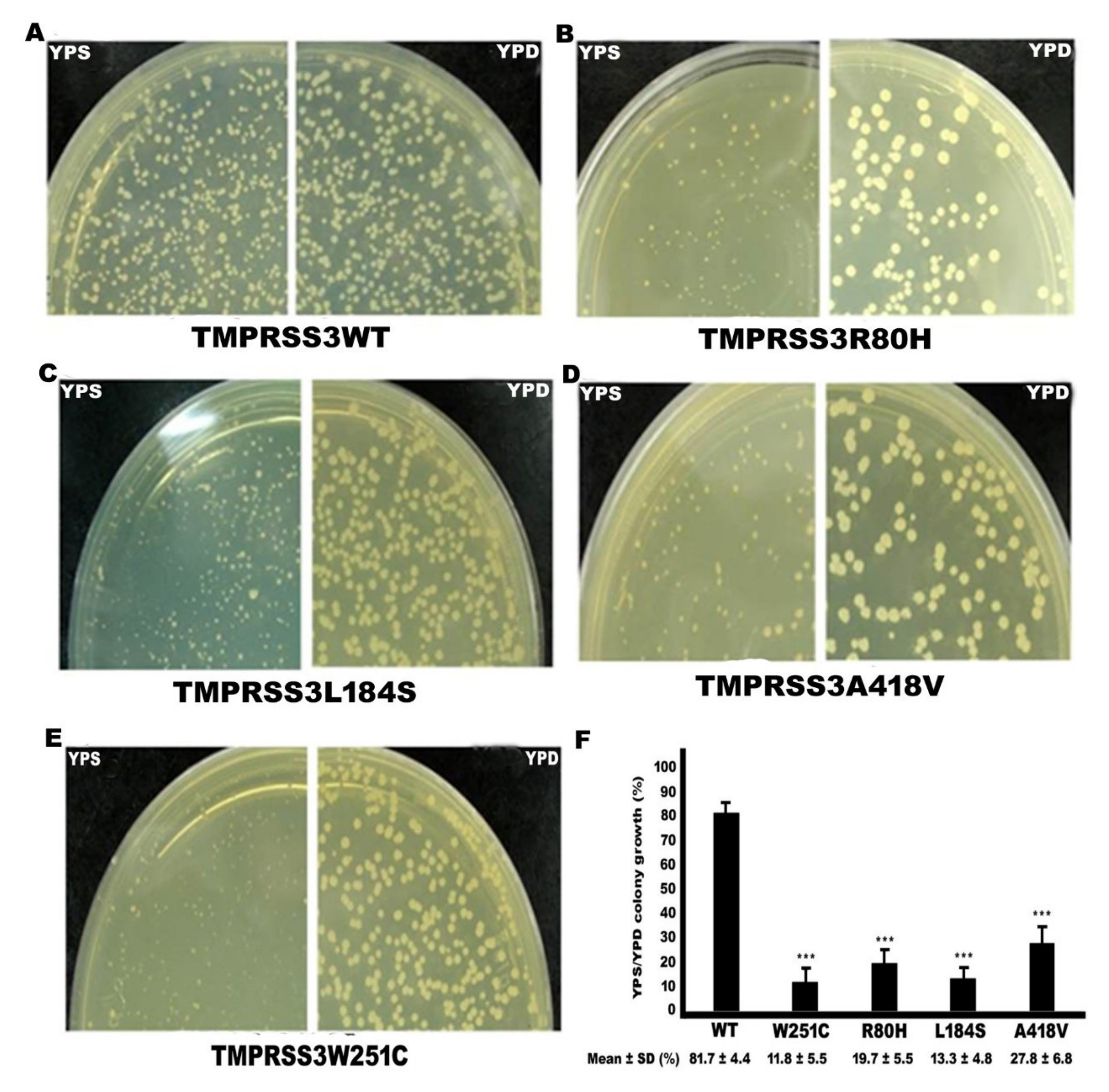
2.3. Functional Study of TMPRSS3 Missense Mutation by sGASP
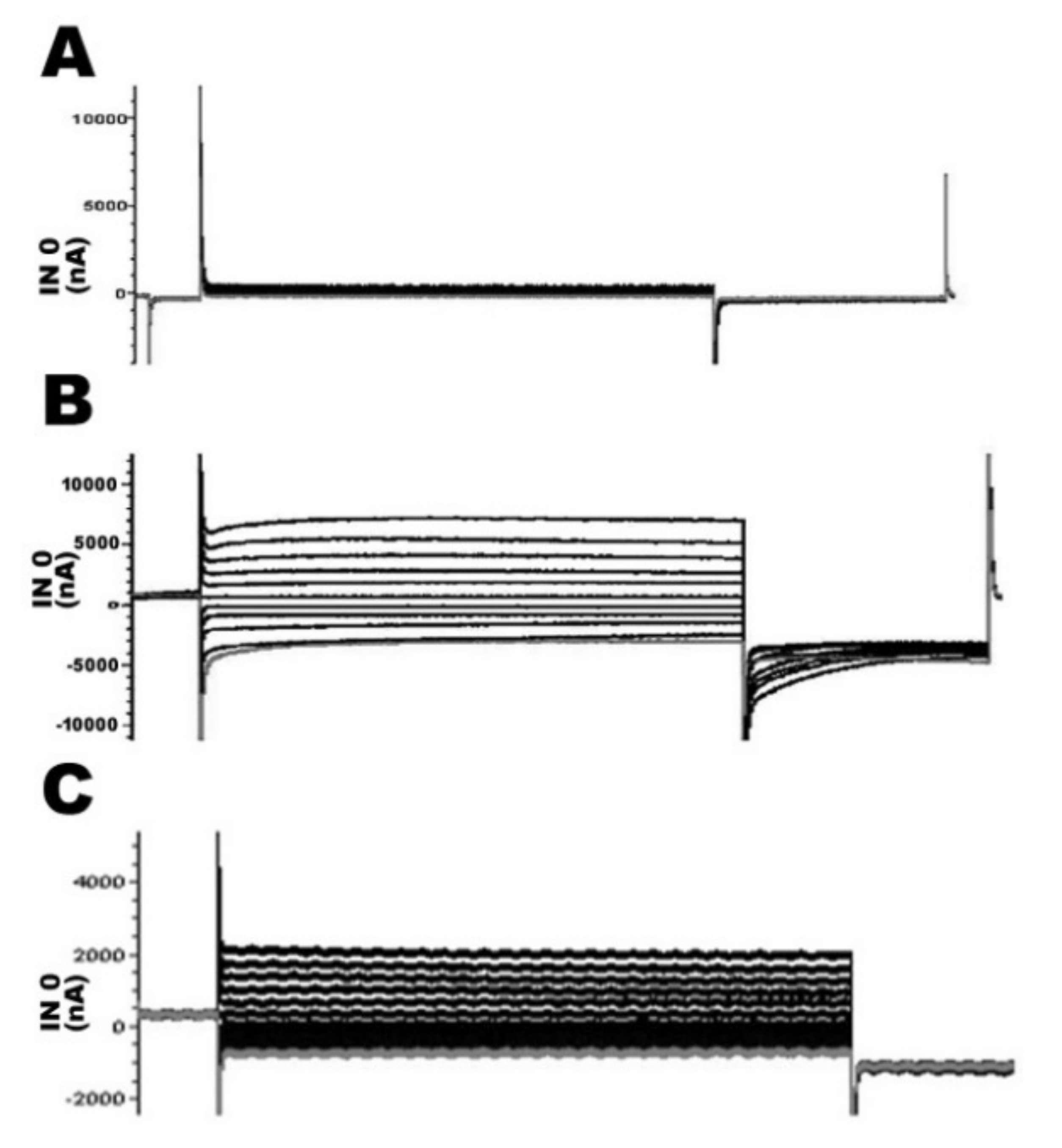
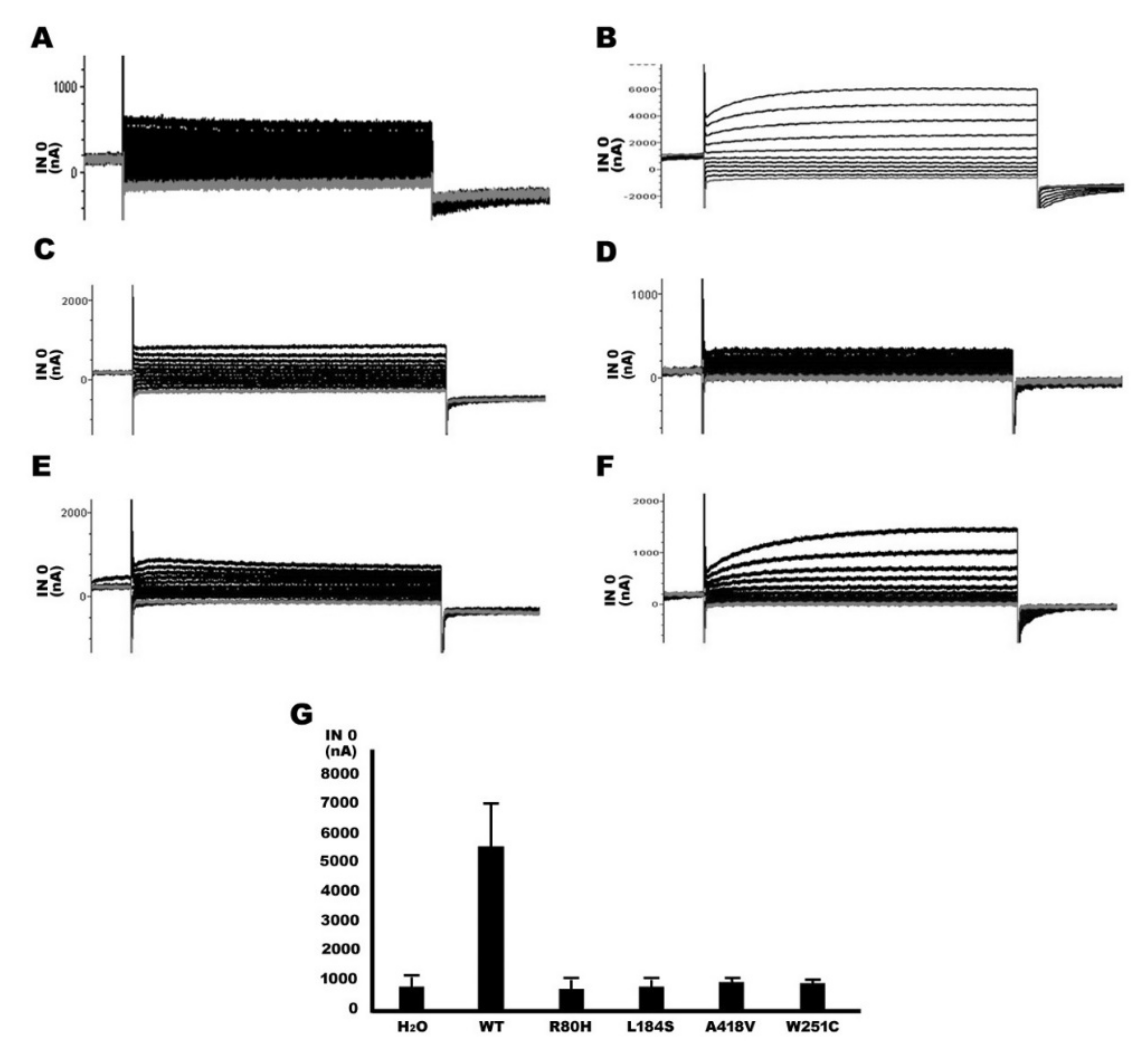
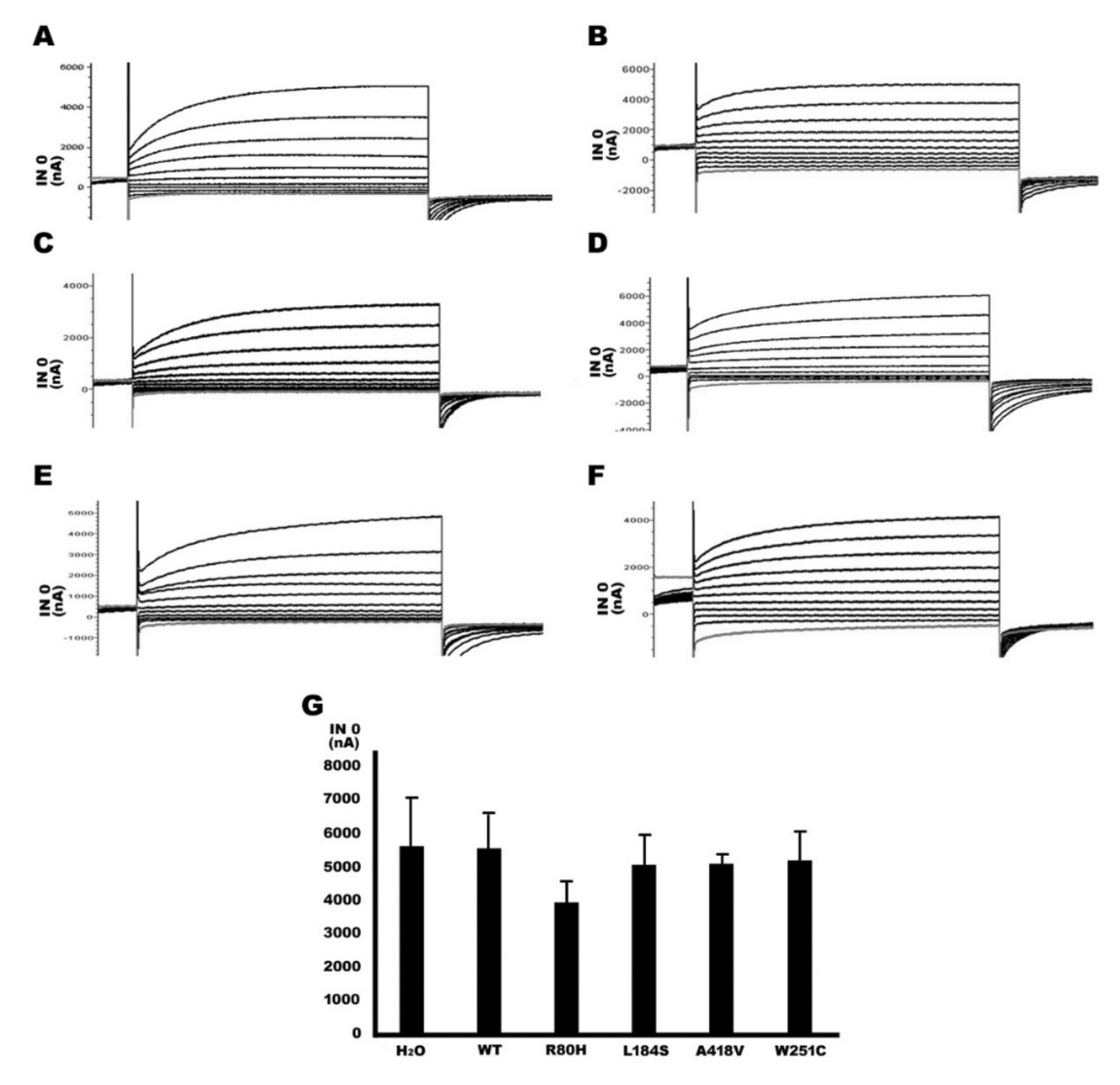
2.4. Functional Study of TMPRSS3 Missense Mutation by Electrophysiological Measurements in Xenopus Oocytes
2.5. Functional Study of TMPRSS3 Heterozygous Missense Mutation by sGASP and Electrophysiological Measurements in Xenopus Oocytes
3. Discussion
4. Materials and Methods
4.1. Research Subjects
4.2. Detection of TMPRSS3 Gene Mutations
4.3. Molecular Cloning and Construct of Wild-Type (WT) and Mutant TMPRSS3 Expression Plasmids for Functional Study
4.4. sGASP Assay (Secretory Genetic Assay of Site-Specific Proteolysis)
4.5. cRNA Preparation for This Study
4.6. Electrophysiological Measurements in Xenopus Oocytes
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Apps, S.A.; Rankin, W.; Kurmis, A.P. Connexin 26 mutations in autosomal recessive deafness disorders: A review. Int. J. Audiol. 2007, 46, 75–81. [Google Scholar] [CrossRef]
- Morton, N.E. Genetic epidemiogy of hearing impairment. Ann. NY Acad. Sci. 1991, 630, 16–31. [Google Scholar] [CrossRef]
- Pallares-Ruiz, N.; Blanchet, P.; Mondain, M.; Claustres, M.; Roux, A.F. A large deletion including most of GJB6 in recessive nonsyndromic deafness: A digenic effect? Eur. J. Hum. Genet. 2002, 10, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Lee, J.S.; Cho, K.J.; Yu, S.; Yoon, J.-H.; Gee, H.Y.; Choi, J.Y. Genetic Predisposition to Sporadic Congenital Hearing Loss in a Pediatric Population. Sci. Rep. 2017, 7, 45973. [Google Scholar] [CrossRef] [Green Version]
- Bosanquet, D.; Williams, N.; Lewis, M. Acquired small bowel diverticular disease: A review. Br. J. Hosp. Med. 2010, 71, 552–555. [Google Scholar] [CrossRef]
- Szabo, R.; Wu, Q.; Dickson, R.B.; Netzel-Arnett, S.; Antalis, T.M.; Bugge, T.H. Type II transmembrane serine proteases. Thromb. Haemost. 2003, 90, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Scott, H.S.; Kudoh, J.; Wattenhofer, M.; Shibuya, K.; Berry, A.; Chrast, R.; Guipponi, M.; Wang, J.; Kawasaki, K.; Asakawa, S.; et al. Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat. Genet. 2001, 27, 59–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, J.D.; Clements, J.A.; Quigley, J.P.; Antalis, T.M. Type II transmembrane serine proteases Insights into an emerging class of cell surface proteolytic enzymes. J. Biol. Chem. 2001, 276, 857–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonné-Tamir, B.; DeStefano, A.L.; Briggs, C.E.; Adair, R.; Franklyn, B.; Weiss, S.; Korostishevsky, M.; Frydman, M.; Baldwin, C.T.; Farrer, L.A. Linkage of congenital recessive deafness (gene DFNB10) to chromosome 21q22.3. Am. J. Hum. Genet. 1996, 58, 1254–1259. [Google Scholar]
- Veske, A.; Oehlmann, R.; Younus, F.; Mohyuddin, A.; Müller-Myhsok, B.; Mehdi, S.Q.; Gal, A. Autosomal Recessive Non-Syndromic Deafness Locus (DFNB8) Maps on Chromosome 21Q22 in a Large Consanguineous Kindred from Pakistan. Hum. Mol. Genet. 1996, 5, 165–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guipponi, M.; Toh, M.-Y.; Tan, J.; Park, D.; Hanson, K.; Ballana, E.; Kwong, D.; Cannon, P.Z.; Wu, Q.; Gout, A.; et al. An integrated genetic and functional analysis of the role of type II transmembrane serine proteases (TMPRSSs) in hearing loss. Hum. Mutat. 2007, 29, 130–141. [Google Scholar] [CrossRef]
- Gao, X.; Huang, S.-S.; Yuan, Y.-Y.; Xu, J.-C.; Gu, P.; Bai, D.; Kang, D.-Y.; Han, M.-Y.; Wang, G.-J.; Zhang, M.-G.; et al. Identification ofTMPRSS3as a Significant Contributor to Autosomal Recessive Hearing Loss in the Chinese Population. Neural Plast. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Yuan, Y.-Y.; Wang, G.-J.; Xu, J.-C.; Su, Y.; Lin, X.; Dai, P. Novel Mutations and Mutation Combinations ofTMPRSS3Cause Various Phenotypes in One Chinese Family with Autosomal Recessive Hearing Impairment. BioMed Res. Int. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Park, D.; Oh, M.; Sellamuthu, S.; Park, W.J. Detection of site-specific proteolysis in secretory pathways. Biochem. Biophys. Res. Commun. 2002, 296, 419–424. [Google Scholar] [CrossRef]
- Lee, Y.J.; Park, D.; Kim, S.Y.; Park, W.J. Pathogenic mutations but not polymorphisms in congenital and childhood onset autosomal recessive deafness disrupt the proteolytic activity of TMPRSS3. J. Med Genet. 2003, 40, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, V.; Dahlmann, A.; Bertog, M.; Korbmacher, C. Trypsin can activate the epithelial sodium channel (ENaC) in microdissected mouse distal nephron. Am. J. Physiol. Physiol. 2008, 295, F1052–F1062. [Google Scholar] [CrossRef] [PubMed]
- Haerteis, S.; Krappitz, A.; Krappitz, M.; Murphy, J.E.; Bertog, M.; Krueger, B.; Nacken, R.; Chung, H.; Hollenberg, M.D.; Knecht, W.; et al. Proteolytic Activation of the Human Epithelial Sodium Channel by Trypsin IV and Trypsin I Involves Distinct Cleavage Sites. J. Boil. Chem. 2014, 289, 19067–19078. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q. Type II transmembrane serine proteases. Cur. Top. Develop. Biol. 2003, 54, 167–206. [Google Scholar]
- Bugge, T.H.; Antalis, T.M.; Wu, Q. Type II Transmembrane Serine Proteases*. J. Boil. Chem. 2009, 284, 23177–23181. [Google Scholar] [CrossRef] [Green Version]
- Guipponi, M.; Vuagniaux, G.; Wattenhofer, M.; Shibuya, K.; Vazquez, M.; Dougherty, L.; Scamuffa, N.; Guida, E.; Okui, M.; Rossier, C.; et al. The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Hum. Mol. Genet. 2002, 11, 2829–2836. [Google Scholar] [CrossRef] [Green Version]
- Ben-Yosef, T.; Wattenhofer, M.; Riazuddin, S.; Ahmed, Z.M.; Scott, H.S.; Kudoh, J.; Shibuya, K.; Antonarakis, S.E.; Bonne-Tamir, B.; Radhakrishna, U.; et al. Novel mutations of TMPRSS3 in four DFNB8/B10 families segregating congenital autosomal recessive deafness. J. Med. Genet. 2001, 38, 396–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masmoudi, S.; Antonarakis, S.E.; Schwede, T.; Ghorbel, A.M.; Gratri, M.; Pappasavas, M.-P.; Drira, M.; Elgaied-Boulila, A.; Wattenhofer, M.; Rossier, C.; et al. Novel missense mutations of TMPRSS3 in two consanguineous Tunisian families with non-syndromic autosomal recessive deafness. Hum. Mutat. 2001, 18, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Wattenhofer, M.; Di Iorio, M.; Rabionet, R.; Dougherty, L.; Pampanos, A.; Schwede, T.; Montserrat-Sentís, B.; Arbones, M.; Iliades, T.; Pasquadibisceglie, A.; et al. Mutations in the TMPRSS3 gene are a rare cause of childhood nonsyndromic deafness in Caucasian patients. J. Mol. Med. 2002, 80, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Wattenhofer, M.; Sahin-Calapoglu, N.; Andreasen, D.; Kalay, E.; Caylan, R.; Braillard, B.; Fowler-Jaeger, N.; Reymond, A.; Rossier, B.C.; Karaguzel, A.; et al. A novel TMPRSS3 missense mutation in a DFNB8/10 family prevents proteolytic activation of the protein. Hum. Genet. 2005, 117, 528–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weegerink, N.J.; Schraders, M.; Oostrik, J.; Huygen, P.L.; Strom, T.M.; Granneman, S.; Pennings, R.J.; Venselaar, H.; Hoefsloot, L.H.; Elting, M.; et al. Genotype phenotype correlation in DFNB8/10 families with TMPRSS3 mutations. J. Assoc. Res. Otolaryngol. 2011, 12, 753–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchin, T.; Coy, N.; Conlon, H.; Telford, E.; Bromelow, K.; Blaydon, D.; Taylor, G.; Coghill, E.; Brown, S.; Trembath, R.; et al. Assessment of the genetic causes of recessive childhood non-syndromic deafness in the UK - implications for genetic testing. Clin. Genet. 2005, 68, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Battelino, S.; Klančar, G.; Kovač, J.; Battelino, T.; Podkrajsek, K.T. TMPRSS3 mutations in autosomal recessive nonsyndromic hearing loss. Eur. Arch. Oto-Rhino-Laryngology 2015, 273, 1151–1154. [Google Scholar] [CrossRef]
- Lee, J.; Baek, J.-I.; Choi, J.Y.; Kim, U.-K.; Lee, S.-H.; Lee, K.-Y. Genetic analysis of TMPRSS3 gene in the Korean population with autosomal recessive nonsyndromic hearing loss. Gene 2013, 532, 276–280. [Google Scholar] [CrossRef]
- Fan, D.; Zhu, W.; Li, D.; Ji, D.; Wang, P. Identification of a Novel Homozygous Mutation, TMPRSS3: C.535G>A, in a Tibetan Family with Autosomal Recessive Non-Syndromic Hearing Loss. PLoS ONE 2014, 9, e114136. [Google Scholar] [CrossRef] [Green Version]
- Guipponi, M.; Antonarakis, S.E.; Scott, H.S. TMPRSS3, a type II transmembrane serine protease mutated in non-syndromic autosomal recessive deafness. Front. Biosci. 2008, 13, 1557–1567. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-J.; Huang, S.-H.; Chou, K.-H.; Liao, P.-J.; Su, C.-C.; Li, S.-Y. Identification of Mutations in Members of the Connexin Gene Family as a Cause of Nonsyndromic Deafness in Taiwan. Audiol. Neurotol. 2007, 12, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-J.; Wang, W.-H.; Lin, Y.-C.; Weng, H.-H.; Yang, J.-T.; Hwang, C.-F.; Wu, C.-M.; Li, S.-Y. Prospective variants screening of connexin genes in children with hearing impairment: Genotype/phenotype correlation. Qual. Life Res. 2010, 128, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.N.; Krawczak, M.; Polychronakos, C.; Tyler-Smith, C.; Kehrer-Sawatzki, H. Where genotype is not predictive of phenotype: Towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Qual. Life Res. 2013, 132, 1077–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlotogora, J. Jo Penetrance and expressivity in the molecular age. Genet. Med. 2003, 5, 347–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Chapelle, A. Genetic predisposition to human disease: Allele-specific expression and low-penetrance regulatory loci. Oncogene 2009, 28, 3345–3348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, F.; Walsh, J.; Mburu, P.; Varela, A.; Brown, K.A.; Antonio, M.; Beisel, K.W.; Steel, K.P.; Brown, S.D.M. A type VII myosin encoded by the mouse deafness gene shaker-1. Nature 1995, 374, 62–64. [Google Scholar] [CrossRef]
- Well, D.; Blanchard, S.; Kaplan, J.; Guilford, P.; Gibson, F.; Walsh, J.; Mburu, P.; Varela, A.; Levilliers, J.; Weston, M.D.; et al. Defective myosin VIIA gene responsible for Usher syndrome type IB. Nature 1995, 374, 60–61. [Google Scholar] [CrossRef]
- Donaudy, F.; Ferrara, A.; Esposito, L.; Hertzano, R.; Ben-David, O.; Bell, R.E.; Melchionda, S.; Zelante, L.; Avraham, K.B.; Gasparini, P. Multiple Mutations of MYO1A, a Cochlear-Expressed Gene, in Sensorineural Hearing Loss. Am. J. Hum. Genet. 2003, 72, 1571–1577. [Google Scholar] [CrossRef] [Green Version]
- Guipponi, M.; Tan, J.; Cannon, P.Z.; Donley, L.; Crewther, P.; Clarke, M.; Wu, Q.; Shepherd, R.K.; Scott, H.S. Mice Deficient for the Type II Transmembrane Serine Protease, TMPRSS1/hepsin, Exhibit Profound Hearing Loss. Am. J. Pathol. 2007, 171, 608–616. [Google Scholar] [CrossRef] [Green Version]
- Kubisch, C.; Schroeder, B.C.; Friedrich, T.; Lutjohann, B.; El-Amraoui, A.; Marlin, S.; Petit, C.; Jentsch, T.J. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 1998, 96, 437–446. [Google Scholar] [CrossRef] [Green Version]
- Cecilia, M.C.; Laurent, S.; Gary, B.; Bernard, T.; Ivan, G.; Jran-Daniel, H.; Bernard, C.R. Amiloride-sensitive epithelial Na+ channel is msde of three homologous subunits. Nature 1994, 367, 463–467. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymorphism | Allele Frequency (%) | |||||
|---|---|---|---|---|---|---|
| TMPRSS3 Protein Domain | Region | Genotype | Amino Acid Change | Hearing Loss | Control | p-Value * |
| (n = 460) | (n = 240) | |||||
| 5′-UTR | −78T>A | ----- | T: 0.997 A: 0.003 | T:0.996 A:0.004 | 0.742658 | |
| −36G>C | ----- | G:0.995 A:0.005 | G:0.996 A: 0.004 | 0.848021 | ||
| TM | Exon 3 | 157G>A | V53I | G:0.750 A:0.250 | G:0.867 A:0.133 | 0.000455 & |
| Intron 5 | 447-13A>G | ----- | A:0.421 G:0.579 | A:0.582 G:0.408 | 0.000035 & | |
| SRCR | Exon 6 | 453G>A | V151V | G:0.608 A:0.392 | G:0.754 A:0.246 | 0.000172 & |
| Intron 7 | 617-4 ins AT | ----- | --:0.663 AT:0.337 | --:0.754 AT:0.246 | 0.016173 & | |
| Serine protease | Exon 8 | 681G>A | Q227Q | G:0.989 A:0.011 | G:0.987 A:0.013 | 0.820762 |
| 757A>G | I253V | A:0.626 G:0.374 | A:0.742 G:0.258 | 0.002906 & | ||
| 763G>T | A255A | G:1.000 T:0.000 | G:0.996 T:0.004 | # | ||
| Exon9 | 945G>A | T315T | G:1.000 A:0.000 | G:0.996 A:0.004 | # | |
| Intron9 | 952 + 18T>C | ----- | T:1.000 C:0.000 | T:0.996 C:0.004 | # | |
| Intron10 | 1048 + 56C>T | ----- | C:0.982 T:0.018 | C:0.987 T:0.013 | 0.568631 | |
| Exon11 | 1121A>G | D374G | A:0.992 G:0.008 | A:0.992 G:0.008 | 0.952574 | |
| 1122C>T | D374D | C:0.992 T:0.008 | C:0.996 T:0.004 | 0.572243 | ||
| Exon12 | 1335C>T | H445H | C:0.976 T:0.024 | C:0.987 T:0.013 | 0.324825 | |
| 3′-UTR | Exon 13 | 1367G>A | ----- | G:0.242 A:0.758 | G:0.517 A:0.483 | <0.000001 & |
| 1399 + 19A>G | ----- | A:0.995 G:0.005 | A:0.992 G:0.008 | 0.641868 | ||
| Location | Nucleotide Change | Domain | Amino Acid Change | Predicted Effect | Individual Found | Percentage (%) |
|---|---|---|---|---|---|---|
| Exon1 | −75 A>G/wt | --- | X | N | 1 | 0.43% |
| Exon2 | 239G>A/WT | LDLRA | R80H | Missense mutation | 1 | 0.43 |
| Intron3 | 205+38 C>T/WT | --- | X | Splicing | 5 | 2.17% |
| Exon6 | 551T>C/WT | SRCR | L184S | Missense mutation | 1 | 0.43% |
| Exon8 | 621T>C/WT | Serine protease | C207C | Neutral (or silent) mutation | 1 | 0.43% |
| Exon9 | 933C>T/WT | Serine protease | A311A | Neutral (or silent) mutation | 1 | 0.43% |
| Intron11 | 1194+15C>A/WT | --- | X | Splicing | 1 | 0.43% |
| Exon12 | 1253 C>T/WT | Serine protease | A418V | Missense mutation | 1 | 0.43% |
| Exon12 | 1269 C>T/WT | Serine protease | I423I | Neutral (or silent) mutation | 1 | 0.43% |
| Intron12 | 1347+11 C>T/WT | --- | X | Splicing | 1 | 0.43% |
| Total | 14 | 6.09% |
| Genotype (Homozygous) | *No Trypsin (INa Current, nA) | *Trypsin (INa Current, nA) |
|---|---|---|
| Water (H2O) | 786 ± 345 | 5607 ± 1456 |
| TMPRSS3WT/TMPRSS3WT | 5550 ± 1463 | 5551 ± 1047 |
| TMPRSS3R80H/TMPRSS3R80H | 702 ± 374 | 3936 ± 615 |
| TMPRSS3L184S/TMPRSS3L184S | 791 ± 261 | 5060 ± 889 |
| TMPRSS3A418V/TMPRSS3A418V | 927 ± 63 | 5084 ± 292 |
| TMPRSS3W251C/TMPRSS3W251C | 888 ± 89 | 5181 ± 889 |
| Genotype (heterozygous) | *No Trypsin (INa current, nA) | *Trypsin (INa current, nA) |
| TMPRSS3WT/ TMPRSS3WT | 5337 ± 1662 | 5351 ± 1368 |
| TMPRSS3R80H/ TMPRSS3WT | 1828 ± 524 | 4130 ± 1075 |
| TMPRSS3L184S/ TMPRSS3WT | 1674 ± 467 | 3910 ± 701 |
| TMPRSS3A418V/TMPRSS3WT | 1811 ± 509 | 3848 ± 717 |
| TMPRSS3W251C/TMPRSS3WT | 1493 ± 606 | 4320 ± 954 |
| Region | Primer Sequence (Oligonucleotide) | Annealing, for PCR Temperature (°C) | Product Size |
|---|---|---|---|
| Exon 1 | F: 5′-CTCTGGTCTCCTTGGCATTGTG-3 R: 5′-CTCTCAAAGCCCTTTCCATTGC-3′ | 61 | 387 bps |
| Exon 2 | F: 5′-GATGCACCTGATGCTACAAG-3′ R: 5′-GGACAGTCAGTCACATTGGTC-3′ | 55 | 286 bps |
| Exon 3 | F: 5′-GGACTGAACTAGAGAATGTGCC-3′ R: 5′-GACAAAGCCATGAGCATGGC-3′ | 63 | 510 bps |
| Exon 4 | F: 5′-AGGGGGACAGTTGTTAGTGTTGC-3′ R: 5′-TACAGATGGGAAGGGTCAGGGTTG-3′ | 62 | 261 bps |
| Exon 5 | F: 5′-TGTGGAGAAACCCCTGCCTATG-3′ R: 5′-GATGTGAGGATGTAATCTGAGAGCG-3′ | 68 | 323 bps |
| Exon 6 | F: 5′-GACTCGCACATCGGTTGAATG-3′ R: 5′-ATACTCCCTCAGGTTCTCACACCC-3′ | 64 | 387 bps |
| Exon 7 | F: 5′-GTGTGACCTCATCCTCATGG-3′ R: 5′-CTCTGAGGGCAAGGAGATAG-3′ | 64 | 293 bps |
| Exon 8 | F: 5′-TAGAGCTGCTGTGAGCTCTG-3′ R: 5′-AGACTCCTCTCCAACTGTAC-3′ | 62 | 438 bps |
| Exon 9 | F: 5′-GGACCACATCTTGCCTGATAACC-3′ R: 5′-AAAGCACACAGCCCACGAAG-3′ | 62 | 694 bps |
| Exon 10 | F: 5′-CTCCTGCTGTGAGCTGATCG-3′ R: 5′-CGAGCAGCTGACATGCACTC-3′ | 61 | 393 bps |
| Exon 11 | F: 5′-GTCTCAGCATGCGCCTTCTG-3′ R: 5′-CCCACGCAGAGCCAGATCAC-3′ | 65 | 407 bps |
| Exon 12 | F: 5′-TGGGTCATCCATTGGGACATC-3′ R: 5′-TCTCTGTTTTCAGCACAAGCGTC-3′ | 64 | 460 bps |
| Exon 13 | F: 5′-TACGGAAGTGACGGAACTGTCG-3′ R: 5′-CTTGAAGGTTGTGCTGGAATCAG-3′ | 64 | 442 bps |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, S.-H.; Yen, Y.-C.; Li, S.-Y.; Yang, J.-J. Novel Mutations in the TMPRSS3 Gene May Contribute to Taiwanese Patients with Nonsyndromic Hearing Loss. Int. J. Mol. Sci. 2020, 21, 2382. https://doi.org/10.3390/ijms21072382
Wong S-H, Yen Y-C, Li S-Y, Yang J-J. Novel Mutations in the TMPRSS3 Gene May Contribute to Taiwanese Patients with Nonsyndromic Hearing Loss. International Journal of Molecular Sciences. 2020; 21(7):2382. https://doi.org/10.3390/ijms21072382
Chicago/Turabian StyleWong, Swee-Hee, Yung-Chang Yen, Shuan-Yow Li, and Jiann-Jou Yang. 2020. "Novel Mutations in the TMPRSS3 Gene May Contribute to Taiwanese Patients with Nonsyndromic Hearing Loss" International Journal of Molecular Sciences 21, no. 7: 2382. https://doi.org/10.3390/ijms21072382