Estrogen Receptors and Endometriosis


 , ,
, ,
Abstract
:1. Introduction
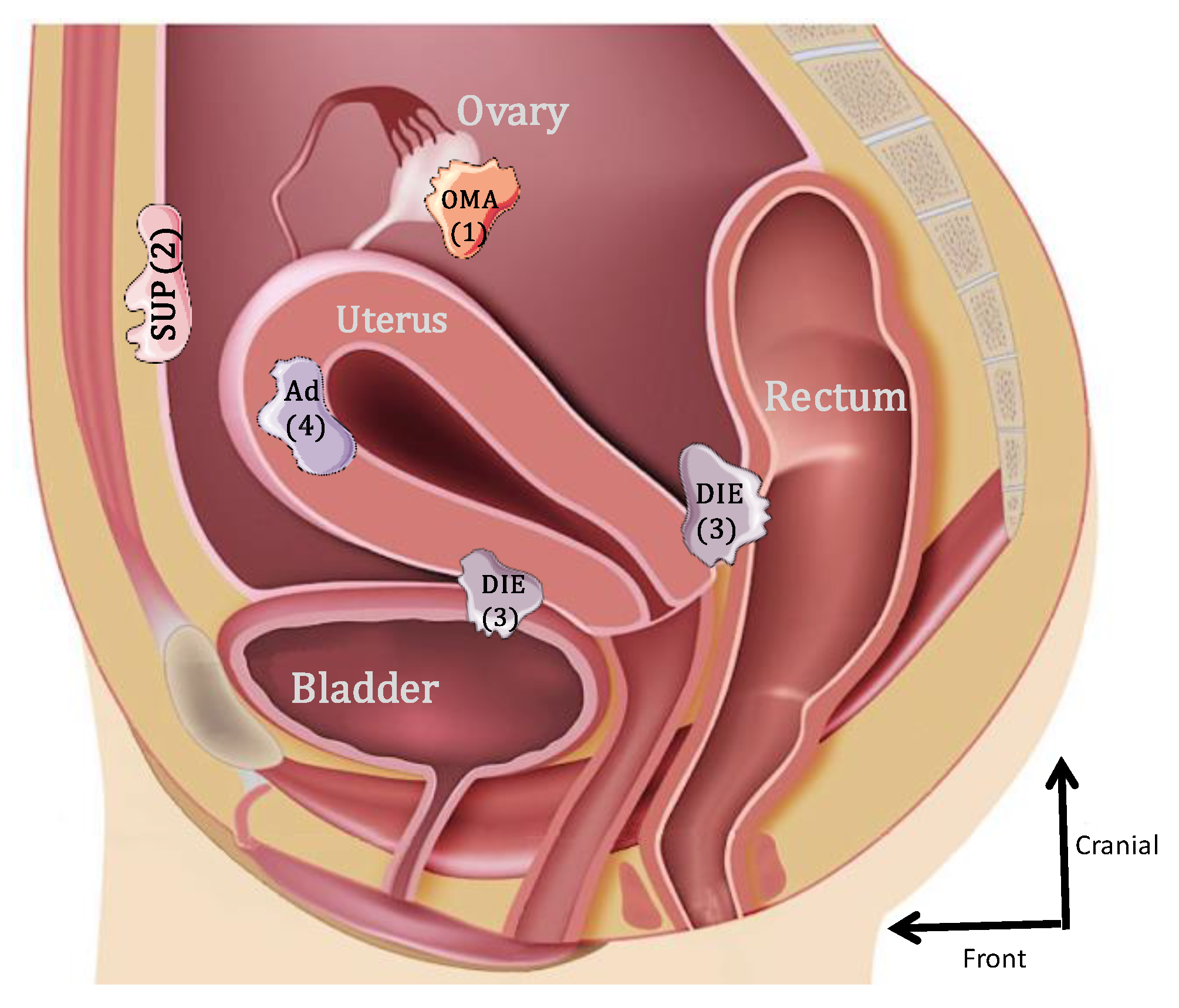
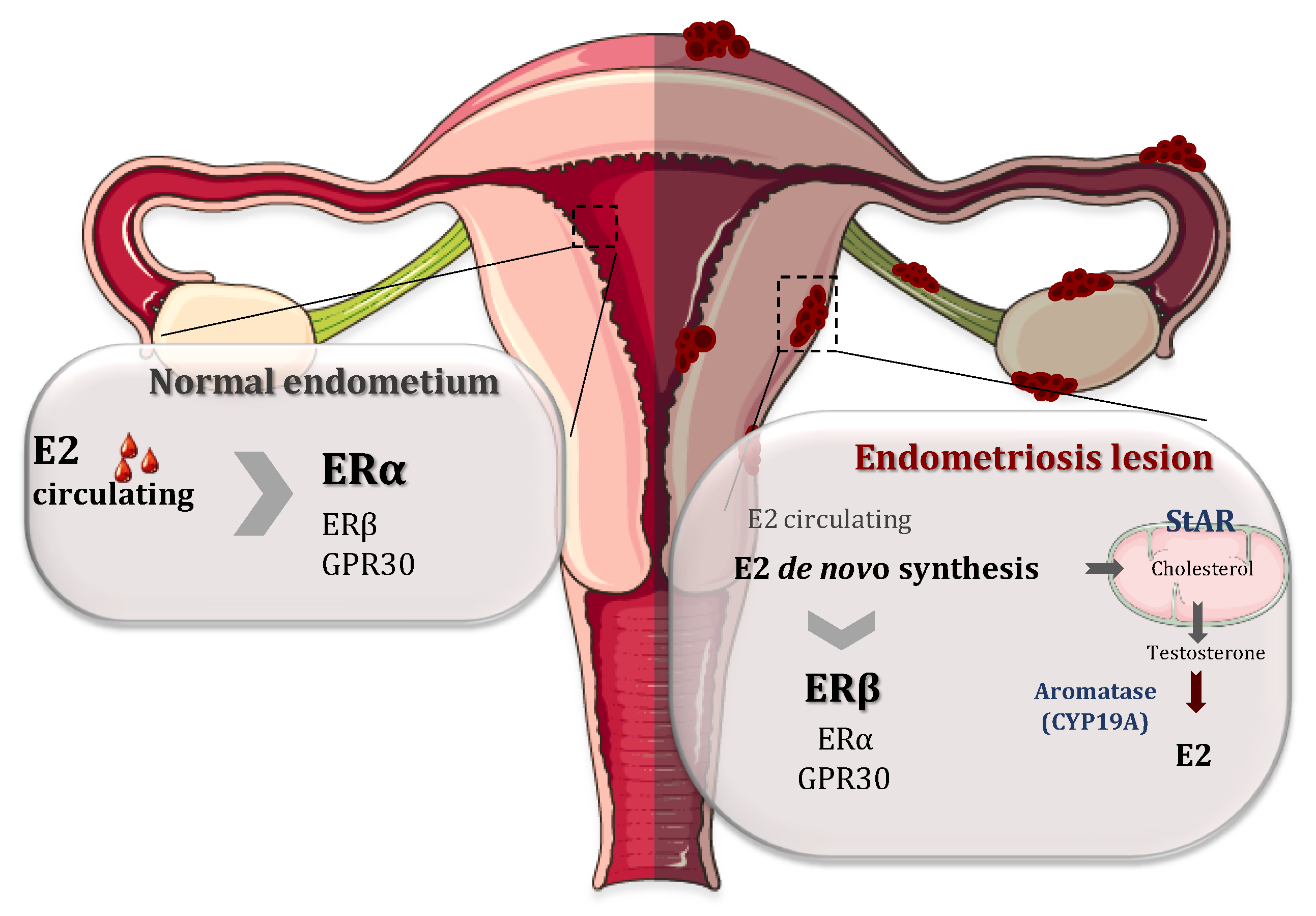
2. Levels of Estradiol and Estrogen Receptors in Endometriosis
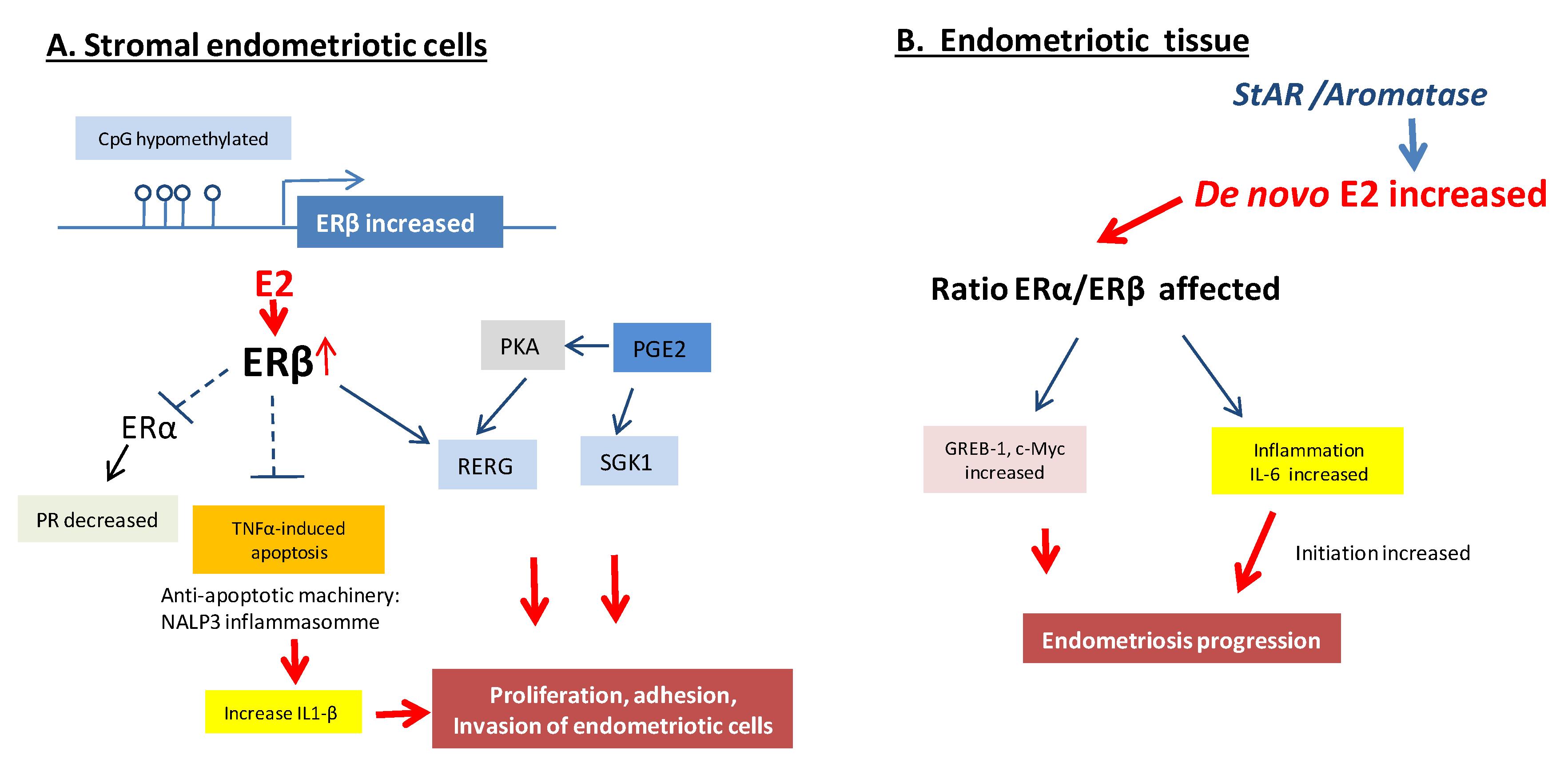
3. The Role of ERs in Endometriosis
4. Treatments and Innovations in Clinical Management Related to ERs
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Giudice, L.C.; Kao, L.C. Endometriosis. Lancet 2004, 364, 1789–1799. [Google Scholar] [CrossRef]
- Bulun, S.E.; Yilmaz, B.D.; Sison, C.; Miyazaki, K.; Bernardi, L.; Liu, S.; Kohlmeier, A.; Yin, P.; Milad, M.; Wei, J. Endometriosis. Endocr. Rev. 2019, 40, 1048–1079. [Google Scholar] [CrossRef]
- Zondervan, K.T.; Becker, C.M.; Koga, K.; Missmer, S.A.; Taylor, R.N.; Vigano, P. Endometriosis. Nat. Rev. Dis. Prim. 2018, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Chantalat, E.; Vidal, F.; Leguevaque, P.; Lepage, B.; Lambaudie, E.; Hebert, T.; Motton, S. Para-aortic workup in locally advanced cervical cancer: Heterogeneity is still the rule. Results from a retrospective multicenter study. Arch. Gynecol. Obstet. 2016, 293, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E. Endometriosis. New Engl. J. Med. 2009, 360, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Chapron, C.; Marcellin, L.; Borghese, B.; Santulli, P. Rethinking mechanisms, diagnosis and management of endometriosis. Nat. Rev. Endocrinol. 2019, 15, 666–682. [Google Scholar] [CrossRef]
- Prescott, J.; Farland, L.V.; Tobias, D.K.; Gaskins, A.J.; Spiegelman, D.; Chavarro, J.E.; Rich-Edwards, J.W.; Barbieri, R.L.; Missmer, S.A. A prospective cohort study of endometriosis and subsequent risk of infertility. Hum. Reprod. 2016, 31, 1475–1482. [Google Scholar] [CrossRef] [Green Version]
- de Ziegler, D.; Borghese, B.; Chapron, C. Endometriosis and infertility: Pathophysiology and management. Lancet 2010, 376, 730–738. [Google Scholar] [CrossRef]
- Pluchino, N.; Wenger, J.M.; Petignat, P.; Tal, R.; Bolmont, M.; Taylor, H.S.; Bianchi-Demicheli, F. Sexual function in endometriosis patients and their partners: Effect of the disease and consequences of treatment. Hum. Reprod. Update 2016, 22, 762–774. [Google Scholar] [CrossRef] [Green Version]
- Nnoaham, K.E.; Hummelshoj, L.; Webster, P.; d’Hooghe, T.; de Cicco Nardone, F.; de Cicco Nardone, C.; Jenkinson, C.; Kennedy, S.H.; Zondervan, K.T.; World Endometriosis Research Foundation Global Study of Women’s Health Consortium. Impact of endometriosis on quality of life and work productivity: A multicenter study across ten countries. Fertil. Steril. 2011, 96, 366–373 e368. [Google Scholar] [CrossRef] [Green Version]
- Canis, M.; Donnez, J.G.; Guzick, D.S.; Halme, J.K.; Rock, J.A.; Schenken, R.S.; Vernon, M.W. Revised American Society for Reproductive Medicine classification of endometriosis: 1996. Fertil. Steril. 1997, 67, 817–821. [Google Scholar] [CrossRef]
- Wang, W.; Li, R.; Fang, T.; Huang, L.; Ouyang, N.; Wang, L.; Zhang, Q.; Yang, D. Endometriosis fertility index score maybe more accurate for predicting the outcomes of in vitro fertilisation than r-AFS classification in women with endometriosis. Reprod. Biol. Endocrinol. 2013, 11, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foti, P.V.; Farina, R.; Palmucci, S.; Vizzini, I.A.A.; Libertini, N.; Coronella, M.; Spadola, S.; Caltabiano, R.; Iraci, M.; Basile, A.; et al. Endometriosis: Clinical features, MR imaging findings and pathologic correlation. Insights Imaging 2018, 9, 149–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facchin, F.; Barbara, G.; Saita, E.; Mosconi, P.; Roberto, A.; Fedele, L.; Vercellini, P. Impact of endometriosis on quality of life and mental health: Pelvic pain makes the difference. J. Psychosom. Obstet. Gynaecol. 2015, 36, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Marki, G.; Bokor, A.; Rigo, J.; Rigo, A. Physical pain and emotion regulation as the main predictive factors of health-related quality of life in women living with endometriosis. Hum. Reprod. 2017, 32, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Minelli, L.; Fanfani, F.; Fagotti, A.; Ruffo, G.; Ceccaroni, M.; Mereu, L.; Landi, S.; Pomini, P.; Scambia, G. Laparoscopic colorectal resection for bowel endometriosis: Feasibility, complications, and clinical outcome. Arch. Surg. 2009, 144, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.W. Recurrence of endometriosis and its control. Hum. Reprod. Update 2009, 15, 441–461. [Google Scholar] [CrossRef]
- Millochau, J.C.; Abo, C.; Darwish, B.; Huet, E.; Dietrich, G.; Roman, H. Continuous Amenorrhea May Be Insufficient to Stop the Progression of Colorectal Endometriosis. J. Minim. Invas. Gynecol. 2016, 23, 839–842. [Google Scholar] [CrossRef]
- Sampson, J.A. Metastatic or Embolic Endometriosis, due to the Menstrual Dissemination of Endometrial Tissue into the Venous Circulation. Am. J. Pathol. 1927, 3, 93–110.43. [Google Scholar]
- Khan, A.W.; Craig, M.; Jarmulowicz, M.; Davidson, B.R. Liver tumours due to endometriosis and endometrial stromal sarcoma. HPB Offic. J. Int. Hepato Pancreat. Biliary Assoc. 2002, 4, 43–45. [Google Scholar] [CrossRef] [Green Version]
- Koninckx, P.R.; Barlow, D.; Kennedy, S. Implantation versus infiltration: The Sampson versus the endometriotic disease theory. Gynecol. Obstet. Invest. 1999, 47, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Eychenne, C.; Suc, B.; Delchier, M.C.; Vidal, F.; Rimailho, J.; Illac, C.; Breibach, F.; Vaysse, C.; Chantalat, E. Hepatic pedicle endometriosis: Case report and review of the literature. J. Obstet. Gynecol. Res. 2019, 45, 2121–2127. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.H.; Monsanto, S.P.; Miller, C.; Singh, S.S.; Thomas, R.; Tayade, C. Pathophysiology and Immune Dysfunction in Endometriosis. BioMed Res. Int. 2015, 2015, 795976. [Google Scholar] [CrossRef] [Green Version]
- Greene, A.D.; Lang, S.A.; Kendziorski, J.A.; Sroga-Rios, J.M.; Herzog, T.J.; Burns, K.A. Endometriosis: Where are we and where are we going? Reproduction 2016, 152, R63–R78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Gong, P.; Chen, Y.; Nwachukwu, J.C.; Srinivasan, S.; Ko, C.; Bagchi, M.K.; Taylor, R.N.; Korach, K.S.; Nettles, K.W.; et al. Dual suppression of estrogenic and inflammatory activities for targeting of endometriosis. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulun, S.E.; Monsavais, D.; Pavone, M.E.; Dyson, M.; Xue, Q.; Attar, E.; Tokunaga, H.; Su, E.J. Role of estrogen receptor-beta in endometriosis. Semin. Reprod. Med. 2012, 30, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.R.; Zhang, R.; Lian, Z.X.; Deng, S.L.; Yu, K. Estrogen-Receptor Expression and Function in Female Reproductive Disease. Cells 2019, 8, 1123. [Google Scholar] [CrossRef] [Green Version]
- Zeitoun, K.M.; Bulun, S.E. Aromatase: A key molecule in the pathophysiology of endometriosis and a therapeutic target. Fertil. Steril. 1999, 72, 961–969. [Google Scholar] [CrossRef]
- Mori, T.; Ito, F.; Koshiba, A.; Kataoka, H.; Takaoka, O.; Okimura, H.; Khan, K.N.; Kitawaki, J. Local estrogen formation and its regulation in endometriosis. Reprod. Med. Biol. 2019, 18, 305–311. [Google Scholar] [CrossRef]
- Zeitoun, K.; Takayama, K.; Sasano, H.; Suzuki, T.; Moghrabi, N.; Andersson, S.; Johns, A.; Meng, L.; Putman, M.; Carr, B.; et al. Deficient 17beta-hydroxysteroid dehydrogenase type 2 expression in endometriosis: Failure to metabolize 17beta-estradiol. J. Clin. Endocrinol. Metab. 1998, 83, 4474–4480. [Google Scholar] [CrossRef] [Green Version]
- Arnal, J.F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Bocedi, A.; Marino, M. Structure-function relationship of estrogen receptor alpha and beta: Impact on human health. Mol. Aspects Med. 2006, 27, 299–402. [Google Scholar] [CrossRef] [PubMed]
- Sagae, S.; Monk, B.J.; Pujade-Lauraine, E.; Gaffney, D.K.; Narayan, K.; Ryu, S.Y.; McCormack, M.; Plante, M.; Casado, A.; Reuss, A.; et al. Advances and Concepts in Cervical Cancer Trials: A Road Map for the Future. Int. J. Gynecol. Cancer 2016, 26, 199–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reslan, O.M.; Khalil, R.A. Vascular effects of estrogenic menopausal hormone therapy. Rev. Recent Clin. Trials 2012, 7, 47–70. [Google Scholar] [CrossRef]
- Feldman, R.D.; Limbird, L.E. GPER (GPR30): A Nongenomic Receptor (GPCR) for Steroid Hormones with Implications for Cardiovascular Disease and Cancer. Ann. Rev. Pharmacol. Toxicol. 2017, 57, 567–584. [Google Scholar] [CrossRef]
- Muka, T.; Vargas, K.G.; Jaspers, L.; Wen, K.X.; Dhana, K.; Vitezova, A.; Nano, J.; Brahimaj, A.; Colpani, V.; Bano, A.; et al. Estrogen receptor beta actions in the female cardiovascular system: A systematic review of animal and human studies. Maturitas 2016, 86, 28–43. [Google Scholar] [CrossRef]
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjold, M.; Gustafsson, J.A. Human estrogen receptor beta-gene structure, chromosomal localization, and expression pattern. J. Clin. Endocrinol. Metab. 1997, 82, 4258–4265. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, C.; Gori, I.; Achtari, C.; Hornung, D.; Chardonnens, E.; Wunder, D.; Fiche, M.; Canny, G.O. The expression of estrogen receptors as well as GREB1, c-MYC, and cyclin D1, estrogen-regulated genes implicated in proliferation, is increased in peritoneal endometriosis. Fertil. Steril. 2012, 98, 1200–1208. [Google Scholar] [CrossRef]
- Matsuzaki, S.; Murakami, T.; Uehara, S.; Canis, M.; Sasano, H.; Okamura, K. Expression of estrogen receptor alpha and beta in peritoneal and ovarian endometriosis. Fertil. Steril. 2001, 75, 1198–1205. [Google Scholar] [CrossRef]
- Xue, Q.; Lin, Z.; Cheng, Y.H.; Huang, C.C.; Marsh, E.; Yin, P.; Milad, M.P.; Confino, E.; Reierstad, S.; Innes, J.; et al. Promoter methylation regulates estrogen receptor 2 in human endometrium and endometriosis. Biol. Reprod. 2007, 77, 681–687. [Google Scholar] [CrossRef]
- Hewitt, S.C.; Harrell, J.C.; Korach, K.S. Lessons in estrogen biology from knockout and transgenic animals. Annu. Rev. Physiol. 2005, 67, 285–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukulmez, O.; Hardy, D.B.; Carr, B.R.; Word, R.A.; Mendelson, C.R. Inflammatory status influences aromatase and steroid receptor expression in endometriosis. Endocrinology 2008, 149, 1190–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, J.; Hirose, R.; Sakaguchi, H.; Tamaya, T. Expression of oestrogen receptor-alpha and -beta in ovarian endometriomata. Mol. Hum. Reprod. 1999, 5, 742–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monsivais, D.; Dyson, M.T.; Yin, P.; Coon, J.S.; Navarro, A.; Feng, G.; Malpani, S.S.; Ono, M.; Ercan, C.M.; Wei, J.J.; et al. ERbeta- and prostaglandin E2-regulated pathways integrate cell proliferation via Ras-like and estrogen-regulated growth inhibitor in endometriosis. Mol. Endocrinol. 2014, 28, 1304–1315. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.W.; Licence, D.; Cook, E.; Luo, F.; Arends, M.J.; Smith, S.K.; Print, C.G.; Charnock-Jones, D.S. Activation of mutated K-ras in donor endometrial epithelium and stroma promotes lesion growth in an intact immunocompetent murine model of endometriosis. J. Pathol. 2011, 224, 261–269. [Google Scholar] [CrossRef]
- Maekawa, R.; Mihara, Y.; Sato, S.; Okada, M.; Tamura, I.; Shinagawa, M.; Shirafuta, Y.; Takagi, H.; Taketani, T.; Tamura, H.; et al. Aberrant DNA methylation suppresses expression of estrogen receptor 1 (ESR1) in ovarian endometrioma. J. Ovar. Res. 2019, 12, 14. [Google Scholar] [CrossRef]
- Trukhacheva, E.; Lin, Z.; Reierstad, S.; Cheng, Y.H.; Milad, M.; Bulun, S.E. Estrogen receptor (ER) beta regulates ERalpha expression in stromal cells derived from ovarian endometriosis. J. Clin. Endocrinol. Metab. 2009, 94, 615–622. [Google Scholar] [CrossRef]
- Dyson, M.T.; Roqueiro, D.; Monsivais, D.; Ercan, C.M.; Pavone, M.E.; Brooks, D.C.; Kakinuma, T.; Ono, M.; Jafari, N.; Dai, Y.; et al. Genome-wide DNA methylation analysis predicts an epigenetic switch for GATA factor expression in endometriosis. PLoS Genet. 2014, 10, e1004158. [Google Scholar] [CrossRef] [Green Version]
- Han, S.J.; O’Malley, B.W. The dynamics of nuclear receptors and nuclear receptor coregulators in the pathogenesis of endometriosis. Hum. Reprod. Update 2014, 20, 467–484. [Google Scholar] [CrossRef] [Green Version]
- Beliard, A.; Noel, A.; Foidart, J.M. Reduction of apoptosis and proliferation in endometriosis. Fertil. Steril. 2004, 82, 80–85. [Google Scholar] [CrossRef]
- Burns, K.A.; Rodriguez, K.F.; Hewitt, S.C.; Janardhan, K.S.; Young, S.L.; Korach, K.S. Role of estrogen receptor signaling required for endometriosis-like lesion establishment in a mouse model. Endocrinology 2012, 153, 3960–3971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, K.A.; Thomas, S.Y.; Hamilton, K.J.; Young, S.L.; Cook, D.N.; Korach, K.S. Early Endometriosis in Females Is Directed by Immune-Mediated Estrogen Receptor alpha and IL-6 Cross-Talk. Endocrinology 2018, 159, 103–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.J.; Jung, S.Y.; Wu, S.P.; Hawkins, S.M.; Park, M.J.; Kyo, S.; Qin, J.; Lydon, J.P.; Tsai, S.Y.; Tsai, M.J.; et al. Estrogen Receptor beta Modulates Apoptosis Complexes and the Inflammasome to Drive the Pathogenesis of Endometriosis. Cell 2015, 163, 960–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.J.; Lee, J.E.; Cho, Y.J.; Park, M.J.; O’Malley, B.W. Genomic Function of Estrogen Receptor beta in Endometriosis. Endocrinology 2019, 160, 2495–2516. [Google Scholar] [CrossRef] [PubMed]
- Monsivais, D.; Dyson, M.T.; Yin, P.; Navarro, A.; Coon, J.S.T.; Pavone, M.E.; Bulun, S.E. Estrogen receptor beta regulates endometriotic cell survival through serum and glucocorticoid-regulated kinase activation. Fertil. Steril. 2016, 105, 1266–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandenberger, A.W.; Lebovic, D.I.; Tee, M.K.; Ryan, I.P.; Tseng, J.F.; Jaffe, R.B.; Taylor, R.N. Oestrogen receptor (ER)-alpha and ER-beta isoforms in normal endometrial and endometriosis-derived stromal cells. Mol. Hum. Reprod. 1999, 5, 651–655. [Google Scholar] [CrossRef] [Green Version]
- Othman, E.E.; Salama, S.; Ismail, N.; Al-Hendy, A. Toward gene therapy of endometriosis: Adenovirus-mediated delivery of dominant negative estrogen receptor genes inhibits cell proliferation, reduces cytokine production, and induces apoptosis of endometriotic cells. Fertil. Steril. 2007, 88, 462–471. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, L.; Yu, Q.; Zhang, Y.; Yan, L.; Chen, Z.J. The estrogen-regulated lncRNA H19/miR-216a-5p axis alters stromal cell invasion and migration via ACTA2 in endometriosis. Mol. Hum. Reprod. 2019, 25, 550–561. [Google Scholar] [CrossRef]
- Noble, L.S.; Takayama, K.; Zeitoun, K.M.; Putman, J.M.; Johns, D.A.; Hinshelwood, M.M.; Agarwal, V.R.; Zhao, Y.; Carr, B.R.; Bulun, S.E. Prostaglandin E2 stimulates aromatase expression in endometriosis-derived stromal cells. J. Clin. Endocrinol. Metab. 1997, 82, 600–606. [Google Scholar] [CrossRef]
- Sun, H.S.; Hsiao, K.Y.; Hsu, C.C.; Wu, M.H.; Tsai, S.J. Transactivation of steroidogenic acute regulatory protein in human endometriotic stromalcells is mediated by the prostaglandin EP2 receptor. Endocrinology 2003, 144, 3934–3942. [Google Scholar] [CrossRef] [Green Version]
- Tamura, M.; Deb, S.; Sebastian, S.; Okamura, K.; Bulun, S.E. Estrogen up-regulates cyclooxygenase-2 via estrogen receptor in human uterine microvascular endothelial cells. Fertil. Steril. 2004, 81, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Daniels, J.; Gray, R.; Hills, R.K.; Latthe, P.; Buckley, L.; Gupta, J.; Selman, T.; Adey, E.; Xiong, T.; Champaneria, R.; et al. Laparoscopic uterosacral nerve ablation for alleviating chronic pelvic pain: A randomized controlled trial. Jama 2009, 302, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Boretto, M.; Cox, B.; Noben, M.; Hendriks, N.; Fassbender, A.; Roose, H.; Amant, F.; Timmerman, D.; Tomassetti, C.; Vanhie, A.; et al. Development of organoids from mouse and human endometrium showing endometrial epithelium physiology and long-term expandability. Development 2017, 144, 1775–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turco, M.Y.; Gardner, L.; Hughes, J.; Cindrova-Davies, T.; Gomez, M.J.; Farrell, L.; Hollinshead, M.; Marsh, S.G.E.; Brosens, J.J.; Critchley, H.O.; et al. Long-term, hormone-responsive organoid cultures of human endometrium in a chemically defined medium. Nat. Cell Biol. 2017, 19, 568–577. [Google Scholar] [CrossRef]
- Boretto, M.; Maenhoudt, N.; Luo, X.; Hennes, A.; Boeckx, B.; Bui, B.; Heremans, R.; Perneel, L.; Kobayashi, H.; Van Zundert, I.; et al. Patient-derived organoids from endometrial disease capture clinical heterogeneity and are amenable to drug screening. Nat. Cell Biol. 2019, 21, 1041–1051. [Google Scholar] [CrossRef]
- Andersson, S.; Sundberg, M.; Pristovsek, N.; Ibrahim, A.; Jonsson, P.; Katona, B.; Clausson, C.M.; Zieba, A.; Ramstrom, M.; Soderberg, O.; et al. Insufficient antibody validation challenges oestrogen receptor beta research. Nat. Commun. 2017, 8, 15840. [Google Scholar] [CrossRef]
- Samartzis, N.; Samartzis, E.P.; Noske, A.; Fedier, A.; Dedes, K.J.; Caduff, R.; Fink, D.; Imesch, P. Expression of the G protein-coupled estrogen receptor (GPER) in endometriosis: A tissue microarray study. Reprod. Biol. Endocrinol. 2012, 10, 30. [Google Scholar] [CrossRef] [Green Version]
- Plante, B.J.; Lessey, B.A.; Taylor, R.N.; Wang, W.; Bagchi, M.K.; Yuan, L.; Scotchie, J.; Fritz, M.A.; Young, S.L. G protein-coupled estrogen receptor (GPER) expression in normal and abnormal endometrium. Reprod. Sci. 2012, 19, 684–693. [Google Scholar] [CrossRef] [Green Version]
- Heublein, S.; Vrekoussis, T.; Kuhn, C.; Friese, K.; Makrigiannakis, A.; Mayr, D.; Lenhard, M.; Jeschke, U. Inducers of G-protein coupled estrogen receptor (GPER) in endometriosis: Potential implications for macrophages and follicle maturation. J. Reprod. Immunol. 2013, 97, 95–103. [Google Scholar] [CrossRef]
- Bedaiwy, M.A.; Allaire, C.; Yong, P.; Alfaraj, S. Medical Management of Endometriosis in Patients with Chronic Pelvic Pain. Semin. Reprod. Med. 2017, 35, 38–53. [Google Scholar] [CrossRef]
- Harris, H.A.; Bruner-Tran, K.L.; Zhang, X.; Osteen, K.G.; Lyttle, C.R. A selective estrogen receptor-beta agonist causes lesion regression in an experimentally induced model of endometriosis. Hum. Reprod. 2005, 20, 936–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falconer, H.; Mwenda, J.M.; Chai, D.C.; Wagner, C.; Song, X.Y.; Mihalyi, A.; Simsa, P.; Kyama, C.; Cornillie, F.J.; Bergqvist, A.; et al. Treatment with anti-TNF monoclonal antibody (c5N) reduces the extent of induced endometriosis in the baboon. Hum. Reprod. 2006, 21, 1856–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, D.L.; Pritts, E.A. Treatment of endometriosis. New Engl. J. Med. 2001, 345, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.W.; Groothuis, P.G. Is it time for a paradigm shift in drug research and development in endometriosis/adenomyosis? Hum. Reprod. Update 2018, 24, 577–598. [Google Scholar] [CrossRef]
- Altintas, D.; Kokcu, A.; Kandemir, B.; Tosun, M.; Cetinkaya, M.B. Comparison of the effects of raloxifene and anastrozole on experimental endometriosis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2010, 150, 84–87. [Google Scholar] [CrossRef]
- Stratton, P.; Sinaii, N.; Segars, J.; Koziol, D.; Wesley, R.; Zimmer, C.; Winkel, C.; Nieman, L.K. Return of chronic pelvic pain from endometriosis after raloxifene treatment: A randomized controlled trial. Obstet. Gynecol. 2008, 111, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Kulak, J., Jr.; Fischer, C.; Komm, B.; Taylor, H.S. Treatment with bazedoxifene, a selective estrogen receptor modulator, causes regression of endometriosis in a mouse model. Endocrinology 2011, 152, 3226–3232. [Google Scholar] [CrossRef]
- Naqvi, H.; Sakr, S.; Presti, T.; Krikun, G.; Komm, B.; Taylor, H.S. Treatment with bazedoxifene and conjugated estrogens results in regression of endometriosis in a murine model. Biol. Reprod. 2014, 90, 121. [Google Scholar] [CrossRef]
- Flores, V.A.; Leone, C.; Taylor, H.S.; Stachenfeld, N.S. Effects of bazedoxifene/conjugated estrogens on reproductive endocrinology and reproductive tract ultrasonographic appearance in premenopausal women: A preliminary study. Gynecol. Endocrinol. 2019, 35, 390–394. [Google Scholar] [CrossRef]
- Khine, Y.M.; Taniguchi, F.; Nagira, K.; Nakamura, K.; Ohbayashi, T.; Osaki, M.; Harada, T. New insights into the efficacy of SR-16234, a selective estrogen receptor modulator, on the growth of murine endometriosis-like lesions. Am. J. Reprod. Immunol. 2018, 80, e13023. [Google Scholar] [CrossRef] [Green Version]
- Streuli, I.; de Ziegler, D.; Santulli, P.; Marcellin, L.; Borghese, B.; Batteux, F.; Chapron, C. An update on the pharmacological management of endometriosis. Expert Opin. Pharmacother. 2013, 14, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Attar, E.; Bulun, S.E. Aromatase inhibitors: The next generation of therapeutics for endometriosis? Fertil. Steril. 2006, 85, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Abushahin, F.; Goldman, K.N.; Barbieri, E.; Milad, M.; Rademaker, A.; Bulun, S.E. Aromatase inhibition for refractory endometriosis-related chronic pelvic pain. Fertil. Steril. 2011, 96, 939–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amsterdam, L.L.; Gentry, W.; Jobanputra, S.; Wolf, M.; Rubin, S.D.; Bulun, S.E. Anastrazole and oral contraceptives: A novel treatment for endometriosis. Fertil. Steril. 2005, 84, 300–304. [Google Scholar] [CrossRef]
- Takayama, K.; Zeitoun, K.; Gunby, R.T.; Sasano, H.; Carr, B.R.; Bulun, S.E. Treatment of severe postmenopausal endometriosis with an aromatase inhibitor. Fertil. Steril. 1998, 69, 709–713. [Google Scholar] [CrossRef]
- Gargiulo, A.R.; Strauss, J.F.; Barbieri, R.L. Yen & Jaffe’s Reproductive Endocrinology E-Book: Physiology, Pathophysiology, and Clinical Management, 8th ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 609–642. [Google Scholar]
- Abdul Karim, A.K.; Shafiee, M.N.; Abd Aziz, N.H.; Omar, M.H.; Abdul Ghani, N.A.; Lim, P.S.; Md Zin, R.R.; Mokhtar, N. Reviewing the role of progesterone therapy in endometriosis. Gynecol. Endocrinol. 2019, 35, 10–16. [Google Scholar] [CrossRef]
- Bedaiwy, M.A.; Alfaraj, S.; Yong, P.; Casper, R. New developments in the medical treatment of endometriosis. Fertil. Steril. 2017, 107, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; De Carolis, C.; Man, G.C.W.; Wang, C.C. The link between immunity, autoimmunity and endometriosis: A literature update. Autoimmun. Rev. 2018, 17, 945–955. [Google Scholar] [CrossRef]
- Ferlita, A.; Battaglia, R.; Andronico, F.; Caruso, S.; Cianci, A.; Purrello, M.; Pietro, C.D. Non-Coding RNAs in Endometrial Physiopathology. Int. J. Mol. Sci. 2018, 19, 2120. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Studies | ER or ERβ | Procedures | Models | Results |
|---|---|---|---|---|
| Enmark et al., 1997 [37] | ERβ/ERα | mRNA by RT-PCR | Rat tissues | Expression ERα was significantly higher than that of ERβ in normal endometrium. |
| Brandenberger et al., 1999 [56] | ERβ/ERα | mRNA by RT-PCR Southern blot Ligand-binding assays | Human normal endometrial and endometriosis-derived stromal cells | Ratio of ERα/ERβ mRNA in stromal cells were decreased in endometriosis as compared to normal endometrium |
| Fujimoto et al., 1999 [43] | ERβ and ERα | mRNA by RT-PCR IHC (Anti-ERα (MC-20) et ERβ –L-20) Southern blot | Human ovarian endometrioma Normal endometrium | In normal endometrium, ERα mRNA were expressed at a higher level than those of ERβ. However, ERβ mRNA expression was higher and over a much greater range in ovarian endometrioma than normal endometrium while ERα expression was lower and more random. |
| Matsuzaki et al., 2001 [39] | ERβ and ERα | mRNA RT-PCR assay TaqMan RT-PCR Nonradioactive in situ hybridization | Human ovarian endometrioma | The predominant expression of ERα in both glandular epithelial and stromal cells might have been essential for the development and growth of peritoneal and ovarian endometriosis The expression of ER was modulated according to the menstrual cycle |
| Beliard et al., 2004 [50] | -No differentiation between ERα and ERβ | Nuclear staining IHC Antibodies used not specified | Human endometriotic tissues (peritoneum) | -No correlation between apoptosis and estrogen receptor levels was found -A lower amount of steroid receptor was found in endometriotic tissues without cyclic modulation compared with the eutopic endometrium |
| Tamura et al., 2004 [61] | ERβ and ERα | mRNA and protein RT-PCR Western Blot | Human uterine microvascular endothelial cells | In uterine microvascular endothelial cells, ERβ mediated estradiol-stimulated COX2 expression and PGE2 production |
| Xue et al., 2007 [40] | ERβ and ERα | mRNA by RT-PCR Western blot: | Human endometrial and endometriotic stromal cells from ovarian endometriomas | -mRNA (34-fold) and protein levels of ERβ were higher in endometriotic stromal cells due to hypomethylation of a CpG island whereas level of ERα was lower in paired endometriotic versus endometrial stromal cells |
| Bukulmez et al., 2008 [42] | ERβ/ERα | mRNA and protein lIHC Histology qRT-PCR Western blot | Human endometriotic tissues | Expression ERβ is significantly higher than that of ERα in ectopic endometrium |
| Trukhacheva et al., 2009 [47] | ERβ and ERα | Si-RNA knockdown RT-PCR IP Western Blot | Human ovarian endometrioma | Overexpression of ERβ in endometriotic stromal cells significantly decreased ERα mRNA and protein levels, and ERβ knock-down significantly decreased proliferation of endometriotic stromal cells |
| Cheng et al., 2011 [45] | ERβ | mRNA by RT-PCR IHC Histology | Mouse: They transplanted steroid-manipulated, menstrual-like endometrium from K-ras(G12V/+)/Ah-Cre(+/+)/ROSA26R-LacZ(+/+)mice into gonad-intact immunocompetent wild-type mice | Elevated levels of ERβ existed in both nuclear and cytoplasmic locations in this mouse model of endometriosis |
| Burns et al., 2012 [51] | ERβ/ERα | mRNA by RT-PCR IHC | Mouse: Uterus samples injected in peritoneal cavity | ERβ gene knockout was less than ERα gene deletion in the suppression of ectopic lesion growth |
| Pellegrini et al., 2012 [38] | ERβ and ERα | mRNA byRT-PCR -IHC | Human endometrium with or without endometriosis | mRNA of ERβ and ERα were upregulated in the eutopic endometrial tissue of patients with endometriosis ERβ and ERα as well as c-myc, cyclin D1 mRNA expression levels were increased in ectopic tissue in comparison with both normal and eutopic endometrium |
| Monsivais et al., 2014 [44] | ERβ | Genome-wide comparative analysis of ERβ binding and gene expression | Human endometriosis and endometrial tissues | Ras-like estrogen-regulated growth inhibitor (RERG) and serum and glucocorticoid-regulated kinase (SGK1) are identified as key ERβ targets |
| Han and al., 2014 (Review) [49] | ERβ and ERα | Gene expression microarray data | Human endometriotic tissues | Aberrant levels of nuclear receptors and nuclear receptors co-regulators in ectopic endometriotic lesions were associated with the progression of endometriosis |
| Zhao et al., 2015 [25] | ERβ/ERα | mRNA by RT-PCR -IHC -Immunofluorescence | -Immunocompetent mice ERβKO and ERαKO -Human endometriotic stromal cells in culture | Both the ERα and the ERβ isoforms were required for the growth of endometriotic-like lesions |
| Han et al., 2015 [54] | ERβ | -IHC | They used mouse overexpressing ERβ and immortalized human endometrial epithelial cells injected in SCID mice | -ERβ also contributed to the epithelial–mesenchymal transition; ERβ overexpression could then increase endometriosis-associated infertility -ERβ played a critical role in endometriosis development, interacted with the apoptotic machinery in the cytoplasm to inhibit TNF-induced apoptosis and with the components of the cytoplasmic inflammasome to increase IL-1β |
| Monsivais et al., 2016 [55] | ERβ | siRNA knockdown of ERβ RT-PCR IHC Western Blot | Human ovarian endometriosis and normal endometrial tissues | Estradiol/ERβ also stimulated SGK1 expression and enzyme activity, leading to increased human endometriotic cell survival |
| Burns et al., 2018 [52] | ERα | mRNA by RT-PCR Flow cytometry Cytokine production | Mouse (WT, αERKO) | E2/ERα/IL-6-mediated cross-talk played a partial role in increasing endometriosis lesion numbers |
| Han et al., 2019 [53] | ERβ | ERβ-transcriptomic and cistromic analyses | New endometrium-specific FLAG-tagged human ERβ overexpression mouse model | ERβ stimulated gene expression associated with IL6/JAK/stat inhibitory signaling in ectopic lesions to enhance progression |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chantalat, E.; Valera, M.-C.; Vaysse, C.; Noirrit, E.; Rusidze, M.; Weyl, A.; Vergriete, K.; Buscail, E.; Lluel, P.; Fontaine, C.; et al. Estrogen Receptors and Endometriosis. Int. J. Mol. Sci. 2020, 21, 2815. https://doi.org/10.3390/ijms21082815
Chantalat E, Valera M-C, Vaysse C, Noirrit E, Rusidze M, Weyl A, Vergriete K, Buscail E, Lluel P, Fontaine C, et al. Estrogen Receptors and Endometriosis. International Journal of Molecular Sciences. 2020; 21(8):2815. https://doi.org/10.3390/ijms21082815
Chicago/Turabian StyleChantalat, Elodie, Marie-Cécile Valera, Charlotte Vaysse, Emmanuelle Noirrit, Mariam Rusidze, Ariane Weyl, Kelig Vergriete, Etienne Buscail, Philippe Lluel, Coralie Fontaine, and et al. 2020. "Estrogen Receptors and Endometriosis" International Journal of Molecular Sciences 21, no. 8: 2815. https://doi.org/10.3390/ijms21082815