The Vascular Involvement in Soft Tissue Fibrosis—Lessons Learned from Pathological Scarring



{kind=link}
Abstract
:1. Introduction
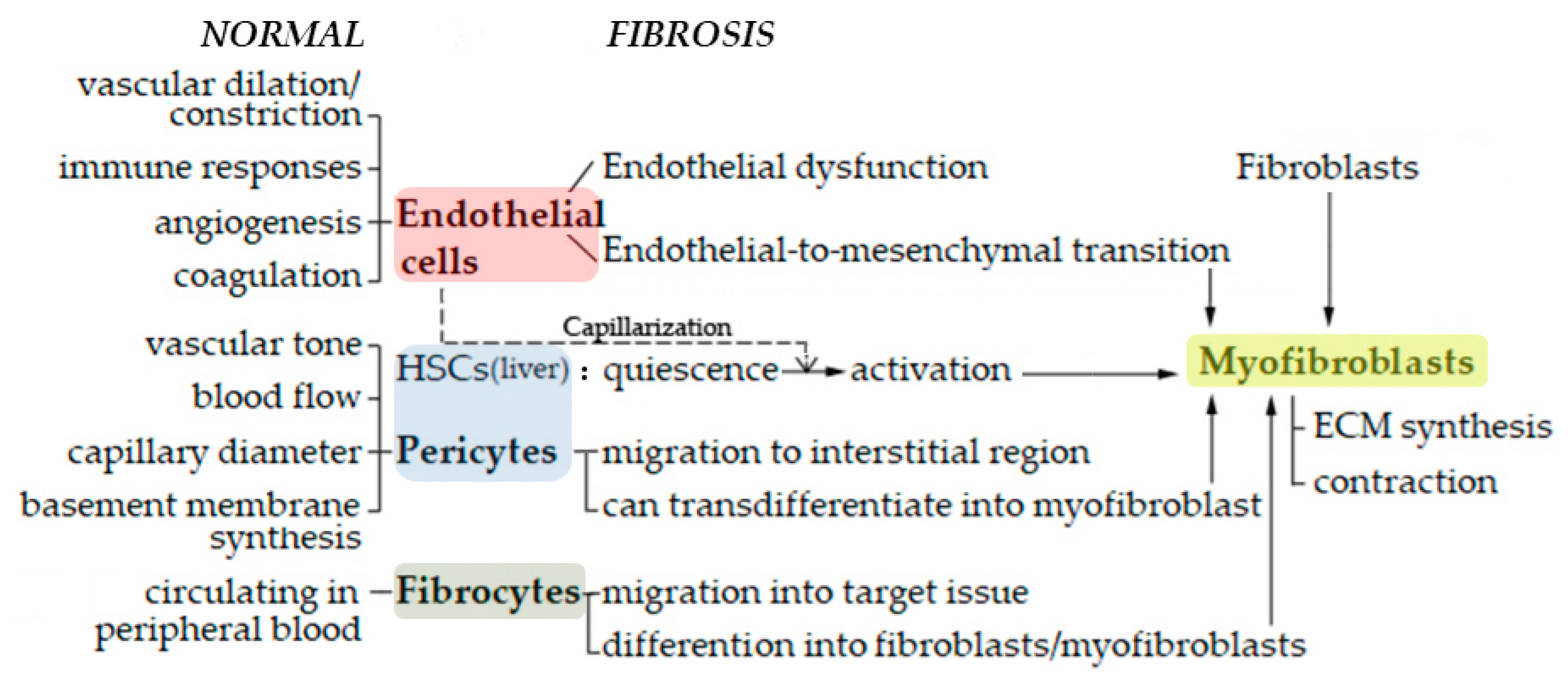
2. Endothelial Cells
2.1. Endothelial Dysfunction
2.1.1. Endothelial Dysfunction in Skin Fibrosis
2.1.2. Endothelial Dysfunction in Liver Fibrosis
2.1.3. Endothelial Dysfunction in Renal Fibrosis
2.1.4. Endothelial Dysfunction in Cardiac Fibrosis
2.2. Endothelial to Mesenchymal Cell Transition (EndoMT)
2.2.1. EndoMT-Derived Myofibroblasts in Skin Fibrosis
2.2.2. EndoMT-Derived Myofibroblasts in Renal Fibrosis
2.2.3. EndoMT-Derived Myofibroblasts in Cardiac Fibrosis
2.2.4. EndoMT-Derived Myofibroblasts in Other Soft Tissue Fibroses
3. Pericytes
3.1. Pericytes in Skin Fibrosis
3.2. Pericytes in Kidney Fibrosis
3.3. Pericytes in Lung Fibrosis
4. Hepatic Stellate Cells (Abbreviated to HSCs in This Section)
5. Fibrocytes
5.1. Fibrocytes in Skin Fibrosis
5.2. Fibrocytes in Lung Fibrosis
5.3. Fibrocytes in Other Soft Tissue Fibroses
6. Interactions between Myofibroblasts and Endothelial Cells
7. Summary
Conflicts of Interest
References
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Liu, L.; You, Z.; Du, Y.; Ogawa, R. Managing keloid scars: From radiation therapy to actual and potential drug deliveries. Int. Wound J. 2019, 16, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, R.; Akaishi, S. Endothelial dysfunction may play a key role in keloid and hypertrophic scar pathogenesis—Keloids and hypertrophic scars may be vascular disorders. Med. Hypotheses 2016, 96, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Arima, J.; Huang, C.; Rosner, B.; Akaishi, S.; Ogawa, R. Hypertension: A systemic key to understanding local keloid severity. Wound Repair Regen. 2015, 23, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ogawa, R. Pharmacological treatment for keloids. Expert Opin. Pharmacother. 2013, 14, 2087–2100. [Google Scholar] [CrossRef]
- Huang, C.; Liu, L.; You, Z.; Zhao, Y.; Dong, J.; Du, Y.; Ogawa, R. Endothelial dysfunction and mechanobiology in pathological cutaneous scarring: Lessons learned from soft tissue fibrosis. Br. J. Dermatol. 2017, 177, 1248–1255. [Google Scholar] [CrossRef]
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular endothelial cell biology: An update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [Green Version]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F., Jr.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Tabit, C.E.; Chung, W.B.; Hamburg, N.M.; Vita, J.A. Endothelial dysfunction in diabetes mellitus: Molecular mechanisms and clinical implications. Rev. Endocr. Metab. Disord. 2010, 11, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Noishiki, C.; Takagi, G.; Kubota, Y.; Ogawa, R. Endothelial dysfunction may promote keloid growth. Wound Repair Regen. 2017, 25, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Poore, S.; Berry, B.; Eidson, D.; McKie, K.T.; Harris, R.A. Evidence of vascular endothelial dysfunction in young patients with cystic fibrosis. Chest 2013, 143, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Miguelez, P.; Thomas, J.; Seigler, N.; Crandall, R.; McKie, K.T.; Forseen, C.; Harris, R.A. Evidence of microvascular dysfunction in patients with cystic fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1479–H1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, M.A.; Fox, B.M.; Seigler, N.; Rodriguez-Miguelez, P.; Looney, J.; Thomas, J.; McKie, K.T.; Forseen, C.; Davison, G.W.; Harris, R.A. Endothelial dysfunction in cystic fibrosis: Role of oxidative stress. Oxid. Med. Cell. Longev. 2019, 2019, 1629638. [Google Scholar] [CrossRef] [Green Version]
- Steib, C.J.; Gerbes, A.L.; Bystron, M.; Op den Winkel, M.; Härtl, J.; Roggel, F.; Prüfer, T.; Göke, B.; Bilzer, M. Kupffer cell activation in normal and fibrotic livers increases portal pressure via thromboxane A(2). J. Hepatol. 2007, 47, 228–238. [Google Scholar] [CrossRef]
- Graupera, M.; García-Pagán, J.C.; Parés, M.; Abraldes, J.G.; Roselló, J.; Bosch, J.; Rodés, J. Cyclooxygenase-1 inhibition corrects endothelial dysfunction in cirrhotic rat livers. J. Hepatol. 2003, 39, 515–521. [Google Scholar] [CrossRef]
- Rodríguez-Vilarrupla, A.; Laviña, B.; García-Calderó, H.; Russo, L.; Rosado, E.; Roglans, N.; Bosch, J.; García-Pagán, J.C. PPARα activation improves endothelial dysfunction and reduces fibrosis and portal pressure in cirrhotic rats. J. Hepatol. 2012, 56, 1033–1039. [Google Scholar] [CrossRef]
- Deng, W.; Zhu, Y.; Lin, J.; Zheng, L.; Zhang, C.; Luo, M. Inhibition of soluble epoxide hydrolase lowers portal hypertension in cirrhotic rats by ameliorating endothelial dysfunction and liver fibrosis. Prostaglandins. Other. Lipid. Mediat 2017, 131, 67–74. [Google Scholar]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Fraser, R.; Bosanquet, A.G.; Day, W.A. Filtration of chylomicrons by the liver may influence cholesterol metabolism and atherosclerosis. Atherosclerosis 1978, 29, 113–123. [Google Scholar] [CrossRef]
- Xu, G.F.; Wang, X.Y.; Ge, G.L.; Li, P.T.; Jia, X.; Tian, D.L.; Jiang, L.D.; Yang, J.X. Dynamic changes of capillarization and peri-sinusoid fibrosis in alcoholic liver diseases. World J. Gastroenterol. 2004, 10, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Okanoue, T.; Sawa, Y.; Hori, N.; Ohta, M.; Kagawa, K. Defenestration of the sinusoidal endothelial cell in a rat model of cirrhosis. Hepatology 1993, 17, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Abraldes, J.G.; Albillos, A.; Bañares, R.; Turnes, J.; González, R.; García-Pagán, J.C.; Bosch, J. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: A randomized controlled trial. Gastroenterology 2009, 136, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Pi, X.; Xie, L.; Patterson, C. Emerging roles of vascular endothelium in metabolic homeostasis. Circ. Res. 2018, 123, 477–494. [Google Scholar] [CrossRef]
- Braet, F.; Wisse, E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: A review. Comp. Hepatol. 2002, 1, 1. [Google Scholar] [CrossRef] [Green Version]
- Fraser, R.; Dobbs, B.R.; Rogers, G.W. Lipoproteins and the liver sieve: The role of the fenestrated sinusoidal endothelium in lipoprotein metabolism, atherosclerosis, and cirrhosis. Hepatology 1995, 21, 863–874. [Google Scholar]
- Pasarín, M.; La Mura, V.; Gracia-Sancho, J.; García-Calderó, H.; Rodríguez-Vilarrupla, A.; García-Pagán, J.C.; Bosch, J.; Abraldes, J.G. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PLoS ONE 2012, 7, e32785. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Wang, X.; Wang, L.; Wang, L.; Atkinson, R.D.; Kanel, G.C.; Gaarde, W.A.; Deleve, L.D. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology 2012, 142, 918–927. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Tiwari, M.M.; Messer, K.J.; Holthoff, J.H.; Gokden, N.; Brock, R.W.; Mayeux, P.R. Peritubular capillary dysfunction and renal tubular epithelial cell stress following lipopolysaccharide administration in mice. Am. J. Physiol. Renal. Physiol. 2007, 292, F261–F268. [Google Scholar] [CrossRef]
- Molitoris, B.A. Therapeutic translation in acute kidney injury: The epithelial/endothelial axis. J. Clin. Investig. 2014, 124, 2355–2363. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Chen, H.; Li, F.; Wang, S.; Guo, J. Hypoxia-induced microRNA-155 promotes fibrosis in proximal tubule cells. Mol. Med. Rep. 2015, 11, 4555–4560. [Google Scholar] [CrossRef] [PubMed]
- Lipphardt, M.; Song, J.W.; Ratliff, B.B.; Dihazi, H.; Müller, G.A.; Goligorsky, M.S. Endothelial dysfunction is a superinducer of syndecan-4: Fibrogenic role of its ectodomain. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H484–H496. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflug. Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef]
- Ni, H.; Chen, J.; Pan, M.; Zhang, M.; Zhang, J.; Chen, P.; Liu, B. FTY720 prevents progression of renal fibrosis by inhibiting renal microvasculature endothelial dysfunction in a rat model of chronic kidney disease. J. Mol. Histol. 2013, 44, 693–703. [Google Scholar] [CrossRef]
- Balint, B.; Jaremek, V.; Thorburn, V.; Whitehead, S.N.; Sposato, L.A. Left atrial microvascular endothelial dysfunction, myocardial inflammation and fibrosis after selective insular cortex ischemic stroke. Int. J. Cardiol. 2019, 292, 148–155. [Google Scholar] [CrossRef]
- Huby, A.C.; Antonova, G.; Groenendyk, J.; Gomez-Sanchez, C.E.; Bollag, W.B.; Filosa, J.A.; Belin de Chantemèle, E.J. Adipocyte-derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. Circulation 2015, 132, 2134–2145. [Google Scholar] [CrossRef]
- Chen, Z.W.; Tsai, C.H.; Pan, C.T.; Chou, C.H.; Liao, C.W.; Hung, C.S.; Wu, V.C.; Lin, Y.H.; TAIPAI Study Group. Endothelial dysfunction in primary aldosteronism. Int. J. Mol. Sci. 2019, 20, 5214. [Google Scholar] [CrossRef] [Green Version]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Lin, Y.C.; Sung, Y.K.; Jiang, X.; Peters-Golden, M.; Nicolls, M.R. Simultaneously targeting myofibroblast contractility and extracellular matrix cross-linking as a therapeutic concept in airway fibrosis. Am. J. Transplant. 2017, 17, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Follonier Castella, L.; Gabbiani, G.; McCulloch, C.A.; Hinz, B. Regulation of myofibroblast activities: Calcium pulls some strings behind the scene. Exp. Cell Res. 2010, 316, 2390–2401. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. The myofibroblast: Paradigm for a mechanically active cell. J. Biomech. 2010, 43, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Mendoza, F.A.; Jimenez, S.A. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of human fibrotic diseases. J. Clin. Med. 2016, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Meng, X.M.; Ng, Y.Y.; Ma, F.Y.; Zhou, S.; Zhang, Y.; Yang, C.; Huang, X.R.; Xiao, J.; Wang, Y.Y.; et al. TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 2016, 7, 8809–8822. [Google Scholar]
- Medici, D.; Kalluri, R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer. Biol. 2012, 22, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Chu, A.S.; Diaz, R.; Hui, J.J.; Yanger, K.; Zong, Y.; Alpini, G.; Stanger, B.Z.; Wells, R.G. Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology 2011, 53, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef] [Green Version]
- Burgy, O.; Königshoff, M. The WNT signaling pathways in wound healing and fibrosis. Matrix Biol. 2018, 68–69, 67–80. [Google Scholar] [CrossRef]
- Lee, W.J.; Park, J.H.; Shin, J.U.; Noh, H.; Lew, D.H.; Yang, W.I.; Yun, C.O.; Lee, K.H.; Lee, J.H. Endothelial-to-mesenchymal transition induced by Wnt 3a in keloid pathogenesis. Wound Repair Regen. 2015, 23, 435–442. [Google Scholar] [CrossRef]
- He, J.; Xu, Y.; Koya, D.; Kanasaki, K. Role of the endothelial-to-mesenchymal transition in renal fibrosis of chronic kidney disease. Clin. Exp. Nephrol. 2013, 17, 488–497. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 2008, 19, 2282–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essawy, M.; Soylemezoglu, O.; Muchaneta-Kubara, E.C.; Shortland, J.; Brown, C.B.; el Nahas, A.M. Myofibroblasts and the progression of diabetic nephropathy. Nephrol. Dial. Transplant. 1997, 12, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedagogos, E.; Hewitson, T.; Fraser, I.; Nicholls, K.; Becker, G. Myofibroblasts and arteriolar sclerosis in human diabetic nephropathy. Am. J. Kidney Dis. 1997, 29, 912–918. [Google Scholar] [CrossRef]
- Li, J.; Qu, X.; Bertram, J.F. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 2009, 175, 1380–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, M.; Maezawa, Y.; Yokote, K.; Joh, K.; Kobayashi, K.; Kawamura, H.; Nishimura, M.; Roberts, A.B.; Saito, Y.; Mori, S. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem. Biophys. Res. Commun. 2003, 305, 1002–1007. [Google Scholar] [CrossRef]
- Li, J.; Qu, X.; Yao, J.; Caruana, G.; Ricardo, S.D.; Yamamoto, Y.; Yamamoto, H.; Bertram, J.F. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 2612–2624. [Google Scholar] [CrossRef] [Green Version]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life. Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [Green Version]
- Widyantoro, B.; Emoto, N.; Nakayama, K.; Anggrahini, D.W.; Adiarto, S.; Iwasa, N.; Yagi, K.; Miyagawa, K.; Rikitake, Y.; Suzuki, T.; et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 2010, 121, 2407–2418. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Tang, H.; Mao, J.; Ye, X.; Zhang, F.; Kerr, W.G.; Zheng, T.; Zhu, Z. SHIP-1, a target of miR-155, regulates endothelial cell responses in lung fibrosis. FASEB J. 2020, 34, 2011–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribera, J.; Pauta, M.; Melgar-Lesmes, P.; Córdoba, B.; Bosch, A.; Calvo, M.; Rodrigo-Torres, D.; Sancho-Bru, P.; Mira, A.; Jiménez, W.; et al. A small population of liver endothelial cells undergoes endothelial-to-mesenchymal transition in response to chronic liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G492–G504. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallone, T.L.; Silldorff, E.P. Pericyte regulation of renal medullary blood flow. Exp. Nephrol. 2001, 9, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Peppiatt, C.M.; Howarth, C.; Mobbs, P.; Attwell, D. Bidirectional control of CNS capillary diameter by pericytes. Nature 2006, 443, 700–704. [Google Scholar] [CrossRef] [Green Version]
- Stratman, A.N.; Schwindt, A.E.; Malotte, K.M.; Davis, G.E. Endothelial-derived PDGF-BB and HB-EGF coordinately regulate pericyte recruitment during vasculogenic tube assembly and stabilization. Blood 2010, 116, 4720–4730. [Google Scholar] [CrossRef] [Green Version]
- Darby, I.A.; Hewitson, T.D. Hypoxia in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 553–562. [Google Scholar] [CrossRef]
- Kischer, C.W.; Thies, A.C.; Chvapil, M. Perivascular myofibroblasts and microvascular occlusion in hypertrophic scars and keloids. Hum. Pathol. 1982, 13, 819–824. [Google Scholar] [CrossRef]
- Dulauroy, S.; Di Carlo, S.E.; Langa, F.; Eberl, G.; Peduto, L. Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat. Med. 2012, 18, 1262–1270. [Google Scholar] [CrossRef]
- Sundberg, C.; Ivarsson, M.; Gerdin, B.; Rubin, K. Pericytes as collagen-producing cells in excessive dermal scarring. Lab. Investig. 1996, 74, 452–466. [Google Scholar]
- Kawakami, T.; Mimura, I.; Shoji, K.; Tanaka, T.; Nangaku, M. Hypoxia and fibrosis in chronic kidney disease: Crossing at pericytes. Kidney Int. Suppl. 2014, 4, 107–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, I.G.; Duffield, J.S. The FOXD1 lineage of kidney perivascular cells and myofibroblasts: Functions and responses to injury. Kidney Int. Suppl. 2014, 4, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campanholle, G.; Ligresti, G.; Gharib, S.A.; Duffield, J.S. Cellular mechanisms of tissue fibrosis. 3. Novel mechanisms of kidney fibrosis. Am. J. Physiol. Cell Physiol. 2013, 304, C591–C603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asada, N.; Takase, M.; Nakamura, J.; Oguchi, A.; Asada, M.; Suzuki, N.; Yamamura, K.; Nagoshi, N.; Shibata, S.; Rao, T.N.; et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J. Clin. Investig. 2011, 121, 3981–3990. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.Y.; Chang, Y.T.; Lin, S.L. Microvascular pericytes in healthy and diseased kidneys. Int. J. Nephrol. Renovasc. Dis. 2014, 7, 39–48. [Google Scholar]
- Hung, C.; Linn, G.; Chow, Y.H.; Kobayashi, A.; Mittelsteadt, K.; Altemeier, W.A.; Gharib, S.A.; Schnapp, L.M.; Duffield, J.S. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 188, 820–830. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Deleve, L.D.; Wang, X.; Guo, Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology 2008, 48, 920–930. [Google Scholar] [CrossRef]
- Liu, L.; You, Z.; Yu, H.; Zhou, L.; Zhao, H.; Yan, X.; Li, D.; Wang, B.; Zhu, L.; Xu, Y.; et al. Mechanotransduction-modulated fibrotic microniches reveal the contribution of angiogenesis in liver fibrosis. Nat. Mater. 2017, 16, 1252–1261. [Google Scholar] [CrossRef]
- Ford, A.J.; Jain, G.; Rajagopalan, P. Designing a fibrotic microenvironment to investigate changes in human liver sinusoidal endothelial cell function. Acta Biomater. 2015, 24, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Marrone, G.; Maeso-Díaz, R.; García-Cardena, G.; Abraldes, J.G.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: Behind the molecular mechanisms of statins. Gut 2015, 64, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Tangkijvanich, P.; Kongtawelert, P.; Pothacharoen, P.; Mahachai, V.; Suwangool, P.; Poovorawan, Y. Serum hyaluronan: A marker of liver fibrosis in patients with chronic liver disease. Asian Pac. J. Allergy Immunol. 2003, 21, 115–120. [Google Scholar] [PubMed]
- Rojas, A.; Chang, F.C.; Lin, S.L.; Duffield, J.S. The role played by perivascular cells in kidney interstitial injury. Clin. Nephrol. 2012, 77, 400–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucala, R.; Spiegel, L.A.; Chesney, J.; Hogan, M.; Cerami, A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med. 1994, 1, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, R.; Donnelly, S.C.; Peng, T.; Bucala, R.; Metz, C.N. Peripheral blood fibrocytes: Differentiation pathway and migration to wound sites. J. Immunol. 2001, 166, 7556–7562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, T.E.; Cowper, S.; Wu, S.P.; Bockenstedt, L.K.; Bucala, R. Circulating fibrocytes: Collagen-secreting cells of the peripheral blood. Int. J. Biochem. Cell Biol. 2004, 36, 598–606. [Google Scholar] [CrossRef]
- Pilling, D.; Fan, T.; Huang, D.; Kaul, B.; Gomer, R.H. Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts. PLoS ONE 2009, 4, e7475. [Google Scholar] [CrossRef]
- Phillips, R.J.; Burdick, M.D.; Hong, K.; Lutz, M.A.; Murray, L.A.; Xue, Y.Y.; Belperio, J.A.; Keane, M.P.; Strieter, R.M. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Investig. 2004, 114, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Kao, H.K.; Chen, B.; Murphy, G.F.; Li, Q.; Orgill, D.P.; Guo, L. Peripheral blood fibrocytes: Enhancement of wound healing by cell proliferation, re-epithelialization, contraction, and angiogenesis. Ann. Surg. 2011, 254, 1066–1074. [Google Scholar] [CrossRef]
- Thevenot, P.T.; Baker, D.W.; Weng, H.; Sun, M.W.; Tang, L. The pivotal role of fibrocytes and mast cells in mediating fibrotic reactions to biomaterials. Biomaterials 2011, 32, 8394–8403. [Google Scholar] [CrossRef] [Green Version]
- Mathangi Ramakrishnan, K.; Meenakshi Janakiraman, M.; Babu, M. Expression of fibrocyte markers by keloid fibroblasts: An insight into fibrosis during burn wound healing—A preliminary study. Ann. Burns Fire Disasters 2012, 25, 148–151. [Google Scholar] [PubMed]
- Hong, K.M.; Belperio, J.A.; Keane, M.P.; Burdick, M.D.; Strieter, R.M. Differentiation of human circulating fibrocytes as mediated by transforming growth factor-beta and peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2007, 282, 22910–22920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.W.; Tsai, Y.T.; Weng, H.; Tang, L. Alternative strategies to manipulate fibrocyte involvement in the fibrotic tissue response: Pharmacokinetic inhibition and the feasibility of directed-adipogenic differentiation. Acta Biomater. 2014, 10, 3108–3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, T.E.; Mino, M.J.; Moffatt, L.T.; Mauskar, N.A.; Prindeze, N.J.; Ghassemi, P.; Ramella-Roman, J.C.; Jordan, M.H.; Shupp, J.W. Biphasic presence of fibrocytes in a porcine hypertrophic scar model. J. Burn Care Res. 2015, 36, e125–e135. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Scott, P.G.; Dodd, C.; Medina, A.; Jiao, H.; Shankowsky, H.A.; Ghahary, A.; Tredget, E.E. Identification of fibrocytes in postburn hypertrophic scar. Wound Repair Regen. 2005, 13, 398–404. [Google Scholar] [CrossRef]
- Iqbal, S.A.; Sidgwick, G.P.; Bayat, A. Identification of fibrocytes from mesenchymal stem cells in keloid tissue: A potential source of abnormal fibroblasts in keloid scarring. Arch. Dermatol. Res. 2012, 304, 665–671. [Google Scholar] [CrossRef]
- Andersson-Sjöland, A.; de Alba, C.G.; Nihlberg, K.; Becerril, C.; Ramírez, R.; Pardo, A.; Westergren-Thorsson, G.; Selman, M. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2008, 40, 2129–2140. [Google Scholar] [CrossRef]
- Phan, S.H. Fibroblast phenotypes in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2003, 29, S87–S92. [Google Scholar]
- Inomata, M.; Kamio, K.; Azuma, A.; Matsuda, K.; Kokuho, N.; Miura, Y.; Hayashi, H.; Nei, T.; Fujita, K.; Saito, Y.; et al. Pirfenidone inhibits fibrocyte accumulation in the lungs in bleomycin-induced murine pulmonary fibrosis. Respir. Res. 2014, 15, 16. [Google Scholar] [CrossRef] [Green Version]
- Kisseleva, T.; Uchinami, H.; Feirt, N.; Quintana-Bustamante, O.; Segovia, J.C.; Schwabe, R.F.; Brenner, D.A. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J. Hepatol. 2006, 45, 429–438. [Google Scholar] [CrossRef]
- Xu, J.; Cong, M.; Park, T.J.; Scholten, D.; Brenner, D.A.; Kisseleva, T. Contribution of bone marrow-derived fibrocytes to liver fibrosis. Hepatobiliary Surg. Nutr. 2015, 4, 34–47. [Google Scholar] [PubMed]
- Hempel, F.; Roderfeld, M.; Savai, R.; Sydykov, A.; Irungbam, K.; Schermuly, R.; Voswinckel, R.; Köhler, K.; Churin, Y.; Kiss, L.; et al. Depletion of Bone Marrow-Derived Fibrocytes Attenuates TAA-Induced Liver Fibrosis in Mice. Cells 2019, 8, 1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozono, Y.; Shide, K.; Toyoshima, F.; Takaishi, Y.; Tsuchimochi, M.; Kamiunten, A.; Kameda, T.; Nakamura, K.; Miike, T.; Kusumoto, K.; et al. Monocyte-derived fibrocytes elimination had little contribution on liver fibrosis. Hepatobiliary Pancreat. Dis. Int. 2019, 18, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Sopel, M.; Falkenham, A.; Oxner, A.; Ma, I.; Lee, T.D.; Légaré, J.F. Fibroblast progenitor cells are recruited into the myocardium prior to the development of myocardial fibrosis. Int. J. Exp. Pathol. 2012, 93, 115–124. [Google Scholar] [CrossRef]
- Xu, J.; Lin, S.C.; Chen, J.; Miao, Y.; Taffet, G.E.; Entman, M.L.; Wang, Y. CCR2 mediates the uptake of bone marrow-derived fibroblast precursors in angiotensin II-induced cardiac fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H538–H547. [Google Scholar] [CrossRef]
- Sakai, N.; Wada, T.; Matsushima, K.; Bucala, R.; Iwai, M.; Horiuchi, M.; Kaneko, S. The renin-angiotensin system contributes to renal fibrosis through regulation of fibrocytes. J. Hypertens 2008, 26, 780–790. [Google Scholar] [CrossRef]
- Xu, W.; Koeck, T.; Lara, A.R.; Neumann, D.; DiFilippo, F.P.; Koo, M.; Janocha, A.J.; Masri, F.A.; Arroliga, A.C.; Jennings, C.; et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc. Natl. Acad. Sci. USA 2007, 104, 1342–1347. [Google Scholar] [CrossRef] [Green Version]
- Sakao, S.; Hao, H.; Tanabe, N.; Kasahara, Y.; Kurosu, K.; Tatsumi, K. Endothelial-like cells in chronic thromboembolic pulmonary hypertension: Crosstalk with myofibroblast-like cells. Respir. Res. 2011, 12, 109. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wang, M.; Zhu, F.; Sun, J.; Xu, H.; Chong Lee Shin, O.L.; Zhao, Z.; Pei, G.; Zhu, H.; Cao, C.; et al. Putative endothelial progenitor cells do not promote vascular repair but attenuate pericyte-myofibroblast transition in UUO-induced renal fibrosis. Stem Cell Res. Ther. 2019, 10, 104. [Google Scholar] [CrossRef]
- Miyazaki, T.; Haraguchi, S.; Kim-Kaneyama, J.R.; Miyazaki, A. Endothelial calpain systems orchestrate myofibroblast differentiation during wound healing. FASEB J. 2019, 33, 2037–2046. [Google Scholar] [CrossRef]
- Nieves Torres, E.C.; Yang, B.; Brahmbhatt, A.; Mukhopadhyay, D.; Misra, S. Blood outgrowth endothelial cells reduce hypoxia-mediated fibroblast to myofibroblast conversion by decreasing proangiogenic cytokines. J. Vasc. Res. 2014, 51, 458–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruart, M.; Chavarria, L.; Campreciós, G.; Suárez-Herrera, N.; Montironi, C.; Guixé-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagán, J.C.; Hernández-Gea, V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J. Hepatol. 2019, 70, 458–469. [Google Scholar] [CrossRef]
- Zhou, T.; Zheng, Y.; Sun, L.; Badea, S.R.; Jin, Y.; Liu, Y.; Rolfe, A.J.; Sun, H.; Wang, X.; Cheng, Z.; et al. Microvascular endothelial cells engulf myelin debris and promote macrophage recruitment and fibrosis after neural injury. Nat. Neurosci. 2019, 22, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y. Endothelial glycocalyx as a critical signalling platform integrating the extracellular haemodynamic forces and chemical signalling. J. Cell. Mol. Med. 2017, 21, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Takemura, G.; Suzuki, K.; Oda, K.; Takada, C.; Hotta, Y.; Miyazaki, N.; Tsujimoto, A.; Muraki, I.; Ando, Y.; et al. Three-dimensional ultrastructure of capillary endothelial glycocalyx under normal and experimental endotoxemic conditions. Crit. Care 2017, 21, 261. [Google Scholar] [CrossRef] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.; Ogawa, R. The Vascular Involvement in Soft Tissue Fibrosis—Lessons Learned from Pathological Scarring. Int. J. Mol. Sci. 2020, 21, 2542. https://doi.org/10.3390/ijms21072542
Huang C, Ogawa R. The Vascular Involvement in Soft Tissue Fibrosis—Lessons Learned from Pathological Scarring. International Journal of Molecular Sciences. 2020; 21(7):2542. https://doi.org/10.3390/ijms21072542
Chicago/Turabian StyleHuang, Chenyu, and Rei Ogawa. 2020. "The Vascular Involvement in Soft Tissue Fibrosis—Lessons Learned from Pathological Scarring" International Journal of Molecular Sciences 21, no. 7: 2542. https://doi.org/10.3390/ijms21072542
APA StyleHuang, C., & Ogawa, R. (2020). The Vascular Involvement in Soft Tissue Fibrosis—Lessons Learned from Pathological Scarring. International Journal of Molecular Sciences, 21(7), 2542. https://doi.org/10.3390/ijms21072542