Inducible Polarized Secretion of Exosomes in T and B Lymphocytes


Abstract
:1. Introduction
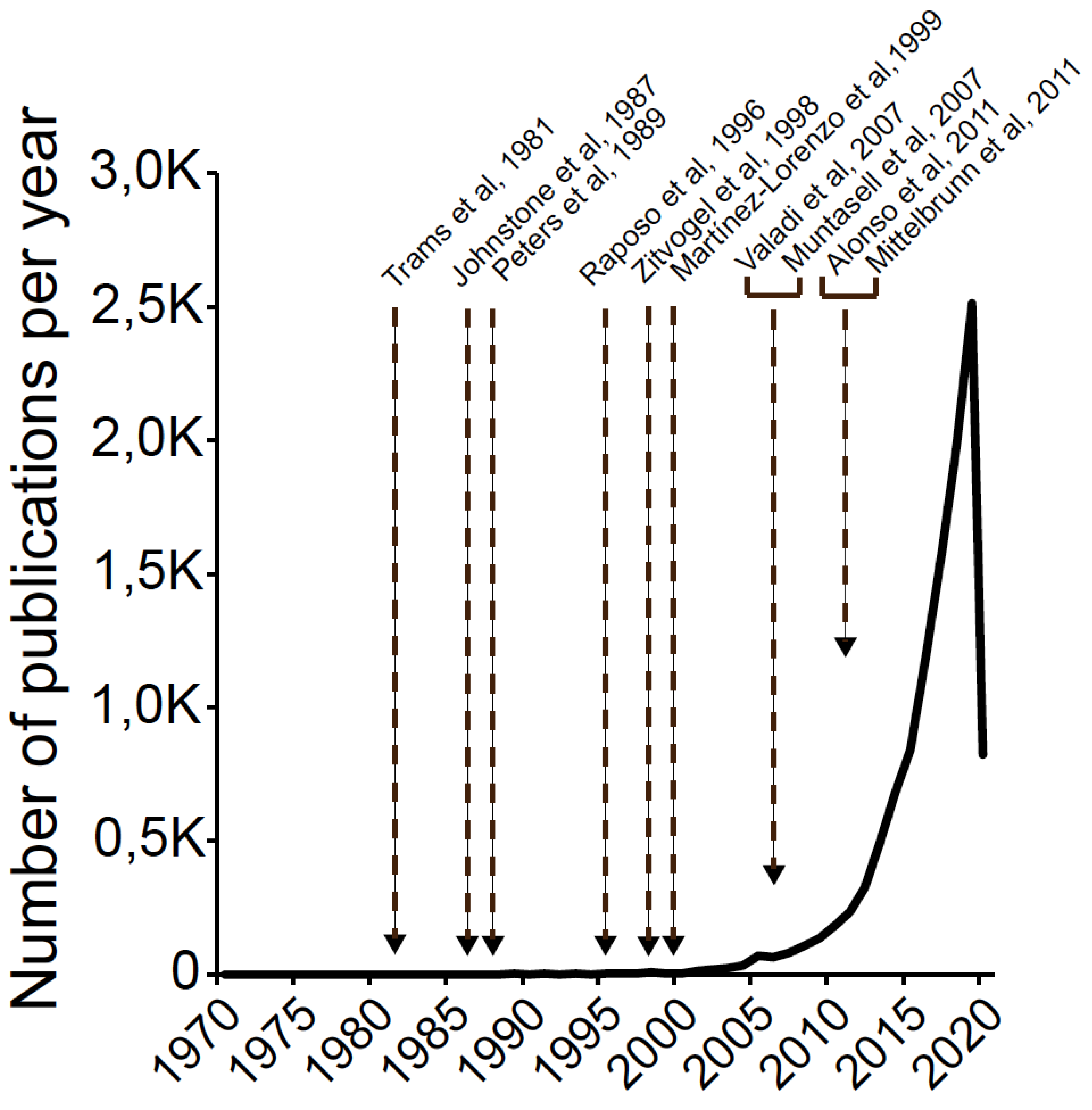
1.1. A Brief History of Exosomes: Exosome Timeline and Relevant Facts
1.2. Immune Synapse and Secretory Traffic
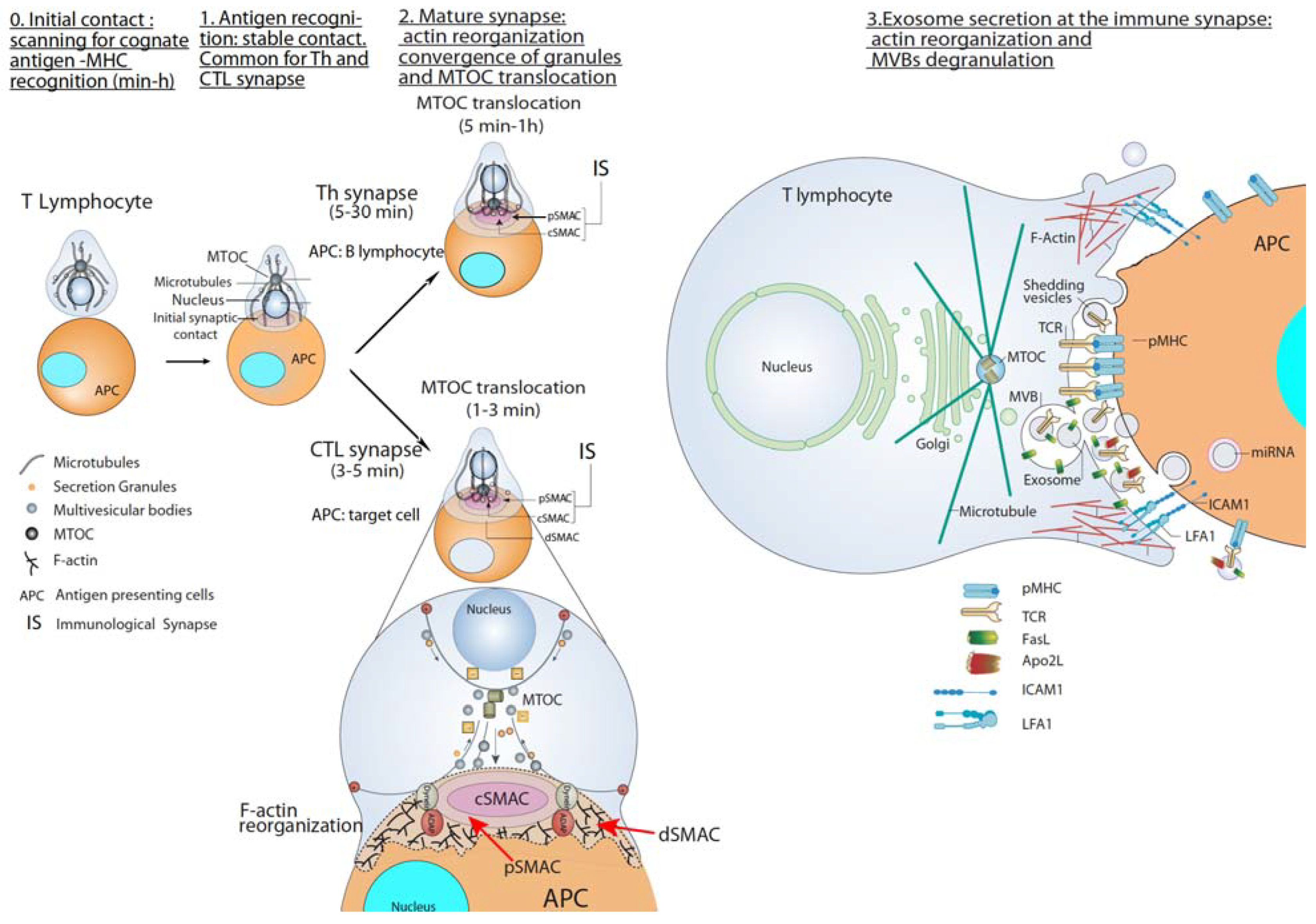
2. T Lymphocyte–APC Immune Synapse: Exosome Secretion by T Lymphocytes
2.1. CTL-Target Cell Immune Synapse
2.2. Th Immune Synapse
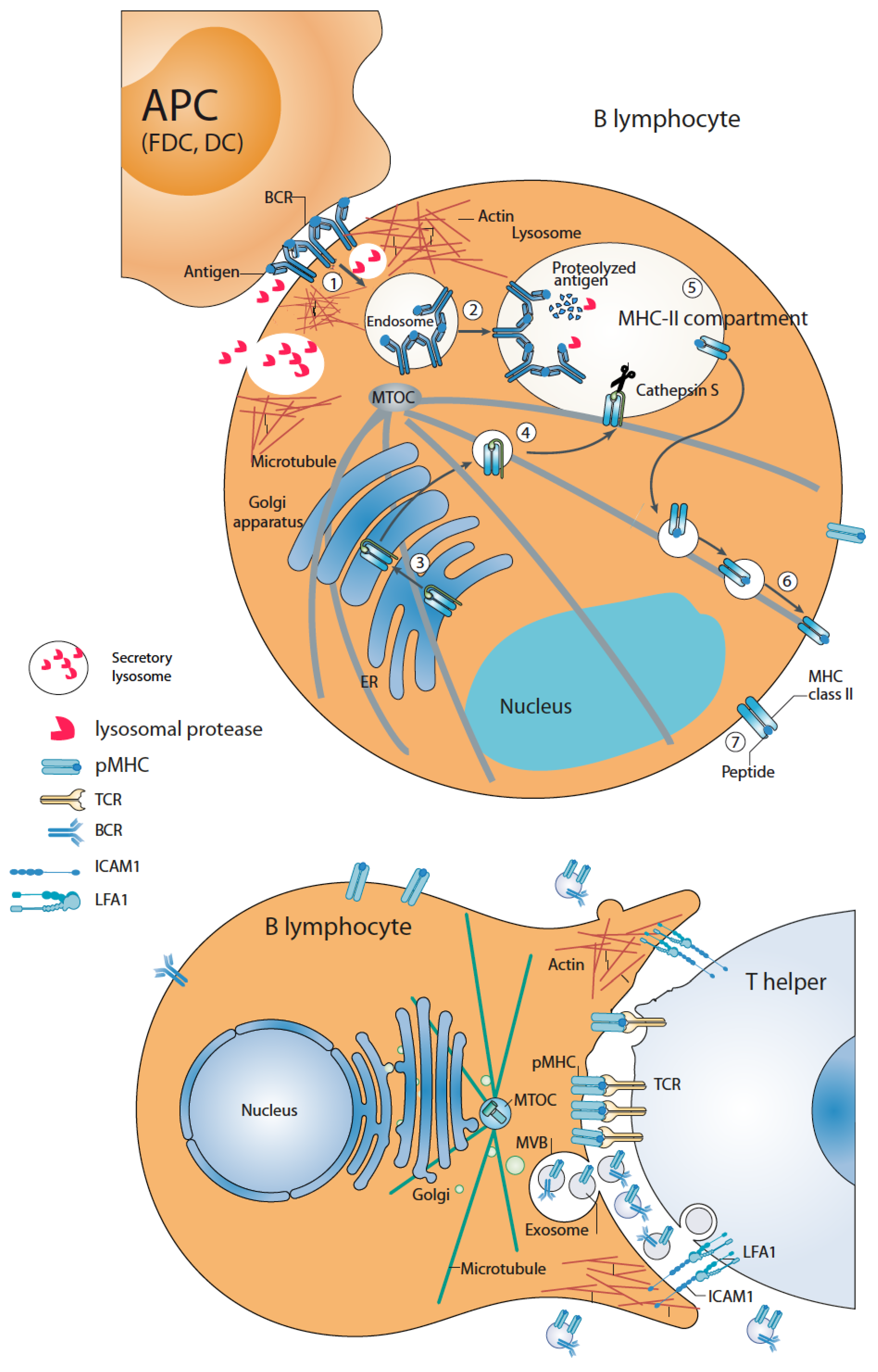
3. B Lymphocyte-APC Immune Synapse: Antigen Capture and Processing by B Lymphocytes
4. B Lymphocyte–Th Immune Synapse: Antigen Presentation and Exosome Secretion by B Lymphocytes
5. Immune Regulation by B and T Lymphocyte-Produced Exosomes
5.1. Role of B-Lymphocyte Produced Exosomes in Antigen Presentation and Immunoregulation
5.2. Role of T Lymphocyte-Produced Exosomes on Apoptosis and Immunoregulation
6. Some Potential Clinical Applications of Immune Cells-Derived Exosomes
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ab | Antibody |
| ADAP | Adhesion and Degranulation Promoting Adapter Protein |
| AICD | Activation-induced cell death |
| APC | Antigen-presenting cell |
| Apo2L | Apo2 ligand (TRAIL) |
| BCR | B-cell receptor for antigen |
| CAR | Chimeric Antigen Receptors |
| cIS | Central region of the immune synapse |
| cSMAC | Central supramolecular activation cluster |
| DC | Dendritic cells |
| CTL | Cytotoxic T lymphocytes |
| DAG | Diacylglycerol |
| DGKα | Diacylglycerol kinase α |
| dSMAC | Distal supramolecular activation cluster |
| EM | Electron microscopy |
| ESCRT | Endosomal sorting complex required for traffic |
| EV | Extracellular vesicle |
| F-actin | Filamentous actin |
| Fact-low | F-actin-low region at the centre of the immune synapse |
| FasL | Fas ligand |
| FDC | Follicular Dendritic cells |
| ILV | Intraluminal vesicles |
| IS | Immune synapse |
| MHC | Major histocompatibility complex |
| miRNA | MicroRNA |
| MVB | Multivesicular bodies |
| MTOC | Microtubule-organizing center |
| NK | Natural killer cell |
| OVA | Ovalbumin |
| PBL | Peripheral blood lymphocytes |
| PKC | Protein kinase C |
| PKCδ | Protein kinase C δ isoform |
| PLC | Phospholipase C |
| pMHC | Peptide/MHC complex |
| pSMAC | Peripheral supramolecular activation cluster |
| SL | Secretory lysosomes |
| SLE | Systemic lupus erythematosus |
| SMAC | Supramolecular activation cluster |
| TCR | T-cell receptor for antigen |
| Th | T helper |
| TLR | Toll-like receptors |
| Treg | T regulatory cells |
References
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [PubMed]
- Johnstone, R.M. The Jeanne Manery-Fisher Memorial Lecture 1991. Maturation of reticulocytes: formation of exosomes as a mechanism for shedding membrane proteins. Biochem. Cell Biol. 1992, 70, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Babst, M. MVB vesicle formation: ESCRT-dependent, ESCRT-independent and everything in between. Curr. Opin. Cell Biol. 2011, 23, 452–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, C.C.; Vacca, F.; Gruenberg, J. Endosome maturation, transport and functions. Semin. Cell Dev. Biol. 2014, 31, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Geuze, H.J. The role of endosomes and lysosomes in MHC class II functioning. Immunol. Today 1998, 19, 282–287. [Google Scholar] [CrossRef]
- Denzer, K.; Kleijmeer, M.J.; Heijnen, H.F.; Stoorvogel, W.; Geuze, H.J. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J. Cell Sci. 2000, 113, 3365–3374. [Google Scholar] [PubMed]
- Pan, B.T.; Teng, K.; Wu, C.; Adam, M.; Johnstone, R.M. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J. Cell Biol. 1985, 101, 942–948. [Google Scholar] [CrossRef] [Green Version]
- Trams, E.G.; Lauter, C.J.; Salem, N. Jr.; Heine, U. Exfoliation of membrane ecto-enzymes in the form of micro-vesicles. Biochim. Biophys. Acta 1981, 645, 63–70. [Google Scholar] [CrossRef]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef]
- Martinez-Lorenzo, M.J.; Anel, A.; Gamen, S.; Monle n, I.; Lasierra, P.; Larrad, L.; Pineiro, A.; Alava, M.A.; Naval, J. Activated human T cells release bioactive Fas ligand and APO2 ligand in microvesicles. J. Immunol. 1999, 163, 1274–1281. [Google Scholar]
- Monleon, I.; Martinez-Lorenzo, M.J.; Monteagudo, L.; Lasierra, P.; Taules, M.; Iturralde, M.; Pineiro, A.; Larrad, L.; Alava, M.A.; Naval, J.; et al. Differential secretion of Fas ligand- or APO2 ligand/TNF-related apoptosis-inducing ligand-carrying microvesicles during activation-induced death of human T cells. J. Immunol. 2001, 167, 6736–6744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Mazzeo, C.; Rodriguez, M.C.; Marsh, M.; Fraile-Ramos, A.; Calvo, V.; Avila-Flores, A.; Merida, I.; Izquierdo, M. Diacylglycerol kinase alpha regulates the formation and polarisation of mature multivesicular bodies involved in the secretion of Fas ligand-containing exosomes in T lymphocytes. Cell Death Differ. 2011, 18, 1161–1173. [Google Scholar] [CrossRef] [Green Version]
- Muntasell, A.; Berger, A.C.; Roche, P.A. T cell-induced secretion of MHC class II-peptide complexes on B cell exosomes. EMBO J. 2007, 26, 4263–4272. [Google Scholar] [CrossRef] [Green Version]
- Peters, P.J.; Geuze, H.J.; Van der Donk, H.A.; Slot, J.W.; Griffith, J.M.; Stam, N.J.; Clevers, H.C.; Borst, J. Molecules relevant for T cell-target cell interaction are present in cytolytic granules of human T lymphocytes. Eur. J. Immunol. 1989, 19, 1469–1475. [Google Scholar] [CrossRef] [Green Version]
- Mittelbrunn, M.; Gutierrez-Vazquez, C.; Villarroya-Beltri, C.; Gonzalez, S.; Sanchez-Cabo, F.; Gonzalez, M.A.; Bernad, A.; Sanchez-Madrid, F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011, 2, 282. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Vazquez, C.; Villarroya-Beltri, C.; Mittelbrunn, M.; Sanchez-Madrid, F. Transfer of extracellular vesicles during immune cell-cell interactions. Immunol. Rev. 2013, 251, 125–142. [Google Scholar] [CrossRef] [Green Version]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Lotvall, J.; Hill, A.F.; Hochberg, F.; Buzas, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Soekmadji, C.; Hill, A.F.; Wauben, M.H.; Buzas, E.I.; Di Vizio, D.; Falcon-Perez, J.M.; Gardiner, C.; Hochberg, F.; Kurochkin, I.V.; et al. Updating the MISEV minimal requirements for extracellular vesicle studies: building bridges to reproducibility. J. Extracell. Vesicles 2017, 6, 1396823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Li, Q.; Wang, H.; Peng, H.; Huyan, T.; Cacalano, N.A. Exosomes: Versatile Nano Mediators of Immune Regulation. Cancers 2019, 11, 1557. [Google Scholar] [CrossRef] [Green Version]
- Willms, E.; Johansson, H.J.; Mager, I.; Lee, Y.; Blomberg, K.E.; Sadik, M.; Alaarg, A.; Smith, C.I.; Lehtio, J.; El Andaloussi, S.; et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep. 2016, 6, 22519. [Google Scholar] [CrossRef]
- van der Vlist, E.J.; Nolte, E.N.; Stoorvogel, W.; Arkesteijn, G.J.; Wauben, M.H. Fluorescent labeling of nano-sized vesicles released by cells and subsequent quantitative and qualitative analysis by high-resolution flow cytometry. Nat. Protoc. 2012, 7, 1311–1326. [Google Scholar] [CrossRef]
- Fooksman, D.R.; Vardhana, S.; Vasiliver-Shamis, G.; Liese, J.; Blair, D.A.; Waite, J.; Sacristan, C.; Victora, G.D.; Zanin-Zhorov, A.; Dustin, M.L. Functional anatomy of T cell activation and synapse formation. Annu. Rev. Immunol. 2010, 28, 79–105. [Google Scholar] [CrossRef] [Green Version]
- de la Roche, M.; Asano, Y.; Griffiths, G.M. Origins of the cytolytic synapse. Nat. Rev. Immunol. 2016, 16, 421–432. [Google Scholar] [CrossRef]
- Griffiths, G.M.; Tsun, A.; Stinchcombe, J.C. The immunological synapse: a focal point for endocytosis and exocytosis. J. Cell Biol. 2010, 189, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Billadeau, D.D.; Nolz, J.C.; Gomez, T.S. Regulation of T-cell activation by the cytoskeleton. Nat. Rev. Immunol. 2007, 7, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Kuokkanen, E.; Sustar, V.; Mattila, P.K. Molecular control of B cell activation and immunological synapse formation. Traffic 2015, 16, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Yuseff, M.I.; Pierobon, P.; Reversat, A.; Lennon-Dumenil, A.M. How B cells capture, process and present antigens: a crucial role for cell polarity. Nat. Rev. Immunol. 2013, 13, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Monks, C.R.F.; Freiberg, B.A.; Kupfer, H.; Sciaky, N.; Kupfer, A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 1998, 395, 82–86. [Google Scholar] [CrossRef]
- Le Floc’h, A.; Huse, M. Molecular mechanisms and functional implications of polarized actin remodeling at the T cell immunological synapse. Cell. Mol. Life Sci. 2015, 72, 537–556. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Tato, C.M.; Davis, M.M. How the immune system talks to itself: the varied role of synapses. Immunol. Rev. 2013, 251, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Yuseff, M.I.; Lankar, D.; Lennon-Dumenil, A.M. Dynamics of membrane trafficking downstream of B and T cell receptor engagement: impact on immune synapses. Traffic 2009, 10, 629–636. [Google Scholar] [CrossRef] [Green Version]
- Ritter, A.T.; Angus, K.L.; Griffiths, G.M. The role of the cytoskeleton at the immunological synapse. Immunol. Rev. 2013, 256, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yin, W.; Jing, Y.; Kang, D.; Yang, L.; Cheng, J.; Yu, Z.; Peng, Z.; Li, X.; Wen, Y.; et al. The Coordination Between B Cell Receptor Signaling and the Actin Cytoskeleton During B Cell Activation. Front. Immunol. 2018, 9, 3096. [Google Scholar] [CrossRef] [Green Version]
- Wurzer, H.; Hoffmann, C.; Al Absi, A.; Thomas, C. Actin Cytoskeleton Straddling the Immunological Synapse between Cytotoxic Lymphocytes and Cancer Cells. Cells 2019, 8, 463. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo, M.; Cantrell, D.A. T-cell activation. Trends Cell Biol. 1992, 2, 268–271. [Google Scholar] [CrossRef]
- Spitaler, M.; Emslie, E.; Wood, C.D.; Cantrell, D. Diacylglycerol and protein kinase D localization during T lymphocyte activation. Immunity 2006, 24, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühn, S.; Geyer, M. Formins as effector proteins of Rho GTPases. Small GTPases 2014, 5, e29513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.; Curado, S.; Mayya, V.; Dustin, M.L. T cell antigen receptor activation and actin cytoskeleton remodeling. Biochim. Biophys. Acta 2014, 1838. [Google Scholar] [CrossRef] [Green Version]
- Stinchcombe, J.C.; Majorovits, E.; Bossi, G.; Fuller, S.; Griffiths, G.M. Centrosome polarization delivers secretory granules to the immunological synapse. Nature 2006, 443, 462–465. [Google Scholar] [CrossRef]
- Ritter, A.T.; Asano, Y.; Stinchcombe, J.C.; Dieckmann, N.M.; Chen, B.C.; Gawden-Bone, C.; van Engelenburg, S.; Legant, W.; Gao, L.; Davidson, M.W.; et al. Actin depletion initiates events leading to granule secretion at the immunological synapse. Immunity 2015, 42, 864–876. [Google Scholar] [CrossRef] [Green Version]
- Carisey, A.F.; Mace, E.M.; Saeed, M.B.; Davis, D.M.; Orange, J.S. Nanoscale Dynamism of Actin Enables Secretory Function in Cytolytic Cells. Curr. Biol. 2018, 28, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Tamzalit, F.; Wang, M.S.; Jin, W.; Tello-Lafoz, M.; Boyko, V.; Heddleston, J.M.; Black, C.T.; Kam, L.C.; Huse, M. Interfacial actin protrusions mechanically enhance killing by cytotoxic T cells. Sci. Immunol. 2019. [Google Scholar] [CrossRef]
- Chemin, K.; Bohineust, A.; Dogniaux, S.; Tourret, M.; Guegan, S.; Miro, F.; Hivroz, C. Cytokine secretion by CD4+ T cells at the immunological synapse requires Cdc42-dependent local actin remodeling but not microtubule organizing center polarity. J. Immunol. 2012, 189, 2159–2168. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, E.; Liu, X.; Huse, M. Actin clearance promotes polarized dynein accumulation at the immunological synapse. PLoS ONE 2019, 14, e0210377. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Kapoor, T.M.; Chen, J.K.; Huse, M. Diacylglycerol promotes centrosome polarization in T cells via reciprocal localization of dynein and myosin II. Proc. Natl. Acad. Sci. USA 2013, 110, 11976–11981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combs, J.; Kim, S.J.; Tan, S.; Ligon, L.A.; Holzbaur, E.L.; Kuhn, J.; Poenie, M. Recruitment of dynein to the Jurkat immunological synapse. Proc. Natl. Acad. Sci. USA 2006, 103, 14883–14888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huse, M. Microtubule-organizing center polarity and the immunological synapse: protein kinase C and beyond. Front. Immunol. 2012, 3, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, H.; Zhou, J.; Xie, J.; Davis, M.M. Distinct Roles of Cytoskeletal Components in Immunological Synapse Formation and Directed Secretion. J. Immunol. 2015, 195, 4117–4125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, S.; Christian, L.; Tan, S.Y.; Ki, S.; Ehrlich, L.I.R.; Poenie, M. Dynein Separately Partners with NDE1 and Dynactin To Orchestrate T Cell Focused Secretion. J. Immunol. 2016, 197, 2090–2101. [Google Scholar] [CrossRef]
- Mentlik, A.N.; Sanborn, K.B.; Holzbaur, E.L.; Orange, J.S. Rapid Lytic Granule Convergence to the MTOC in Natural Killer Cells Is Dependent on Dynein But Not Cytolytic Commitment. Mol. Biol. Cell 2010, 21, 2241–2256. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.-T.; Mace, E.M.; Carisey, A.F.; Viswanath, D.I.; Christakou, A.E.; Wiklund, M.; Önfelt, B.; Orange, J.S. NK cells converge lytic granules to promote cytotoxicity and prevent bystander killing. J. Cell Biol. 2016, 215, 875–889. [Google Scholar] [CrossRef]
- de Saint Basile, G.; Menasche, G.; Fischer, A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat. Rev. Immunol. 2010, 10, 568–579. [Google Scholar] [CrossRef]
- Mazzeo, C.; Calvo, V.; Alonso, R.; Merida, I.; Izquierdo, M. Protein kinase D1/2 is involved in the maturation of multivesicular bodies and secretion of exosomes in T and B lymphocytes. Cell Death Differ. 2016, 23, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Carroll-Portillo, A.; Surviladze, Z.; Cambi, A.; Lidke, D.S.; Wilson, B.S. Mast cell synapses and exosomes: membrane contacts for information exchange. Front. Immunol. 2012, 3, 46. [Google Scholar] [CrossRef] [Green Version]
- Calvo, V.; Izquierdo, M. Imaging Polarized Secretory Traffic at the Immune Synapse in Living T Lymphocytes. Front. Immunol. 2018, 9, 684. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; den Boer, A.T.; Gunzer, M. Tuning immune responses: diversity and adaptation of the immunological synapse. Nat. Rev. Immunol. 2005, 5, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Peters, P.J.; Borst, J.; Oorschot, V.; Fukuda, M.; Krahenbuhl, O.; Tschopp, J.; Slot, J.W.; Geuze, H.J. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J. Exp. Med. 1991, 173, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Vignaux, F.; Vivier, E.; Malissen, B.; Depraetere, V.; Nagata, S.; Golstein, P. TCR/CD3 coupling to Fas-based cytotoxicity. J. Exp. Med. 1995, 181, 781–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossi, G.; Griffiths, G.M. Degranulation plays an essential part in regulating cell surface expression of Fas ligand in T cells and natural killer cells. Nat. Med. 1999, 5, 90–96. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis by death factor. Cell 1997, 88, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Alonso, R.; Rodriguez, M.C.; Pindado, J.; Merino, E.; Merida, I.; Izquierdo, M. Diacylglycerol kinase alpha regulates the secretion of lethal exosomes bearing Fas ligand during activation-induced cell death of T lymphocytes. J. Biol. Chem. 2005, 280, 28439–28450. [Google Scholar] [CrossRef] [Green Version]
- Blanchard, N.; Lankar, D.; Faure, F.; Regnault, A.; Dumont, C.; Raposo, G.; Hivroz, C. TCR activation of human T cells induces the production of exosomes bearing the TCR/CD3/zeta complex. J. Immunol. 2002, 168, 3235–3241. [Google Scholar] [CrossRef] [Green Version]
- Quann, E.J.; Merino, E.; Furuta, T.; Huse, M. Localized diacylglycerol drives the polarization of the microtubule-organizing center in T cells. Nat. Immunol. 2009, 10, 627–635. [Google Scholar] [CrossRef]
- Sanjuan, M.A.; Jones, D.R.; Izquierdo, M.; Merida, I. Role of diacylglycerol kinase alpha in the attenuation of receptor signaling. J. Cell Biol. 2001, 153, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Chauveau, A.; Le Floc’h, A.; Bantilan, N.S.; Koretzky, G.A.; Huse, M. Diacylglycerol kinase alpha establishes T cell polarity by shaping diacylglycerol accumulation at the immunological synapse. Sci. Signal. 2014, 7, ra82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco, S.; Merida, I. Diacylglycerol, when simplicity becomes complex. Trends Biochem. Sci. 2007, 32, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.S.; Monu, N.; Shen, D.T.; Mecklenbrauker, I.; Radoja, N.; Haydar, T.F.; Leitges, M.; Frey, A.B.; Vukmanovic, S.; Radoja, S. Protein kinase Cdelta regulates antigen receptor-induced lytic granule polarization in mouse CD8+ CTL. J. Immunol. 2007, 178, 7814–7821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.S.; Haydar, T.F.; Radoja, S. Protein kinase C delta localizes to secretory lysosomes in CD8+ CTL and directly mediates TCR signals leading to granule exocytosis-mediated cytotoxicity. J. Immunol. 2008, 181, 4716–4722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, C.; Sattentau, Q.J. Regulated secretion from CD4+ T cells. Trends Immunol. 2007, 28, 474–481. [Google Scholar] [CrossRef]
- Huse, M.; Lillemeier, B.F.; Kuhns, M.S.; Chen, D.S.; Davis, M.M. T cells use two directionally distinct pathways for cytokine secretion. Nat. Immunol. 2006, 7, 247–255. [Google Scholar] [CrossRef]
- Huse, M.; Quann, E.J.; Davis, M.M. Shouts, whispers and the kiss of death: directional secretion in T cells. Nat. Immunol. 2008, 9, 1105–1111. [Google Scholar] [CrossRef]
- Desai, D.M.; Newton, M.E.; Kadlecek, T.; Weiss, A. Stimulation of the phosphatidylinositol pathway can induce T-cell activation. Nature 1990, 348, 66–69. [Google Scholar] [CrossRef]
- Izquierdo, M.; Ruiz-Ruiz, M.C.; Lopez-Rivas, A. Stimulation of phosphatidylinositol turnover is a key event for Fas-dependent, activation-induced apoptosis in human T lymphocytes. J. Immunol. 1996, 157, 21–28. [Google Scholar]
- Montoya, M.C.; Sancho, D.; Bonello, G.; Collette, Y.; Langlet, C.; He, H.T.; Aparicio, P.; Alcover, A.; Olive, D.; Sanchez-Madrid, F. Role of ICAM-3 in the initial interaction of T lymphocytes and APCs. Nat. Immunol. 2002, 3, 159–168. [Google Scholar] [CrossRef]
- Bello-Gamboa, A.; Izquierdo, J.M.; Velasco, M.; Moreno, S.; Garrido, A.; Meyers, L.; Palomino, J.C.; Calvo, V.; Izquierdo, M. Imaging the Human Immunological Synapse. J. Vis. Exp. 2019. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Mazzeo, C.; Merida, I.; Izquierdo, M. A new role of diacylglycerol kinase alpha on the secretion of lethal exosomes bearing Fas ligand during activation-induced cell death of T lymphocytes. Biochimie 2007, 89, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainero, E.; Caswell, P.T.; Muller, P.A.; Grindlay, J.; McCaffrey, M.W.; Zhang, Q.; Wakelam, M.J.; Vousden, K.H.; Graziani, A.; Norman, J.C. Diacylglycerol kinase alpha controls RCP-dependent integrin trafficking to promote invasive migration. J. Cell Biol. 2012, 196, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Herranz, G.; Aguilera, P.; Davila, S.; Sanchez, A.; Stancu, B.; Gomez, J.; Fernandez-Moreno, D.; de Martin, R.; Quintanilla, M.; Fernandez, T.; et al. Protein Kinase C delta Regulates the Depletion of Actin at the Immunological Synapse Required for Polarized Exosome Secretion by T Cells. Front. Immunol. 2019, 10, 851. [Google Scholar] [CrossRef]
- Choudhuri, K.; Llodra, J.; Roth, E.W.; Tsai, J.; Gordo, S.; Wucherpfennig, K.W.; Kam, L.C.; Stokes, D.L.; Dustin, M.L. Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature 2014, 507, 118–123. [Google Scholar] [CrossRef]
- Saliba, D.G.; Cespedes-Donoso, P.F.; Balint, S.; Compeer, E.B.; Korobchevskaya, K.; Valvo, S.; Mayya, V.; Kvalvaag, A.; Peng, Y.; Dong, T.; et al. Composition and structure of synaptic ectosomes exporting antigen receptor linked to functional CD40 ligand from helper T cells. Elife 2019. [Google Scholar] [CrossRef]
- Lankar, D.; Vincent-Schneider, H.; Briken, V.; Yokozeki, T.; Raposo, G.; Bonnerot, C. Dynamics of major histocompatibility complex class II compartments during B cell receptor-mediated cell activation. J. Exp. Med. 2002, 195, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Siemasko, K.; Eisfelder, B.J.; Williamson, E.; Kabak, S.; Clark, M.R. Cutting edge: signals from the B lymphocyte antigen receptor regulate MHC class II containing late endosomes. J. Immunol. 1998, 160, 5203–5208. [Google Scholar]
- Arita, S.; Baba, E.; Shibata, Y.; Niiro, H.; Shimoda, S.; Isobe, T.; Kusaba, H.; Nakano, S.; Harada, M. B cell activation regulates exosomal HLA production. Eur. J. Immunol. 2008, 38, 1423–1434. [Google Scholar] [CrossRef]
- Yuseff, M.I.; Reversat, A.; Lankar, D.; Diaz, J.; Fanget, I.; Pierobon, P.; Randrian, V.; Larochette, N.; Vascotto, F.; Desdouets, C.; et al. Polarized secretion of lysosomes at the B cell synapse couples antigen extraction to processing and presentation. Immunity 2011, 35, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Lindenbergh, M.F.S.; Stoorvogel, W. Antigen Presentation by Extracellular Vesicles from Professional Antigen-Presenting Cells. Annu. Rev. Immunol. 2018, 36, 435–459. [Google Scholar] [CrossRef]
- Denzer, K.; van Eijk, M.; Kleijmeer, M.J.; Jakobson, E.; de Groot, C.; Geuze, H.J. Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J. Immunol. 2000, 165, 1259–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Mohan, V.; Krishnaswamy, V.R.; Solomonov, I.; Sagi, I. Exosomes as a storehouse of tissue remodeling proteases and mediators of cancer progression. Cancer Metastasis Rev. 2019, 38, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Duchez, S.; Rodrigues, M.; Bertrand, F.; Valitutti, S. Reciprocal polarization of T and B cells at the immunological synapse. J. Immunol. 2011, 187, 4571–4580. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Morphew, M.K.; McIntosh, J.R.; Davis, M.M. CD4+ T-cell synapses involve multiple distinct stages. Proc. Natl. Acad. Sci. USA 2011, 108, 17099–17104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quann, E.J.; Liu, X.; Altan-Bonnet, G.; Huse, M. A cascade of protein kinase C isozymes promotes cytoskeletal polarization in T cells. Nat. Immunol. 2011, 12, 647–654. [Google Scholar] [CrossRef]
- Gomez, T.S.; Kumar, K.; Medeiros, R.B.; Shimizu, Y.; Leibson, P.J.; Billadeau, D.D. Formins Regulate the Actin-Related Protein 2/3 Complex-Independent Polarization of the Centrosome to the Immunological Synapse. Immunity 2007, 26, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Na, B.R.; Kwon, M.S.; Chae, M.W.; Kim, H.R.; Kim, C.H.; Jun, C.D.; Park, Z.Y. Transgelin-2 in B-Cells Controls T-Cell Activation by Stabilizing T Cell B Cell Conjugates. PLoS ONE 2016, 11, e0156429. [Google Scholar] [CrossRef]
- Schnyder, T.; Castello, A.; Feest, C.; Harwood, N.E.; Oellerich, T.; Urlaub, H.; Engelke, M.; Wienands, J.; Bruckbauer, A.; Batista, F.D. B cell receptor-mediated antigen gathering requires ubiquitin ligase Cbl and adaptors Grb2 and Dok-3 to recruit dynein to the signaling microcluster. Immunity 2011, 34, 905–918. [Google Scholar] [CrossRef] [Green Version]
- Ibanez-Vega, J.; Del Valle Batalla, F.; Saez, J.J.; Soza, A.; Yuseff, M.I. Proteasome Dependent Actin Remodeling Facilitates Antigen Extraction at the Immune Synapse of B Cells. Front. Immunol. 2019, 10, 225. [Google Scholar] [CrossRef] [Green Version]
- Obino, D.; Farina, F.; Malbec, O.; Saez, P.J.; Maurin, M.; Gaillard, J.; Dingli, F.; Loew, D.; Gautreau, A.; Yuseff, M.I.; et al. Actin nucleation at the centrosome controls lymphocyte polarity. Nat. Commun. 2016, 7, 10969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torralba, D.; Martin-Cofreces, N.B.; Sanchez-Madrid, F. Mechanisms of polarized cell-cell communication of T lymphocytes. Immunol. Lett. 2019, 209, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Mittelbrunn, M.; Sanchez-Madrid, F. Intercellular communication: diverse structures for exchange of genetic information. Nat. Rev. Mol. Cell Biol. 2012, 13, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Messina, L.; Gutierrez-Vazquez, C.; Rivas-Garcia, E.; Sanchez-Madrid, F.; de la Fuente, H. Immunomodulatory role of microRNAs transferred by extracellular vesicles. Biol. Cell 2015, 107, 61–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rialland, P.; Lankar, D.; Raposo, G.; Bonnerot, C.; Hubert, P. BCR-bound antigen is targeted to exosomes in human follicular lymphoma B-cells. Biol. Cell 2006, 98, 491–501. [Google Scholar] [CrossRef]
- Saunderson, S.C.; Schuberth, P.C.; Dunn, A.C.; Miller, L.; Hock, B.D.; MacKay, P.A.; Koch, N.; Jack, R.W.; McLellan, A.D. Induction of exosome release in primary B cells stimulated via CD40 and the IL-4 receptor. J. Immunol. 2008, 180, 8146–8152. [Google Scholar] [CrossRef] [Green Version]
- Knight, A.M. Regulated release of B cell-derived exosomes: do differences in exosome release provide insight into different APC function for B cells and DC? Eur. J. Immunol. 2008, 38, 1186–1189. [Google Scholar] [CrossRef]
- Saunderson, S.C.; McLellan, A.D. Role of Lymphocyte Subsets in the Immune Response to Primary B Cell-Derived Exosomes. J. Immunol. 2017, 199, 2225–2235. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Li, R.; Yang, Y.; Shi, C.; Shen, Y.; Lu, C.; Chen, Y.; Zhou, W.; Lin, A.; Yu, L.; et al. Specific Decrease in B-Cell-Derived Extracellular Vesicles Enhances Post-Chemotherapeutic CD8(+) T Cell Responses. Immunity 2019, 50, 738.e7–750.e7. [Google Scholar] [CrossRef] [Green Version]
- Lenardo, M.J. The molecular regulation of lymphocyte apoptosis. Semin. Immunol. 1997, 9, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Dhein, J.; Walczak, H.; Baumler, C.; Debatin, K.M.; Krammer, P.H. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95). Nature 1995, 373, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Brunner, T.; Mogil, R.J.; LaFace, D.; Yoo, N.J.; Mahboubi, A.; Echeverri, F.; Martin, S.J.; Force, W.R.; Lynch, D.H.; Ware, C.F.; et al. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature 1995, 373, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Ju, S.T.; Panka, D.J.; Cui, H.; Ettinger, R.; el-Khatib, M.; Sherr, D.H.; Stanger, B.Z.; Marshak-Rothstein, A. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature 1995, 373, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Alderson, M.R.; Tough, T.W.; Davis-Smith, T.; Braddy, S.; Falk, B.; Schooley, K.A.; Goodwin, R.G.; Smith, C.A.; Ramsdell, F.; Lynch, D.H. Fas ligand mediates activation-induced cell death in human T lymphocytes. J. Exp. Med. 1995, 181, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caby, M.P.; Lankar, D.; Vincendeau-Scherrer, C.; Raposo, G.; Bonnerot, C. Exosomal-like vesicles are present in human blood plasma. Int. Immunol. 2005, 17, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Yang, J.; Xie, R.; Gao, L.; Yang, Y.; Fan, H.; Qian, K. Exosomal-like vesicles with immune-modulatory features are present in human plasma and can induce CD4+ T-cell apoptosis in vitro. Transfusion 2011, 51, 1002–1011. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, H.; Li, W.; Deng, Y.; Munegowda, M.A.; Chibbar, R.; Qureshi, M.; Xiang, J. Dendritic cells recruit T cell exosomes via exosomal LFA-1 leading to inhibition of CD8+ CTL responses through downregulation of peptide/MHC class I and Fas ligand-mediated cytotoxicity. J. Immunol. 2010, 185, 5268–5278. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Xie, Y.; Li, W.; Chibbar, R.; Xiong, S.; Xiang, J. CD4(+) T cell-released exosomes inhibit CD8(+) cytotoxic T-lymphocyte responses and antitumor immunity. Cell. Mol. Immunol. 2011, 8, 23–30. [Google Scholar] [CrossRef]
- Li, L.; Jay, S.M.; Wang, Y.; Wu, S.W.; Xiao, Z. IL-12 stimulates CTLs to secrete exosomes capable of activating bystander CD8(+) T cells. Sci. Rep. 2017, 7, 13365. [Google Scholar] [CrossRef]
- Wu, S.W.; Li, L.; Wang, Y.; Xiao, Z. CTL-Derived Exosomes Enhance the Activation of CTLs Stimulated by Low-Affinity Peptides. Front. Immunol. 2019, 10, 1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Okoye, I.S.; Coomes, S.M.; Pelly, V.S.; Czieso, S.; Papayannopoulos, V.; Tolmachova, T.; Seabra, M.C.; Wilson, M.S. MicroRNA-Containing T-Regulatory-Cell-Derived Exosomes Suppress Pathogenic T Helper 1 Cells. Immunity 2014, 41, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuler, P.J.; Saze, Z.; Hong, C.S.; Muller, L.; Gillespie, D.G.; Cheng, D.; Harasymczuk, M.; Mandapathil, M.; Lang, S.; Jackson, E.K.; et al. Human CD4+ CD39+ regulatory T cells produce adenosine upon co-expression of surface CD73 or contact with CD73+ exosomes or CD73+ cells. Clin. Exp. Immunol. 2014, 177, 531–543. [Google Scholar] [CrossRef]
- Smyth, L.A.; Ratnasothy, K.; Tsang, J.Y.; Boardman, D.; Warley, A.; Lechler, R.; Lombardi, G. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur. J. Immunol. 2013, 43, 2430–2440. [Google Scholar] [CrossRef] [PubMed]
- Aiello, S.; Rocchetta, F.; Longaretti, L.; Faravelli, S.; Todeschini, M.; Cassis, L.; Pezzuto, F.; Tomasoni, S.; Azzollini, N.; Mister, M.; et al. Extracellular vesicles derived from T regulatory cells suppress T cell proliferation and prolong allograft survival. Sci. Rep. 2017, 7, 11518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, A.; Fanelli, G.; Letizia, M.; Tung, S.L.; Boardman, D.; Lechler, R.; Lombardi, G.; Smyth, L.A. Regulatory T cell-derived exosomes: possible therapeutic and diagnostic tools in transplantation. Front. Immunol. 2014, 5, 555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anel, A.; Gallego-Lleyda, A.; de Miguel, D.; Naval, J.; Martinez-Lostao, L. Role of Exosomes in the Regulation of T-cell Mediated Immune Responses and in Autoimmune Disease. Cells 2019, 8, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulivieri, C.; Baldari, C.T. Regulation of T Cell Activation and Differentiation by Extracellular Vesicles and Their Pathogenic Role in Systemic Lupus Erythematosus and Multiple Sclerosis. Molecules 2017, 22, 225. [Google Scholar] [CrossRef]
- Wen, C.; Seeger, R.C.; Fabbri, M.; Wang, L.; Wayne, A.S.; Jong, A.Y. Biological roles and potential applications of immune cell-derived extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1400370. [Google Scholar] [CrossRef] [Green Version]
- Syn, N.L.; Wang, L.; Chow, E.K.; Lim, C.T.; Goh, B.C. Exosomes in Cancer Nanomedicine and Immunotherapy: Prospects and Challenges. Trends Biotechnol. 2017, 35, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Veerman, R.E.; Gucluler Akpinar, G.; Eldh, M.; Gabrielsson, S. Immune Cell-Derived Extracellular Vesicles—Functions and Therapeutic Applications. Trends Mol. Med. 2019, 25, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Lei, C.; Liu, S.; Cui, Y.; Wang, C.; Qian, K.; Li, T.; Shen, Y.; Fan, X.; Lin, F.; et al. CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity. Nat. Commun. 2019, 10, 4355. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.J.; Sun, X.Y.; Huang, K.M.; Zhang, L.; Yang, Z.S.; Zou, D.D.; Wang, B.; Warnock, G.L.; Dai, L.J.; Luo, J. Therapeutic potential of CAR-T cell-derived exosomes: a cell-free modality for targeted cancer therapy. Oncotarget 2015, 6, 44179–44190. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.G.; Liu, C.; Su, K.; Yu, S.; Zhang, L.; Zhang, S.; Wang, J.; Cao, X.; Grizzle, W.; Kimberly, R.P. A membrane form of TNF-alpha presented by exosomes delays T cell activation-induced cell death. J. Immunol. 2006, 176, 7385–7393. [Google Scholar] [CrossRef]
- Tavasolian, F.; Moghaddam, A.S.; Rohani, F.; Abdollahi, E.; Janzamin, E.; Momtazi-Borojeni, A.A.; Moallem, S.A.; Jamialahmadi, T.; Sahebkar, A. Exosomes: Effectual players in rheumatoid arthritis. Autoimmun. Rev. 2020, 102511. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, J.K.; Lee, E.Y.; Lee, E.B.; Song, Y.W. Circulating exosomes from patients with systemic lupus erythematosus induce an proinflammatory immune response. Arthritis Res. Ther. 2016, 18, 264. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Wu, H.; Liu, Y.; Zhao, M.; Li, D.; Lu, Q. Recent advances of exosomes in immune modulation and autoimmune diseases. Autoimmunity 2016, 49, 357–365. [Google Scholar] [CrossRef]
- Colletti, M.; Galardi, A.; De Santis, M.; Guidelli, G.M.; Di Giannatale, A.; Di Luigi, L.; Antinozzi, C. Exosomes in Systemic Sclerosis: Messengers Between Immune, Vascular and Fibrotic Components? Int. J. Mol. Sci. 2019, 20, 4337. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; Kalluri, R. Exosomes Exercise Inhibition of Anti-Tumor Immunity during Chemotherapy. Immunity 2019, 50, 547–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| CTL/APC | Th/APC | B/APC | B/Th | |
|---|---|---|---|---|
| Exosome secretion | + [17] | + [15] | Unknown 1 | + [16] |
| MTOC polarization | + [45] | + [95] | + [90] | + [94] |
| Secretory granules polarization | Lytic granules, MVB [29] | Lymphokine-containing granules, MVB [15,49] | SL, MHC-II+ compartment [90] | MHC-II+ compartment [94] |
| DAG/DGK-control of MTOC polarization | + [69] | + [15,69] | Unknown | Unknown |
| PKC/PKD control of secretory granules/MVB traffic | + (PKCδ, PKCθ) 2 [34,73,96] | + (PKCδ) [84] (PKD1/2) [59] | + (PKCζ) [88,90] (PKD1/3) [59] | Unknown |
| F-actin reorganization at IS | + (CDC42/WASP/ARP2/3, Formins: FMNL1, Dia1) [31] | + (CDC42/WASP/ARP2/3) (Formins: FMNL1, Dia1) [49,97] | + (CDC42/WASP/ARP2/3) Ezrin, Moesin) [32] | Unknown (TAGNL2?) [98] |
| F-actin reduction at cSMAC and MTOC polarization | + [46] | + (PKCδ dynein) [50,51,52,84] | + (dynein, proteasome) [99,100] | Unknown |
| F-actin reduction at MTOC and MTOC polarization | Unknown | Unknown | + (Proteasome) [100,101] | Unknown |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calvo, V.; Izquierdo, M. Inducible Polarized Secretion of Exosomes in T and B Lymphocytes. Int. J. Mol. Sci. 2020, 21, 2631. https://doi.org/10.3390/ijms21072631
Calvo V, Izquierdo M. Inducible Polarized Secretion of Exosomes in T and B Lymphocytes. International Journal of Molecular Sciences. 2020; 21(7):2631. https://doi.org/10.3390/ijms21072631
Chicago/Turabian StyleCalvo, Victor, and Manuel Izquierdo. 2020. "Inducible Polarized Secretion of Exosomes in T and B Lymphocytes" International Journal of Molecular Sciences 21, no. 7: 2631. https://doi.org/10.3390/ijms21072631
APA StyleCalvo, V., & Izquierdo, M. (2020). Inducible Polarized Secretion of Exosomes in T and B Lymphocytes. International Journal of Molecular Sciences, 21(7), 2631. https://doi.org/10.3390/ijms21072631