Abstract
The therapeutic concept of unleashing a pre-existing immune response against the tumor by the application of immune-checkpoint inhibitors (ICI) has resulted in long-term survival in advanced cancer patient subgroups. However, the majority of patients do not benefit from single-agent ICI and therefore new combination strategies are eagerly necessitated. In addition to conventional chemotherapy, kinase inhibitors as well as tumor-specific vaccinations are extensively investigated in combination with ICI to augment therapy responses. An unprecedented clinical outcome with chimeric antigen receptor (CAR-)T cell therapy has led to the approval for relapsed/refractory diffuse large B cell lymphoma and B cell acute lymphoblastic leukemia whereas response rates in solid tumors are unsatisfactory. Immune-checkpoints negatively impact CAR-T cell therapy in hematologic and solid malignancies and as a consequence provide a therapeutic target to overcome resistance. Established biomarkers such as programmed death ligand 1 (PD-L1) and tumor mutational burden (TMB) help to select patients who will benefit most from ICI, however, biomarker negativity does not exclude responses. Investigating alterations in the antigen presenting pathway as well as radiomics have the potential to determine tumor immunogenicity and response to ICI. Within this review we summarize the literature about specific combination partners for ICI and the applicability of artificial intelligence to predict ICI therapy responses.
Keywords:
kinase inhibitor; vaccination; CAR-T cell; radiomics; PD-1; PD-L1; tumor neoantigen; HLA; resistance mechanism; T cell exhaustion 1. Introduction
It has been recognized for a long time that tumor cell evasion from the immune system is a hallmark of malignant cancers [1] and several mechanisms have been identified by which tumor cells shape an immunosuppressive microenvironment, that are reviewed elsewhere [2,3,4]. One of the best-studied and most relevant mechanisms is the suppression of T cells through activation of negative regulatory pathways by tumor cells, so-called immune-checkpoints. Several immune-checkpoint molecules have been identified in the last years with cytotoxic T-lymphocyte protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) being the best studied systems [5]. These molecules play a crucial role in controlling the physiological immune response and in preventing over-activation of the immune system [6,7].
A magnitude of clinical trials investigating immune-checkpoint inhibitors (ICI) as monotherapy, combination therapy or in combination with cytotoxic agents as well as with targeted therapy has demonstrated improved clinical outcome across various types of cancer and in turn led to the respective approval status by the Food and Drug Administration (FDA) as depicted in Figure 1.
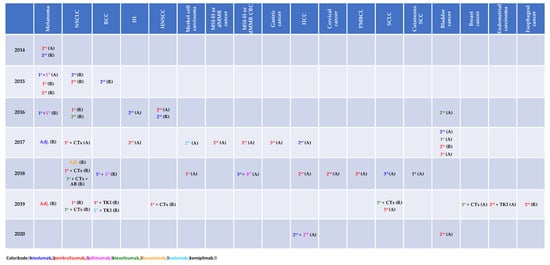
Figure 1.
Immune-checkpoint inhibitor approval status by the Food and Drug Administration (access date: 03/13/2020). A: accelerated, AB: antibody, CTx: chemotherapy, dMMR: mismatch repair deficiency, HCC: hepatocellular carcinoma, HL: Hodgkin’s lymphoma, HNSCC: head and neck squamous cell carcinoma, MSI-H: microsatellite instability, NSCLC: non-small cell lung cancer, PMBCL: primary mediastinal B cell lymphoma, R: regular, RCC: renal cell carcinoma, TKI: tyrosine kinase inhibitor.
Although immune-checkpoint blockade derives long-term overall survival (OS) in a subset of cancer patients [8,9], identification of patients who will not benefit from ICI remains challenging. Extensively investigated biomarkers such as programmed cell death-ligand 1 (PD-L1) [10,11,12,13] help predicting clinical outcome with ICI whereas negativity does not exclude responses [14,15]. Tumor immunogenicity is a prerequisite for reversing T cell exhaustion by ICI. Although alterations in the antigen presenting machinery have a relevant impact on the therapeutic success of immune-checkpoint blockade [16,17], the latter findings have not influenced clinical decision-making so far. Radiomics—extracting information about tumor biological processes from imaging studies—may serve as an alternative to tumor tissue-based biomarkers and may facilitate response prediction to ICI. Apart from combining ICI with chemotherapy, other ICI or monoclonal antibodies, growing evidence provides the rationale for combination strategies with kinase inhibitors [15,18] or tumor-specific vaccinations [19]. While treatment with chimeric antigen receptor (CAR)-T cells, T cells genetically modified ex vivo to express a new surface antigen receptor, led to unprecedented response rates in hematologic malignancies such as relapsed/refractory B cell acute lymphoblastic leukemia (B-ALL) [20] and diffuse large B cell lymphoma (DLBCL) [21,22], therapeutic success in solid cancers is poor [23,24]. Up-regulation of immune-checkpoints and in turn ineffective T cell function have been identified as primary and secondary resistance mechanisms to CAR-T cell therapy and therefore provide the rationale for combinations with ICI [25,26].
Within this review, we summarize and discuss the biological background of immune-checkpoints (with focus on PD-1), specific promising combination partners for immune-checkpoint blockade (kinase inhibitors, tumor-specific vaccinations, CAR-T cells) and response prediction to ICI by artificial intelligence (with focus on the antigen presenting pathway) including the applicability of radiomics.
2. Programmed Cell Death Protein 1 (PD-1) and its Key Role in T Cell Exhaustion
In search for proteins that mediate programmed cell death in T cells upon cytokine deprivation, around 30 years ago Ishida and colleagues identified a protein they termed PD-1, which was inducibly expressed in T cell lines undergoing apoptosis [27]. Structurally, the 50–55-kDa type I transmembrane glycoprotein PD-1 is a monomeric member of the immunoglobulin gene superfamily with an IgV domain homologous to CD28, CTLA-4 and ICOS in the extracellular region. While further research did not confirm a direct participation of PD-1 in programmed cell death of T cells, knockout mice revealed that loss of PD-1 increases the risk of severe T cell mediated autoimmune pathologies and lupus-like syndromes, showing that PD-1 negatively regulates T cell immune responses as an “immune-checkpoint” molecule [28,29,30]. It soon turned out that PD-1 on T cells is important for induction of peripheral but not central T cell tolerance. While interaction of T cells with resting dendritic cells results in tolerance induction, this interaction leads to efficient T cell priming in the absence of PD-1 [31]. In line with this, PD-1 was found to be a key molecule in chronic viral infections in mice. Mice infected with different lymphocytic choriomeningitis virus strains can either suffer an acute infection which is efficiently cleared within weeks or otherwise, the virus is not effectively combated and persists chronically [32]. In the latter case, high numbers of virus specific PD-1 positive T cells accumulate in the host and blocking PD-1 (by blocking antibodies or PD-1 knockout) results in T cell reactivation and clearance of the virus [33]. Based on these experiments, the term T cell exhaustion was coined for antigen specific, primed, PD-1 positive T cells, unable to fight off target cells. In search for ligands for PD-1, two transmembrane glycoproteins were discovered, termed PD-L1 and PD-L2 [34,35]. Unlike PD-1, which is expressed only in distinct immune cell subsets [36], PD-L1/2 are expressed on a wide variety of tissues (Table 1). Particularly, high PD-L1 expression was noticed on some tumor cells and it was shown that its expression suppresses the cytolytic activity of cancer-specific T cells. Analogous to virus experiments, blocking PD-1/PD-L1 interactions can result in the reinvigoration of efficient anti-cancer immune responses, both in mouse models and in patients, proving that T cell exhaustion significantly contributes to immunological tolerance towards tumor cells [37,38,39]. While PD-L1/2 have only short cytoplasmic tails with signaling competence so far only reported in B cells [40], the cytoplasmic domain of PD-1 comprises two conserved signaling motifs, immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM). Generally, PD-L1/2 interaction with PD-1 induces phosphorylation of ITIM and ITSM, leading to recruitment of the phosphatases SHP-1 and SHP-2. Subsequent dephosphorylation of the T cell receptor (TCR) activation signals CD-3ζ and zeta chain-associated protein kinase 70 (ZAP70) leads to inhibition of the downstream phosphatidylinositol 3-kinase (PI3K)/AkT/Ras signaling pathway and shuts down cytokine production and effector activities [41,42,43]. Importantly, PD-1 expression is not restricted to exhausted T cells but generally up-regulated upon T cell activation and expressed in various T cell subsets, such as regulatory T cells (Treg), T follicular helper (TFH) cells, T follicular regulatory (TFR) cells and memory T cells. In addition, it is expressed in several other cell types including B cells, natural killer (NK) cells, some myeloid cells and cancer cells upon activation [36]. Consequently, downstream signaling pathways may be different in the respective cell subsets and vastly depend on co-signals from the microenvironment as well as differentiation, metabolic and hypoxic states. Notably, profiling of exhausted T cells revealed expression of several other exhaustion-related immune-checkpoint receptors, such as T cell membrane protein 3 (TIM-3) or lymphocyte-activation gene 3 (LAG-3) [44] and the outcome of ICI therapies may depend on the exact composition of PD-1 positive immune cell subsets as well as their complex spatiotemporal dynamics in their interaction with tumor cells. Hence, to maximize durable clinical responses to ICI, it will be crucial to find effective combination treatments, which are discussed in this review.

Table 1.
Expression pattern of PD-1 and its ligands. Adapted from [43,45].
3. Kinase Inhibitors as Combination Treatments to Increase T Cell Activation
Although immune-checkpoint inhibition by blocking antibodies efficiently unleashes effective anti-cancer immune responses in some patients, current studies show that targeting T cells by additional (anti-cancer) compounds may potentiate immune-checkpoint therapies. In this context, drugs were shown either to modulate expression of inhibitory receptors on T cells or to interfere with T cell function or differentiation, both synergizing with immune-checkpoint therapies (Figure 2). The serine/threonine kinase glycogen synthase kinase 3 (GSK-3) is a central regulator of PD-1 transcription in CD8+ T cells. Silencing or pharmacological inhibition of GSK-3 in mice resulted in up-regulation of T-box transcription factor 21 (tbx21) in CD8+ T cells, which in turn led to down-regulation of PD-1 and to enhanced cytolytic CD8+ T cell function [46]. In murine cancer models, GSK-3 inhibition was similarly effective as PD-1/PD-L1 blockage in reinvigorating anti-cancer immunity and a mild synergistic effect was noticed [47,48].
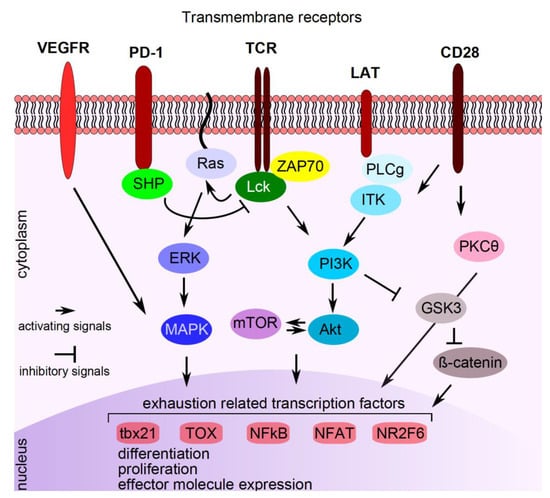
Figure 2.
Pathways interfering with PD-1 signaling. Signaling compounds are indicated in the cytoplasm, transcription factors/repressors are indicated in the nucleus. See text for explanations. VEGFR: vascular endothelial growth factor receptor, PD-1: programmed cell death protein 1, TCR: T cell receptor, LAT: linker for activation of T cells, CD: cluster of differentiation, SHP: small heterodimer partner, Lck: lymphocyte-specific protein tyrosine kinase, ZAP70: zeta chain-associated protein kinase 70, PLCg: phospholipase C gamma 1, ITK: interleukin-2 inducible T cell kinase, ERK: extracellular signal-regulated kinase, PI3K: phosphatidylinositol 3-kinase, PKCӨ: protein kinase C theta, MAPK: mitogen-activated protein kinase, mTOR: mechanistic target of rapamycin, Akt: protein kinase B, GSK3: serine/threonine kinase glycogen synthase kinase 3, tbx21: T-box transcription factor 21, TOX: thymocyte selection-associated high mobility group box protein TOX, NFKB: nuclear factor kappa-light-chain-enhancer of activated B cells, NFAT: nuclear factor of activated T cells, NR2F6: nuclear receptor subfamily 2 group F member 6.
The mechanistic target of rapamycin (mTOR) kinase functions in the context of multiprotein signaling complexes mTORC1/2, which are implicated in diverse metabolic, stress and immunological pathways [49]. Although mTOR inhibition by sirolimus or everolimus is largely immune suppressive and used to impede host versus graft rejection after organ transplantation, intermediate doses of the mTOR inhibitor vistusertib selectively promote effector T cell function and potentiate anti-PD-1, anti-PD-L1 and anti-CTLA-4 therapy in a colorectal mouse tumor model [50].
Interference with cell cycle progression has become an attractive concept for cancer therapy and recently, several cell cycle inhibitors, particularly inhibitors of cyclin dependent kinase 4 and 6 (CDK4/6) (abemaciclib, palbociclib and ribociclib) have been approved for advanced estrogen receptor positive breast cancer [51,52,53]. However, CDK4/6 inhibitors have additional immune activating effects, by increasing antigen presentation in cancer cells and by mediating increased nuclear factor of activated T cells (NFAT) activation in response to TCR engagement and repressed DNA methyltransferase 1 in T cells, which augments anti-cancer immunity upon PD-1 blockade [54,55,56]. Ibrutinib, a Bruton’s tyrosine kinase (BTK) inhibitor, has become standard of care for relapsed and high-risk chronic lymphocytic leukemia (CLL) patients [57]. Although ibrutinib was developed to specifically inhibit BTK dependent B cell receptor (BCR) signaling, it also binds interleukin (IL)-2-inducible T cell kinase (ITKs) in T cells, which leads to increased T cell numbers and function due to impaired activation-induced cell death through ITK inhibition [58] and due to Th1 polarization [59]. Concomitantly, in mouse experiments, ibrutinib enhanced T cell anti-tumor immunity [60], prompting the initiation of a phase 2 study on ibrutinib and PD-1 blockade (pembrolizumab) combination therapy in high-risk CLL (ClinicalTrials.gov Identifier: NCT03514017). In line with this, another BCR signaling inhibitor, acting on PI3K (idelalisib) was also shown to decrease proliferation and effector function of Tregs in vitro. This effect was mediated by inhibition of the PI3K/Akt/nuclear factor kappa-light-chain-enhancer of activated B cells (NFKB) axis, whereupon a synergistic effect of idelalisib with immune-checkpoint inhibition was proposed [61,62].
Recently, the transcription factor TOX was identified to be responsible for driving an exhaustion specific transcriptional profile in T cells. TOX is activated by vascular endothelial growth factor (VEGF)-A upon binding to VEGF receptor (VEGFR) on T cells and inhibition of VEGFR potentiated anti-tumor immunity in mice treated with ICI [63]. Hence, specific kinase inhibitors interfering with VEGF-A/VEGFR downstream signaling could act synergistically with PD-1 blockade in reinvigorating exhausted T cells. In line with this, a specific mitogen-activated protein kinase (MAPK) inhibitor (G-38963, which is similar to cobimetinib) counteracts TCR-induced apoptosis of tumor infiltrating cytotoxic T cells in mice, thereby potentiating anti-tumor immunity. Moreover, combination with anti-PD-L1 treatment results in durable and synergistic tumor regression [64].
Finally, the transcriptional repressor nuclear receptor subfamily 2 group F member 6 (NR2F6) was recently found to be an additional key player of fine-tuning of T cell effector functions. NR2F6 directly occupies promoter regions of important cytokine gene loci, thereby impeding activation-induced binding of NFAT/AP-1 transcription factors [65]. In mouse studies, it could be shown that loss of NR2F6 leads to enhanced T cell activation upon PD-1 blockade and to increased tumor eradication [66]. As NR2F6 is inactivated by protein kinase C (PKC)-theta dependent phosphorylation, specific compounds promoting PKC-theta activity or interfering with NR2F6 dephosphorylation may be useful to potentiate immune-checkpoint therapies [65].
4. Combination of Vaccination Strategies with Immune-Checkpoint Blockade
Despite the success of vaccinations against microbes or viral diseases therapeutic vaccinations against cancer cells have not yielded similar success so far. The major obstacle is to achieve a strong enough immune response, which kills millions of tumor cells to achieve a clinical benefit in an exhausted immune system, which is common in cancer patients. Therefore, vaccinations against hepatitis B virus and human papillomavirus as common causes of cancer are the only effective cancer vaccinations so far.
Nevertheless, several improvements on the way to an effective therapeutic cancer vaccination have been achieved in the last decade. Tumor-associated antigens, which are self-proteins that are abnormally expressed by malignant cells, have been used as target antigens in the past. Neoantigens arising from mutations or oncogenic viral antigens may represent more specific and more efficient targets for vaccination strategies [67]. Due to the immune-stimulating effect, ICI may appear as the ideal combination partner for a vaccination against malignant cells to overcome the exhaustion of the patients’ immune system and possible evasion strategies of the cancer cells. Several preclinical studies could demonstrate this synergistic effect of ICI and vaccination so far [68,69,70] and recently a clinical phase II trial showed the feasibility and efficacy in patients with incurable human papillomavirus 16–related cancer [19]. The combination of nivolumab and the vaccine ISA101 targeting the viral proteins E6 and E7 resulted in encouraging progression-free survival (PFS) and OS in 24 patients compared to already published efficacy data of ICI in similar populations. Nevertheless, as in all other ICI trials before, the majority of patients had no response to immunotherapy in this trial resulting in a median PFS of 2.3 months. Therefore, there is great interest in co-stimulatory molecules expressed on T cells of the patients e.g.,: CD28, ICOS, CD27, 4-1BB, OX40 and CD40L, which may enhance immune response [71]. This has already been shown in preclinical models for agonist OX40 or anti-CD40 antibodies, where the addition to the combination of an anti-CTLA-4 antibody and a vaccine enhanced tumor response in a mouse model [72,73].
5. Augmenting Immune Response by Combining CAR-T Cell Therapy and Immune-Checkpoint Blockade
CAR-T cells are a form of cellular immunotherapy, where T cells are genetically modified ex vivo to express a new surface antigen receptor [74]. Most currently used CAR constructs consist of a single-chain variable fragment (scFV) antigen-recognition domain of an antibody linked to a CD3-derived T cell activation domain and a costimulatory domain (most commonly CD28, 4.1BB, or both) [74]. This allows for major histocompatibility complex (MHC) independent tumor cell recognition and killing.
CAR-T cells targeting the CD19 surface antigen expressed in various B cell malignancies have led to unprecedented results in B-ALL and DLBCL, which resulted in the approval of Axicabtagene ciloleucel (Yescarta®) and Tisagenlecleucel (Kymriah®) for relapsed/refractory B cell lymphomas and B-ALL [20,21,22]. The overall response rate (ORR, i.e., patients achieving a complete (CR)- or partial (PR) remission) in clinical trials with anti-CD19 CAR-T cells for aggressive B cell lymphomas and B-ALL ranges from 52% to 83% [20,21,22]. However, the ORR in patients with CLL treated with anti-CD19 CAR-T cells is substantially lower at about 30% [75], indicating that targeting the same antigen in different malignancies results in heterogeneous therapy responses probably due to the unique nature and microenvironment of different tumor types. CAR-T cells have also been tested in various solid tumors (i.e., against human epidermal growth factor receptor 2 (HER2) or mesothelin) [23,24], however, the results have been far less promising compared to hematologic malignancies, indicating that solid tumors can escape and/or suppress CAR-T cells. There are a bundle of steps for successful CAR-T cell treatment: (1) CAR-T cells have to migrate (i.e., home) to the tumor site (2) recognize a tumor specific antigen (3) exert an immune response against the tumor cell to facilitate killing (4) resist immunosuppressive signals in the tumor microenvironment and (5) persist for a certain period of time for long term disease control. There is an accumulating body of evidence, that the immunosuppressive tumor microenvironment suppresses tumor infiltrating lymphocytes (TILs) and also CAR-T cells. Here we want to summarize mechanisms within the tumor microenvironment that hinder CAR-T cell therapy and strategies to overcome these.
5.1. Mechanisms of Resistance to CAR-T Cell Therapy within the Tumor Microenvironment
Several different tumor types express PD-L1 either due to up-regulation after mutations in the PD-L1 gene (CD274) or as a result of adaptive up-regulation after stimulation with inflammatory cytokines (i.e., interferon-gamma (IFNγ)) present in the microenvironment [76,77]. Binding of PD-L1 to PD-1 generates an inhibitory signal that attenuates the activity of T cells leading to an exhausted phenotype [78,79]. Exhausted T cells are characterized by loss of effector and memory phenotypes, inability to produce cytokines like IFNγ, tumor necrosis factor alpha (TNFα) and IL-2 that inhibits effector functions [78,80].
CAR-T cells, like their physiologic counterparts, express these checkpoint molecules and are therefore equally prone to immunosuppressive signals. Early evidence of this hypothesis was published by Beatty et al. in 2014 [26]. In a mesothelioma mouse model treatment with anti-mesothelin CAR-T cells did not lead to objective responses. After ruling out antigen loss on the tumor cells or lack of CAR-T cell infiltration into the tumor they observed that the CAR-T cells harvested from the tumor site had lost their cytotoxic potential in vitro (i.e., lack of IFNγ production). This was reversible by resting the CAR-T cells ex vivo for 24 h away from the tumor. The CAR-T cells displayed increased expression of the checkpoint molecules PD-1, TIM-3 and LAG-3, which was also reversible after resting the cells ex vivo. These results indicate that CAR-T cells become exhausted and hypofunctional after prolonged exposure to tumor cells due to suppression via checkpoint pathways. Moon et al. confirmed these observations in similar experiments. They injected mesothelioma tumor cell lines into the flanks of NSG mice and treated the mice with anti-mesothelin second generation CAR-T cells. They observed regression of tumor growth but no cures. After excluding antigen loss or lack of CAR expression, they could show that CAR-T cells after antigen encounter in vivo where no longer able to kill mesothelin positive tumor cells in vitro. CAR-T cells that had been exposed to the antigen in vivo, showed a significant up-regulation of PD-1, LAG-3 and TIM-3 indicating CAR-T cell exhaustion [25]. Cherkassky et al. injected anti-mesothelin CAR-T cells into the pleura of mesothelin positive tumor bearing mice and then performed ex vivo stimulation of harvested tumor infiltrating CAR-T cells. Pre-infusion CAR-T cells were used as control. Compared to the control, CAR-T cells exposed to the antigen in vivo had lower levels of cytolytic function and displayed decreased Th1 cytokine secretion in vitro. They could also show that tumor infiltrating CAR-T cells in mice with progressive tumors had high levels of PD-1, TIM-3 and LAG-3 expression indicating that an immunosuppressive microenvironment leads to CAR-T cell hypofunction and favors tumor escape [81]. Taken together, these studies indicate, that CAR-T cells display an exhausted phenotype after prolonged antigen binding in vivo. Gargett et al. evaluated, whether CAR-T cells might already show an exhausted phenotype before infusion. Therefore, they tracked the expression of CD25, CD69, PD-1 and LAG-3 during the manufacturing process of disialoganglioside (GD2) specific CAR-T cells. They observed an up-regulation of PD-1 and LAG-3 upon viral transduction, which declined to normal levels when the cells were cryopreserved. After thawing and in vitro re-stimulation with either anti-CD3/CD28 antibodies or CAR specific antibodies, they observed that re-stimulation via the CAR receptor resulted in higher up-regulation of PD-1 than via CD3/CD28, however, this did not result in a decrease in cytokine production. This shows that GD2 specific CAR-T cells are not functionally exhausted before infusion. When co-culturing the GD2 specific CAR-T cells with melanoma cell lines repetitively, the authors found that the percentage of viable CAR-T cells decreased with each stimulation. Co-cultering with pembrolizumab saved the CAR-T cells from activation-induced cell death, indicating a protective effect of ICI on CAR-T cell viability. Excitingly, when stimulating CAR-T cells in vitro, the authors also found PD-L1 expression on days 3 to 7 after stimulation. The PD-L1 positive cells had lower PD-1 and LAG-3 expression than PD-L1 negative CAR-T cells. When they analyzed blood samples from patients with melanoma treated with the anti-GD2 CAR-T cells in the CARPETS phase I study, they found that, compared to the infused CAR-T cell product, harvested CAR-T cells had up-regulated PD-1 and PD-L1, while normal peripheral CD8+ T cells from the same patient had normal PD-1 expression. They concluded that antigen encounter via the CAR receptor leads to an exhausted phenotype with an associated lack of effector function [82].
Zolov et al. compared the effects of PD-1 signaling of different CAR-T cells. They produced three different CD123 targeting T cells (one with a 4.1BB costimulatory domain, one with CD28 and one without costimulatory domain). They co-cultured these cells with CD123 and PD-L1 positive acute myeloid leukemia (AML) cell lines and found that the CAR-T cells with the CD28 costimulatory domain showed diminished proliferative capacity and cytokine production as compared to the other CAR constructs, indicating that CD28 CAR-T cells might be more prone to exhaustion than their 4.1BB counterpart [83].
All these preclinical models were further corroborated by translational research in the major clinical trials with CD19 CAR-T cells. As an example, Schuster et al. analyzed outcomes of patients with relapsed/refractory aggressive lymphoma from the JULIET trial according to pre-therapeutic CAR-T cell biomarkers. They were able to show, that patients with the highest PD-1/PD-L1 interaction scores as well as patients with the highest percentage of LAG-3 positive T cells had no or short responses and no long-term cures were observed in this cohort of patients [1].
Fraietta et al. reported findings of 41 CLL patients treated with CD19 CAR-T cells. They were able to show that CR patients, compared with PR or non-responding patients, had significantly lower percentages of CAR-T cells with a CD8+PD-1+ phenotype. CD19 CAR-T cells with co-expression of PD-1 and LAG-3 or TIM-3 were associated with poor responses, whereas individuals who had complete and durable remissions were infused with products containing significantly lower frequencies of these cells [84].
To sum up, there is a robust body of evidence from preclinical models and translational research, that CAR-T cell function and persistence can be suppressed by the engagement of checkpoint molecules. In order to cure more patients with this exciting new treatment, strategies to overcome CAR-T cell hypofunction have been explored.
5.2. Overcoming Resistance
The discovery of immune-checkpoints and the subsequent development of checkpoint inhibitors against PD-1, PD-L1 or CTLA-4 has revolutionized immune-oncologic treatment approaches in the last years. As mentioned earlier, suppression of CAR-T cells by the tumor microenvironment leading to an exhausted or senescent phenotype seems to play a major role in treatment failure. Therefore, several approaches to augment the immune response to CAR-T cells via different ways of checkpoint inhibition have been explored.
5.3. CAR-T Cells Combined with Infused Checkpoint Inhibitors
Back in 2013 John et al. already hypothesized that the combination of PD-1 blockade with CAR-T cells could overcome immunosuppression by the microenvironment. Using anti-HER2 CAR-T cells in combination with a PD-1 blocking antibody in a mouse xenograft breast cancer model, they were able to show that the CAR-T cells up-regulate PD-1 after binding to the tumor cells. Mice treated with the combination of CAR-T cells and the PD-1 antibody displayed the strongest reduction in tumor mass and had the longest survival compared to either treatment alone. On a molecular level the combination of the PD-1 antibody and the CAR-T cells led to increased IFNγ and granzyme B production, indicating enhanced effector cell function [85]. Three years later, Cherkassky et al. reported similar findings in an orthotopic mouse model of pleural mesothelioma. They injected subsequently lower doses of mesothelin-specific CAR-T cells (with either a 4.1BB or CD28 costimulatory domain) into the pleura and observed increasing CAR-T cell exhaustion and decreasing cytolytic function at lower CAR-T cell doses. The repeated antigen encounter led to a decrease in effector mechanisms due to up-regulation of the immune-checkpoints PD-1, TIM-3 and LAG-3. In order to overcome this exhaustion, they injected a PD-1 antibody into the peritoneum on day 30 after CAR-T cell treatment, which led to relevant tumor shrinkage. To circumvent repeated antibody infusions, they genetically engineered the CAR-T cell with either small hairpin (sh)RNA blockade or PD-1 CAR-T cells dominant negative receptors to create PD-1 resistant CAR-T cells. Treatment with the PD-1 resistant CAR-T cell led to enhanced tumor burden control and increased survival compared to non-engineered CAR-T cells [81].
Yin et al. tested humanized IL-13Ra2 targeting second generation CAR-T cells in combination with different checkpoint blockades (anti-PD-1, anti-CTLA-4 and anti-TIM-3) in comparison with humanized epidermal growth factor receptor variant III (EGFRvIII) CAR-T cells in a glioma animal model. In an orthotopic mouse model, they injected glioma cells that expressed both IL-13Ra2 and EGFRvIII. When they looked at the expression levels of PD-1, CTLA-4 and TIM-3 on the different CAR-T cells, they observed a different up-regulation after target binding (i.e., CTLA-4 expression was higher in IL-13Ra2 targeting CAR-T cells than in the EGFRvIII CAR-T cells), indicating, that CAR-T cells targeting different antigens rely on different checkpoint molecules. In line with this observation, they were able to show that the combination of the IL-13Ra2 CAR-T cells with a CTLA-4 checkpoint inhibitor resulted in significantly better tumor killing as compared with the combination of a CTLA-4 antibody with the EGFRvIII CAR-T cell [86].
These mechanistic insights have generated a broad range of clinical studies evaluating CAR-T cells in combination with infused ICI.
In 2017, Heczey et al. reported on a phase I clinical trial with anti-GD2 CAR-T cell therapy in patients with neuroblastoma. Eleven patients with a median age of 6.5 years with relapsed or refractory neuroblastoma were treated in three cohorts. Cohort one only received the GD2 CAR-T cells, cohort two received a prior lymphodepletion with cyclophosphamide/fludarabine and cohort three received the same lymphodepletion and pembrolizumab on days 1 and 21. They were able to show increased T cell expansion in the cyclophosphamide/fludarabine cohort, but no effect of the ICI on CAR-T cell expansion was seen. In addition, patients in cohorts two and three had better OS compared to cohort one. Differences between groups 2 and 3 were not reported, probably due to the small sample size [87].
In the same year, anecdotal evidence regarding the efficacy of this combination approach was published by Chong et al. They reported on a 35-year old male with relapsed DLBCL treated with a CD19 CAR (4.1BB costimulatory domain) who showed progression within one month after CAR-T cell infusion. As a result of high PD-L1 expression within the tumor, pembrolizumab was given on day 26 and the patient achieved a remission with pembrolizumab continued every three weeks for one year. The infusion of pembrolizumab led to an increase in CAR-T cell numbers and a decreased expression of PD-1 on CAR-T cells [88].
In the same year Maude et al. reported on four children with relapsed B-ALL that did not show a sufficient response to anti-CD-19 CAR-T cell treatment, who were treated with pembrolizumab. Pembrolizumab treatment resulted in a prolonged detection of circulating CAR-T cells and led to non-lasting objective responses [89].
At the 2018 ASH meeting, Li et al. reported on their single institution experience with the combination of checkpoint blocking antibodies and anti-CD19 CAR-T cells at the Children’s Hospital of Philadelphia. Fourteen patients with relapsed B-ALL or B-lymphoblastic lymphoma, who demonstrated early CAR-T cell loss or lack of response received an ICI no sooner than 14 days after CAR-T cell infusion. Responses were observed in patients with early B cell recovery and extramedullary disease, with some patients displaying ongoing tumor control with ongoing pembrolizumab infusions [90].
At the 2019 ASH meeting Ardeshna et al. reported on the first results of the Alexander trial. Here, patients with relapsed/refractory DLBCL were treated with a bicistronic anti-CD19/anti-CD22 CAR-T cell (AUTO-3) followed by pembrolizumab for three doses every three weeks starting at day 14 after CAR-T cell infusion. Of the 24 patients that have been enrolled, 11 were treated with AUTO-3 and 7 received the combination with pembrolizumab. There were no dose limiting toxicities (DLTs) and no treatment related deaths. Cytokine release syndrome (CRS) grade 1 occurred in 27% of patients and no higher-grade CRS was reported. There was only one case of grade 3 neurotoxicity. Early response rates are promising with an ORR of 57% (29% CR rate) in this ongoing trial [91].
Currently, there are several ongoing trials evaluating the combination of CAR-T cell treatment with infused ICI as shown in Table 2.
Table 2.
Ongoing trials with chimeric antigen receptor (CAR)-T cells in combination with infused checkpoint inhibitors (www.clinicaltrials.gov, access date: 01/30/2020).
5.4. Built in CARs
The combination of CAR-T cell treatment with repetitive intravenous infusions of checkpoint blocking antibodies has several disadvantages, including (1) the need for repeated infusions with an associated increase in treatment cost, (2) the risk of immune related adverse events and (amongst other reasons) (3) lack of penetration of the antibody to the tumor site. Therefore researches have focused on building CARs that either secrete checkpoint inhibitors locally at the tumor site or build CARs with cell intrinsic checkpoint resistance.
5.5. CARs that Secrete Checkpoint Inhibitors In Situ
In 2016, Suarez et al. first reported on a built-in CAR in renal cell carcinoma (RCC) cell lines and in vivo xenograft models. They cloned an anti-PD-L1 antibody sequence into a bicistronic lentiviral vector encoding for an anti carbonic anhydrase IX (anti-CAIX) CAR-T cell. Treatment with this anti-PD-L1 secreting CAR-T cell led to a 50% decrease in T cell exhaustion markers (LAG-3, TIM-3 and PD-1) compared to treatment with a non-secreting CAR-T cell and a three times profounder reduction in tumor mass [92].
One year later, Li et al. reported similar outcomes with an anti-CD-19/PD-1 secreting CAR-T cell. After co-cultering these CAR-T cells with H292-CD19 or SKOV3-CD19 target cells (both with high PD-L1 expression), they found that after 24 h the IFNγ production was similar between the anti-CD19 CAR and the anti-CD19/PD-1 secreting CAR. However, after 72 h, IFNγ production was markedly higher in the anti-CD19/PD-1 secreting CAR indicating prolongation of effector functions. Furthermore, the proliferation rate upon antigen recognition was higher for the anti-CD-19/PD-1 secreting CAR and PD-1 expression was lower, indicating higher proliferative potential and protection from exhaustion. In a xenograft model using the same tumor cell lines, they were able to show, that the anti-CD19/PD-1 secreting CAR had better anti-tumor activity than either the anti-CD19 CAR alone or the anti-CD19 CAR combined with an infused PD-1 antibody. In addition, in vivo expansion was best for the anti-CD19/PD-1 secreting CAR-T cell compared to the other two modalities [93].
Rafiq et al. made similar observations in a mouse lymphoma and ovarian cancer cell model. They generated a second-generation CAR targeting either CD19 or MUC16 that secretes a PD-1 blocking scFV of an antibody. This led to an autocrine binding of the PD-1 antibody to the CAR-T cell but also to bystanding T cells. Similar to the studies above, the authors were able to show an increase in survival in mice treated with anti-PD-1 secreting CAR-T cell compared to CAR-T cell treatment alone [94].
5.6. Inhibiting Checkpoint Signaling in the CAR-T Cell
Another approach is to engineer the PD-1 receptor in order to inhibit intracellular signaling in the CAR-T cell. As an example, Chen et al. genetically engineered CAR-T cells to overexpress a PD-1 dominant negative receptor lacking the intracellular signaling domain. This CAR-T cell exhibited increased proliferation, cytotoxicity, better tumor control and prolonged survival in their mesothelioma mouse model compared to non-engineered CAR-T cells [95]. Using CRISPR/Cas9 technology, Rupp et al. generated PD-1 deficient anti-CD19 CAR-T cells. PD-L1 positive tumor cells rendered their normal CD19 CAR-T cells hypofunctional. With the use of PD-1 deficient CAR-T cells they were able to show enhanced tumor cell killing in a xenograft model of CD19 and PD-L1 positive AML. All animals that received the PD-1 deficient CAR-T cells cleared the tumors within 28 days, whereas this was only achieved in 17% of the mice treated with control CAR-T cells [96].
With the goal of overcoming PD-L1 effects on CAR-T cells, Hu et al. evaluated an anti-mesothelin second generation CAR-T cell with knocked down PD-1 against mesothelin positive triple negative breast cancer (TNBC) cells. They generated mesothelin targeting 4.1BB CAR-T cells that were able to kill mesothelin positive TNBC cells in vitro. As a next step, they compared PD-1 positive anti-mesothelin CAR-T cells to the same CAR-T cells after PD-1 disruption using CRISP/Cas9. The PD-1 disrupted CAR-T cells showed significantly higher antitumor activity in vitro (indicated by higher IFNγ and IL-2 production and cytotoxicity). Interestingly, they also added an anti-PD-1 antibody to rescue the PD-1 positive CAR-T cells. However, PD-1 disruption by CRISP/Cas9 exhibited higher cytotoxicity than the combination of an antibody plus PD-1 positive CAR-T cells. This effect was also observed in a mouse xenograft model, where treatment with PD-1 disrupted CARs lead to a significantly higher reduction of tumor burden than the other combination strategies [97].
Others used this technology to knockout different checkpoint molecules. As an example, Zhang et al. generated LAG-3 deficient CD19 CAR-T cells. These CAR-T cells displayed robust antigen-specific anti-tumor activity in cell culture and in murine xenograft models. However, the anti-tumor effects of the LAG-3 knockout CD19 CAR-T cells were similar to standard CAR-T cells, probably indicating that LAG-3 is not the primary checkpoint by which lymphoma cells induce T cell exhaustion [98]. Therefore, Zou et al. explored whether simultaneous knockout of three checkpoint molecules (PD-1, TIM-3 and LAG-3) in CAR-T cells targeting HER2 leads to increased efficacy. They were able to show that knock down of all three inhibitory receptors led to the highest cytotoxicity and IFNγ production compared to CAR-T cells with knockdown of one of the receptors or no knockdown. Furthermore, they were able to show that the triple knockdown CAR-T cells up-regulated CD56, which correlated with enhanced infiltration of the CAR-T cells into the tumor tissue [99].
Currently, most CAR-T cell treatment is done with autologous T cells, which is a time consuming and costly method. Therefore, Ren et al. used multiplex genome editing using CRISPR/Cas9 to generate allogeneic CAR-T cells with disrupted PD-1, TCR and human leukocyte antigen (HLA)-I against different target antigens. In a mouse xenograft prostate cancer model, they were able to show that PD-1 disrupted CAR-T cells displayed significantly enhanced antitumor activity compared to regular CAR-T cell therapy. They did not observe relevant alloreactivity or graft-versus-host disease (GVHD) with the allogeneic TCR and HLA-I deficient CAR-T cells, showing proof of concept of this approach [100].
5.7. Targeting the Microenvironment
Another possibility is to target the microenvironment with the CAR-T cell. As an example, Zhao et al. constructed a bispecific CAR-T cell that targets the human trophoblast cell surface antigen (Trop2) and PD-L1 at the same time in a gastric cancer model. They showed that the Trop2/PD-L1 CAR-T cells specifically killed Trop 2 and PD-L1 positive gastric cancer cells. The bispecific CAR produced much higher amounts of IFNγ than either the Trop2 or PD-L1 CAR-T cells. This also translated in vivo. The bispecific CAR inhibited tumor growth better than either of the single-target CAR-T cells [101]. Another very exciting approach undertaken by Xie et al. is to generate nanobody-based CAR-T cells that target the tumor microenvironment directly. The variable regions of heavy-chain-only antibodies (VHHs or nanobodies) are small, stable single-domain antibody fragments with affinities comparable to traditional scFVs that can access antigens differently due to their small size. They constructed such nanobody-based CAR-T cells against different molecules found in the tumor microenvironment. Using an anti-PD-L1 CAR they could reduce tumor growth in a melanoma xenograft model. In addition, they generated a CAR-T cell targeting the tumor stroma and vasculature through the EIIIB+ fibronectin splice variant, which is expressed by multiple tumor types. These CAR-T cells successfully delayed tumor growth and improved survival. These results form the basis for different combination strategies in the future [102].
Liu et al. generated chimeric switch receptor CAR-T cells that contain the extracellular domain of PD-1 fused to the transmembrane and cytoplasmic domain of CD28. When this switch receptor binds to PD-L1 it transmits an activating signal via CD28 instead of an inhibitory signal seen with the normal PD-1/PD-L1 interaction. When co-culturing these CAR-T cells with PD-L1 positive tumors, they observed an increased killing efficacy compared to CARs without the switch receptor, indicating that addition of a switch receptor can convert an inhibitory signal into an activating signal [103].
All these fascinating preclinical experiments are translated into in-human use with a number of early phase clinical trials that have been opened recently as summarized in Table 3.
Table 3.
Overview of clinical trials with genetically engineered CAR-T cells (www.clinicaltrials.gov, access date: 01/30/2020).
CAR-T cell treatment has revolutionized the treatment of hematologic malignancies. As we are gaining more knowledge of mechanisms that are responsible for treatment failure and with the advances in genetic engineering strategies to overcome resistance are being explored. Results of clinical trials evaluating approaches as outlined in this review are eagerly awaited and are likely to further improve treatment outcomes especially in the field of solid tumors.
6. Toxicity Associated with Immune-Checkpoint Blockade Combination Strategies
The encouraging and exciting activity of ICI therapy comes at the cost of immune related adverse events (IRAE). IRAE are thought to arise from an “over-activation” of the immune system leading to autoimmune inflammatory events affecting virtually any organ, most commonly the skin, gastrointestinal tract, liver, endocrine system and lung [104,105]. Excellent guidelines on management of these conditions have been recently published and can be found elsewhere [106]. In this review, we have highlighted potential combination strategies with ICI. We have seen in the past that the combination of different ICI (i.e., nivolumab with ipilimumab) results in an increased rate of severe IRAE as reviewed recently in a meta-analysis [107]. Consequently, there is a relevant concern that the combination of ICI with abovementioned potent therapies may lead to excessive toxicity. Overall, clinical experience with the combination strategies discussed in this review is limited. However, a recent phase 3 trial comparing the combination of the tyrosine kinase inhibitor axitinib and pembrolizumab with sunitinib for the treatment of RCC reported an increased rate of grade 3-5 liver toxicity in the combination arm [15]. Similar evidence for the potential of additive toxicity comes from a phase II trial evaluating nivolumab in combination with ibrutinib for patients with advanced CLL [108]. In the latter study, diarrhea was the most commonly reported adverse event, probably indicating additive toxicity caused by the two drug classes. However, none of the eleven fatal adverse events that have been reported in this trial where deemed to be drug-related [108].
Regarding CAR-T cell treatment, the most relevant drug-specific adverse events are CRS and neurotoxicity [109]. Since CRS results from an over-activation of immune effector cells, combination with ICI causes significant concerns regarding excessive toxicity. Up to now, clinical experience with combination strategies of ICI and CAR-T cells is very limited. In the preliminary studies discussed above, the rate and severity of reported CRS was similar to CAR-T cell monotherapy and no life threatening CRS was reported. Overall, larger randomized studies will be required to evaluate the actual risk for severe adverse events with ICI combinations.
7. Predicting Response to Immune-Checkpoint Inhibition by Artificial Intelligence
7.1. Alterations in the Antigen Presenting Pathway
The cancer immunity cycle highlights a cascade of steps which are necessitated to produce anti-tumor responses by the immune system [110]. However, a magnitude of escape mechanisms prevent tumor neoantigen recognition and in turn abolish the effect of ICI. These escape mechanisms are found at the DNA level (e.g., loss of neoantigens due to chromosomal instability), at the RNA level (e.g., decreased neoantigen expression due to promoter hypermethylation) as well as at the protein level (e.g., gene mutations affecting HLA heterozygosity) [111]. Currently available and/or already established predictive markers for ICI such as PD-L1 [10,11,12,13] and tumor mutational burden (TMB) [112] only depict the tip of the iceberg of the cancer immunity cycle. Mutant tumor peptides have to be intracellularly processed into nine to eleven amino acid peptides, which must fit and be presented in the groove of one of the patients’ surface MHC I molecules [113]. Aspects of the MHC I processing and presentation pathway in order to predict tumor neoantigens, binding affinity of these tumor neoantigens to MHC I, as well as the TCR repertoire have come into the focus of immune-checkpoint blocking strategies. Despite a magnitude of evolving biomarkers for ICI and greatest interest in the gut microbiome [114], antibiotic treatment status [115,116] and T cell exhaustion markers [117], within this subsection we review the literature about tumor neoantigen presentation and prediction with regard to the application of ICI for cancer treatment.
A high TMB has been shown to be a positive predictive marker for clinical outcome with ICI across various tumor entities [118,119,120]. A higher tumor neoantigen burden is associated with improved clinical outcome in advanced NSCLC [120] and advanced melanoma [121] patients undergoing immune-checkpoint blockade and shows a strong correlation with TMB. However, mounting evidence suggests that especially patients with a high clonal neoantigen burden and a low intratumoral neoantigen heterogeneity benefit from ICI [122].
Among 77,803 identified tumor neoantigens, Rizvi et al. only found 28 (0.04%) in more than one melanoma patient [120]. Comparable findings (99% unique neoantigens) were reported among gastrointestinal tumors [123]. These data corroborate that tumor neoantigens appear to be private events. Neoantigen binding to MHC I is the most selective step leading to peptide presentation. Only 3–4% of predicted tumor neoantigens turn out to be MHC I binders and in turn form neoepitopes [124,125]. Bjerregaard et al. investigated natural T cell responses to predicted tumor neoepitopes. Among 1948 predicted neopeptide-MHC I combinations from 13 publications, the vast majority showed a strong binding affinity to MHC I. However, only 53 neoepitopes (3%) were able to elicit T cell responses [126].
Tumor neoantigen prediction models (as summarized in Table 4) could be of special interest for the application of ICI and key questions to be answered by these models are: which mutated proteins are processed into eight to eleven amino acid peptides by the proteasome, and are transferred into the endoplasmatic reticulum by the transporter associated with antigen processing (TAP), and are loaded onto one out of six MHC I molecules in the individual patient (about 12,000 HLA alleles identified in the human population [127]), and are shuttled to the cell surface by chaperone proteins in order to be recognized by cytotoxic T-lymphocytes [128].
Table 4.
Overview of (tumor) neoantigen prediction models.
Each of the aforementioned steps is crucial for proper tumor neoantigen presentation. Down-regulation of TAP1 (e.g., by promoter methylation) is associated with a lower infiltration of TILs and with an inferior clinical outcome in early colorectal cancer (CRC) [150] and genetic variants of TAP are associated with the development of high-grade cervical neoplasia [151]. Lower expression of HLA class I genes as well as of beta-2 microglobulin (β2m) are immune escape mechanisms in NSCLC [122,152,153] and melanoma [153] patients undergoing immune-checkpoint blockade. HLA class I loss has been shown to prevent continuous T cell recognition in a human melanoma model [154]. HLA-A down-regulation is mediated e.g., by the RNA-binding protein MEX3B [155], by loss of function mutations in the genes encoding the interferon-receptor associated Janus kinase 1 (JAK1) or Janus kinase 2 (JAK2) [156,157,158] or by truncating mutations in the gene encoding β2m [156]. A major impact of HLA class I genotype on clinical outcome with ICI has been corroborated by Chowell et al. HLA-I homozygosity in at least one locus was associated with an inferior survival in two independent cancer cohorts undergoing immune-checkpoint blockade and proved as an independent predictor of survival in multivariate analysis. The combined effect of HLA class I genotype and TMB on survival was greater than the effect of TMB alone [16]. In a similar approach, Goodman et al. reported a better discrimination of survival among TMB high cancer patients undergoing immune checkpoint blockade by considering the MHC I genotype [17]. Prediction models such as the Loss of Heterozygosity in Human Leukocyte Antigen (LOHHLA) bioinformatics tool enable estimation of allele-specific HLA loss from sequencing data and improve neoantigen prediction accuracy [146]. Hopkins et al. examined the role of the peripheral TCR repertoire in immunotherapy treated pancreatic adenocarcinoma. A low baseline clonality as well as a high number of expanded clones following treatment with anti-CTLA-4 targeting ipilimumab was associated with a statistically significantly longer survival. The latter results were not reproducible with anti-PD-1 targeting therapy [159]. Comparable findings concerning TCR repertoire dynamics [160] and clinical outcome [160,161] with anti-CTLA-4 and anti-PD-1 targeting therapy were reported in advanced melanoma patients [160]. Despite the limited number of patients included in the aforementioned retrospective analyses, the opposite impact of baseline TCR clonality on clinical outcome with anti-CTLA-4 and anti-PD-1 targeting therapy is hypothesis generating and suggests sequential immunotherapy strategies of anti-CTLA-4 followed by anti-PD-1 targeting therapy.
A high false positive rate remains a major drawback of tumor neoantigen prediction algorithms. MHC class I binding affinity (calculated as the wild-type peptide binding affinity relative to the mutant peptide binding affinity) was demonstrated to be a major determinant of cancer peptide immunogenicity and outperformed TMB as well as neoantigen burden for clinical outcome in melanoma and NSCLC patients undergoing immune-checkpoint blockade [162]. In an integrative approach, Kalaora et al. combined whole-exome and RNA sequencing with MHC-peptidomics (analysis of peptide binding to MHC I by liquid chromatography and tandem mass spectrometry) and the neoantigen prediction tool NETMHCpan in advanced melanoma patients. In a direct comparison, this prediction tool, which integrates binding affinity data and mass spectrometry data, outperformed other neoantigen prediction alogorithms [144]. The latter approach highlights the advantage of combining bioinformatic neoantigen prediction with MHC-peptidomics in order to reduce the rate of false positive neoepitopes, especially in cases of rare HLA allotypes [125,163].
However, peptides with a predicted high MHC I binding affinity are not necessarily immunogenic. In neoepitope prediction strategies, attempts such as the integration of information concerning the hydrophobicity of the TCR contact region [149,164], amino acid characteristics [140] or binding differences between wild-type and mutant epitopes [149] yield at increasing the probability to identify clinically relevant neoepitopes [149]. Calis et al. reported two common properties of neopeptide-MHC combinations, which cause differences in T cell recognition: (1) the composition of amino acids in the position 4-6 of the presented peptide as well as (2) the size and absence/presence of aromatic side chains [140]. Neopepsee, a machine-learning-based neoantigen prediction program, integrates nine immunogenicity features including the aforementioned features and was able to determine immunogenic neoantigens in melanoma and CLL. Furthermore, the presence of immunogenic neoantigens determined by Neopepsee was associated with a better prognosis in patients with gastric cancer [149]. Luksza et al. combined estimations of the probability that a neoantigen will be presented on MHC I and the probability that presented neoantigens will be recognized by the TCR repertoire based on tumor clonality, MHC I binding affinity and microbial epitope homology. This model was applied to two melanoma cohorts and one NSCLC cohort undergoing anti-CTLA-4 and anti-PD-1 targeting therapy, respectively, and predicted survival in each cohort [147]. Snyder et al. developed a bioinformatic pipeline incorporating MHC class I binding probability, TCR binding probability, patient specific HLA genotype and epitope-homology analysis in order to identify putative neoepitopes associated with clinical outcome in advanced melanoma patients undergoing anti-CTLA-4 targeting therapy. Among predicted neoantigens, conserved stretches of amino acids were identified that were shared by patients with clinical benefit exceeding six months. These neoepitope signatures were significantly associated with survival in the discovery as well as in the validation set [165]. Published studies evaluating the antigen presenting pathway and TCR repertoire by artificial intelligence and the impact on clinical outcome in patients undergoing immune-checkpoint blockade are summarized in Table 5.
Table 5.
Impact of the antigen presenting pathway and T cell receptor (TCR) repertoire on clinical outcome with immune-checkpoint inhibitors (ICI).
A plethora of previous studies have focused on individual factors affecting the success of immune-checkpoint blockade in immuno-oncology. However, a comprehensive analysis incorporating multiple factors is of utmost importance. Apart from the antigen presenting pathway, future models predicting clinical outcome with ICI necessitate the integration of additional factors affecting the tumor-host interaction such as PD-L1 expression, gut microbiota composition, patient germline genetics, immune microenvironment composition as well as absence/presence of soluble inhibitory molecules as proposed in several cancer immunograms [117,166,167]. For such an approach, DNA sequencing data of the tumor, RNA sequencing data of the microenvironment and germline DNA sequencing will be required. In this regard, Xie et al. developed a multifactorial deep learning model integrating microsatellite instability (MSI-H) burden, somatic copy number alteration (SCNA) burden and modified TMB (mTMB) into four genomic clusters. Data were derived from 8,646 samples of The Cancer Genome Atlas (TCGA) across 29 tumor types. Interestingly, the abovementioned genomic features only showed a weak to moderate correlation, suggesting that each feature has a distinct impact on tumor biology. The authors used TCGA RNA sequencing data to characterize the tumor microenvironment of each genomic cluster by the level of TIL infiltration, expression of immune genes and status of immune pathways. Each cluster was associated with a unique immune landscape. Genomic clusters discriminated patients with different risk for OS in the entire cohort as well as in multiple cancer types. When applying these four genomic clusters to two anti-CTLA-4 treated melanoma cohorts, cluster 4 (MSIhigh, SCNAhigh, mTMBlow) showed the lowest rate of clinical benefit and the shortest OS [168].
However, prospective validation and reproducibility in a real-world setting will be prerequisites for applying such prediction models in clinical practice.
7.2. Radiomics
In general, the assessment of predictive biomarkers for ICI is frequently limited by the availability of tumor tissue, intralesional as well as interlesional tumor heterogeneity [169] and by expression dynamics during the course of disease [170] and necessitates invasive procedures with relevant periprocedural risks [171,172] in often comorbid cancer patients.
Due to the availability of routinely performed imaging studies and correlations of images with underlying biological processes radiomics may serve a new predictive tool in immuno-oncology in the near future. Apart from non-invasive identification of potential responders to ICI, addressing resistance mechanisms as well as visualization of drug distribution and of the tumor microenvironment are major goals of radiomics in immuno-oncology. Radiomics is based on common imaging modalities such as computed tomography (CT), positron emission tomography (PET) and magnetic resonance tomography (MRT) and necessitates the following steps: image acquisition, identification of the target volumes, segmentation, feature extraction and analysis [173].
7.2.1. Assessment of Mutation Status by Radiomics
CT-based radiomic features are associated with molecular aberrations [174,175,176,177] in various types of cancer. Yang et al. found a highly statistically significant association between a CT-based radiomic signature and KRAS/NRAS/BRAF mutations in a test cohort of 61 CRC patients (area under curve (AUC): 0.869, p < 0.001) and confirmed the results in a validation cohort [174]. In the light of the recently reported positive predictive value of KRAS mutations for pembrolizumab monotherapy response in the KEYNOTE-042 study [178] such a radiomic approach could be of clinical relevance for treatment decisions in advanced non-squamous NSCLC. Mismatch repair deficient (dMMR) tumors harbor high numbers of mutation-associated neoantigens and are considered sensitive to ICI [179]. The latter finding in turn has led to the tissue/site-agnostic approval of pembrolizumab in dMMR solid tumors by the FDA. Huang et al. demonstrated the feasibility to assess the mismatch repair status by a CT-based radiomic signature in a test cohort of 140 CRC patients (AUC: 0.914, p < 0.001) and confirmed the good discrimination in a validation cohort including 114 CRC patients (AUC: 0.702, p = 0.012) [175]. Due to the low frequency of dMMR solid tumors in advanced stages [179], this radiomic approach will only identify a minority of potential responders to ICI. NSCLC harboring activating EGFR mutations are insensitive to ICI monotherapy [180,181]. Yip et al. showed the potential of quantitative CT imaging to predict the EGFR mutation status in operable NSCLC patients in the perioperative setting (AUC: 0.67) [176]. Comparable findings based on FDG-PET CT imaging were described by Gevaert et al. in stage 1–4 NSCLC patients (AUC: 0.89) [177].
7.2.2. PD-1/PD-L1 Expression and Heterogeneity Assessed by Radiomics
CT based radiomic features are capable of separating patients with NSCLC [182,183,184,185,186] as well as head and neck squamous cell carcinoma (HNSCC) [183] with differing risk profiles for survival. Furthermore, CT based radiomic approaches allow prediction of dichotomous PD-L1 expression on tumor cells (tumor proportion score: TPS) [184,185] and density of CD3+ [184] or CD8+ [187] TILs in NSCLC. Successful anti-PD-1/anti-PD-L1 receptor-ligand-pair imaging by PET scans in mice with subcutaneously injected melanoma cells was demonstrated by Hettich et al. [188]. In a similar approach, Xing et al. [189] and Niemeijer et al. [190] investigated the correlation between PD-L1/PD-1 expression based on single photon emission computed tomography (SPECT), PD-L1/PD-1 PET and PD-L1/PD-1 expression assessed by immunohistochemistry (IHC) in NSCLC patients. Xing et al. used the anti-PD-L1 antibody NM-01, site-specifically labeled with technetium-99m, for SPECT imaging in 16 NSCLC patients (including squamous and non-squamous histology) in order to correlate tumor uptake with PD-L1 IHC. Patients with a PD-L1 expression ≤1% demonstrated statistically significantly lower tumor to peripheral blood tracer uptake ratios (mean 1.89 vs. 2.49, p = 0.0048) with a corresponding AUC of 0.88. It is noteworthy that four out of twelve patients with lymph nodes metastases showed considerable intra-patient differences (>20%) of PD-L1 expression [189]. Niemeijer et al. reported a statistically significant correlation between radiotracer uptake (18F-BMS-986192, standardized uptake value: SUV) and PD-L1 expression based on IHC (PD-L1 ≥50%: SUVpeak 8.2 versus PD-L1 <50%: SUVpeak 2.9, p = 0.018). The observed heterogeneous intrapatient and interpatient radiotracer uptake highlights the challenge to adequately assess tumor PD-L1 expression by core needle biopsies [190]. The latter two studies prove the feasibility to assess locoregional differences of PD-L1 expression in primary tumors and distant metastases. The assessment of intrapatient PD-L1 expression heterogeneity by radiomics may facilitate treatment decisions concerning intensity of therapy (ICI monotherapy versus ICI combined with chemotherapy) in clinical practice.
7.2.3. Radiomics Predict Clinical Outcome with ICI Therapy
By combining CT images and RNA-sequencing genomic data from tumor biopsies of patients with advanced solid tumors (MOSCATO trial) [191], Sun et al. developed a radiomic signature that could discriminate between high (>median) and low (<median) density of CD8+ TILs (AUC: 0.74, p < 0.0001) [187] and validated the findings in three independent advanced solid tumor cohorts: TCGA validation set [192], tumor immune phenotype validation set [193] and immunotherapy-treated validation set [194]. Patients with a high radiomic score (CD8+ TILs > median) showed a statistically significantly increased median OS (24.3 versus 11.5 months, p = 0.0081) in the immunotherapy-treated validation set and the radiomic score proved to be the strongest independent prognosticator for OS in multivariate analysis (hazard ratio (HR): 0.52, p = 0.0022) [187]. Bensch et al. found a better correlation between clinical outcome and PD-L1 status assessment by PET imaging (89Zr-atezolizumab) in comparison to PD-L1 evaluation by IHC or RNA-sequencing data in 22 patients undergoing treatment with atezolizumab for bladder cancer, NSCLC or TNBC [195]. Khorrami et al. evaluated changes in the radiomic texture during two to three cycles of ICI therapy and reported the “delta-radiomic risk-score“ to predict response as well as OS with ICI in NSCLC [196]. Trebeschi et al. developed a radiomic signature based on pre-treatment CT images on a lesional level in advanced NSCLC and melanoma patients undergoing anti-PD-1 therapy. These radiomic features were significantly associated with response in pulmonary and nodal NSCLC metastases, whereas the model performed poorly on pulmonary and hepatic melanoma metastases. However, the model statistically significantly predicted OS in both tumor types (NSCLC: AUC: 0.76, p < 0.01; melanoma: AUC: 0.77, p < 0.01) [197]. Correlations of CT-based radiomic features and therapy response were also reported for patients with advanced ovarian cancer [198] and bladder cancer [199] undergoing immune-checkpoint blockade. Table 6 summarizes radiomics studies predicting clinical outcome with immune-checkpoint blockade.
Table 6.
Prediction of clinical outcome by radiomics in cancer patients undergoing immune-checkpoint blockade.
On the one hand, a subset of advanced cancer patients derives long-term survival from immune-checkpoint blockade, on the other hand, up to nine per cent of patients experience hyperprogressive disease with rapid fatal outcome upon initiation of anti-PD-1/anti-PD-L1 therapy [203]. In a clinical-radiomic approach Tunali et al. were able to identify patients with a time to progression < 2 months or hyperprogressive disease within an advanced NSCLC cohort treated with single agent or double agent immunotherapy [200]. The latter finding is of utmost importance in clinical practice as such cancer patients should not be treated with ICI monotherapy or with ICI at all. Apart from predicting clinical outcome with immunotherapy, radiomics also has the potential to predict immune-related adverse event. In a small series of 32 advanced cancer patients, Colen et al. found radiomic features that identified the two patients who experienced immunotherapy-induced pneumonitis (accuracy: 100%, p = 0.0033) [204].
The abovementioned findings corroborate the potential of radiomics to visualize drug distribution, tumor characteristics as well as tumor heterogeneity and the feasibility to predict clinical outcome with ICI. However, a major caveat remains the standardization of imaging acquisition, validation in prospective clinical trials and reproducibility in a real-world setting. ICI trials in advanced solid tumors such as the “INSPIRE” trial (NCT02644369) are prospectively investigating changes in radiomic imaging parameters as well as correlations between tumor genomic profiles and radiomic imaging signatures.
8. Conclusions
ICI represent a promising therapeutic strategy to overcome T cell exhaustion in order to reinvigorate T cell responses against cancer cells. CTLA-4, PD-1 and PD-L1 are the most extensively investigated and targeted immune-checkpoints, however, several other immune-checkpoint molecules such as LAG-3 and TIM-3 are therapeutically targeted in ongoing trials. In recent years, a magnitude of ICI has been approved as monotherapy or as combination therapy for the treatment of solid and hematologic malignancies. Combination strategies as for example with tyrosine kinase inhibitors in metastatic RCC improve clinical outcome but come at the cost of increased grade 3–5 hepatotoxicity. CDK4/6 inhibitors, which are considered as therapeutic standard in combination with endocrine therapy in metastatic hormone receptor positive breast cancer, have been demonstrated to increase antigen presentation in cancer cells and as consequence might serve as potent combination drugs for ICI.
The success with CAR-T cells in hematologic malignancies has revolutionized the therapeutic landscape in DLBCL and B-ALL. Up-regulation of immune-checkpoints drives resistance to CART-T cell therapy in hematologic and solid malignancies, which can be overcome by combination strategies with ICI without increasing CRS or neurotoxicity rates. The latter therapeutic approach can be accomplished by separately infusing ICI, by CAR-T cells that secrete checkpoint inhibitors locally as well as by CAR-T cells with cell intrinsic checkpoint resistance.
Although there is a positive correlation between TMB and survival with ICI across various tumor types, high TMB does not necessarily result in immunogenicity. A plethora of steps is crucial for proper tumor neoantigen presentation and T cell recognition. Alterations in the antigen presenting pathway give rise to resistance mechanisms that in turn abolish the effect of ICI. Tumor neoantigen prediction models have been shown to identify cancer patients who benefit most from immune-checkpoint blockade. However, a high false positive rate is a drawback of these models. Individual immunograms including tumor neoantigen prediction, factors affecting the tumor-host interaction such as PD-L1 expression, gut microbiota composition, patient germline genetics, immune microenvironment composition as well as absence/presence of soluble inhibitory molecules may help to distinguish responders from non-responders to ICI.
The predictive value of already established biomarkers such as PD-L1 is considerably heterogeneous across various malignancies and negativity does not exclude responses. Furthermore, intratumoral and intrapatient heterogeneity complicate tumor tissue-based biomarker assessment. Radiomics offers the opportunity to evaluate biomarkers (including intrapatient heterogeneity) based on imaging studies without the necessity to perform tumor tissue biopsies. Several radiomics studies have shown to predict clinical outcome with ICI. Radiomics might also help to identify patients who are at risk for hyperprogressive disease upon initiation of anti-PD-1/anti-PD-L1 therapy and patients who are at risk for high grade IRAE. However, standardization of imaging acquisition and validation of findings in prospective clinical trials will be necessitated before implementation in clinical practice.
Author Contributions
Everyone who worked substantively on the review is listed as an author and all listed authors contributed to the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
N.Z.’s work is supported by a grant from the Paracelsus Medical University (PMU-FFF E-19-29-156-ZAB). R. Geisberger is supported by a grant from the Austrian Science Fund (FWF; P28201).
Conflicts of Interest
F.H.: travel support: Roche, Merck, BMS and Pfizer. M.L.: honoraria: Takeda, BMS, Pfizer; travel support: Novartis, Roche. T.M.: honoraria from Merck and MSD. G.R.: advisory role: Roche and Pfizer; Speakers’ Bureau: AstraZeneca, BMS, Roche, Pfizer; travel support: Roche, Pfizer, BMS. R. Greil: honoraria: Roche, Merck, AstraZeneca, BMS; advisory role: Roche, BMS, AstraZeneca, Merck; research funding: Merck, AstraZeneca, BMS, Roche; travel support: Roche, AstraZeneca, BMS. The remaining authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| AB | antibody |
| Akt | protein kinase B |
| AML | acute myeloid leukemia |
| APC | antigen presenting cell |
| AUC | area under curve |
| β2m | beta-2 microglobulin |
| B-ALL | B cell acute lymphoblastic leukemia |
| BCMA | B cell maturation antigen |
| BCR | B cell receptor |
| BTK | Bruton’s tyrosine kinase |
| CAIX | carbonic anhydrase IX |
| CAR | chimeric antigen receptor |
| CD | cluster of differentiation |
| CDK4/6 | cyclin dependent kinase 4 and 6 |
| CLL | chronic lymphocytic leukemia |
| CR | complete remission |
| CRC | colorectal cancer |
| CRS | cytokine release syndrome |
| CT | computed tomography |
| CTLA-4 | cytotoxic T-lymphocyte protein 4 |
| CTx | chemotherapy |
| DLBCL | diffuse large B cell lymphoma |
| DLT | dose limiting toxicity |
| dMMR | mismatch repair deficiency |
| EGFR | epidermal growth factor receptor |
| EGFRvIII | epidermal growth factor receptor variant III |
| ERK | extracellular signal-regulated kinase |
| FDA | Food and Drug Administration |
| GD2 | disialoganglioside |
| GVHD | graft-versus-host disease |
| GSK-3 | serine/threonine kinase glycogen synthase kinase 3 |
| HCC | hepatocellular carcinoma |
| HER2 | human epidermal growth factor receptor 2 |
| HL | Hodgkin’s lymphoma |
| HLA | human leukocyte antigen |
| HNSCC | head and neck squamous cell carcinoma |
| HR | hazard ratio |
| ICI | immune-checkpoint inhibitors |
| IHC | immunohistochemistry |
| IL | interleukin |
| IFNγ | interferon-gamma |
| IRAE | immune-related adverse events |
| ITK | interleukin-2 inducible T cell kinase |
| ITIM | immunoreceptor tyrosine-based inhibitory motif |
| ITSM | immunoreceptor tyrosine-based switch motif |
| JAK | Janus kinase |
| LAG-3 | lymphocyte-activation gene 3 |
| LAT | linker for activation of T cells |
| Lck | lymphocyte-specific protein tyrosine kinase |
| LOH | loss of heterozygosity |
| LOHHLA | Loss of Heterozygosity in Human Leukocyte Antigen |
| MAPK | mitogen-activated protein kinase |
| MCL | mantle cell lymphoma |
| MHC | major histocompatibility complex |
| MRT | magnetic resonance tomography |
| MSI-H | microsatellite instability |
| mTOR | mechanistic target of rapamycin |
| MUC1 | mucin 1 |
| NFAT | nuclear factor of activated T cells |
| NK cell | natural killer cell |
| NKT cell | natural killer T cell |
| NFKB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NHL | non-Hodgkin lymphoma |
| NR2F6 | nuclear receptor subfamily 2 group F member 6 |
| NSCLC | non-small cell lung cancer |
| OS | overall survival |
| ORR | overall response rate |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed cell death-ligand 1 |
| PET | positron emission tomography |
| PFS | progression-free survival |
| PI3K | phosphatidylinositol 3-kinase |
| PKC | protein kinase C |
| PLCg | phospholipase C gamma 1 |
| PMBCL | primary mediastinal B cell lymphoma |
| PR | partial remission |
| RCC | renal cell carcinoma |
| RGMb | repulsive guidance molecule B |
| SBRT | stereotactic body radiotherapy |
| scFV | single-chain variable fragment |
| SCNA | somatic copy number alterations |
| SHP | small heterodimer partner |
| shRNA | small hairpin RNA |
| SPECT | single photon emission computed tomography |
| SUV | standardized uptake value |
| TAP | transporter associated with antigen processing |
| tbx21 | T-box transcription factor 21 |
| TCGA | The Cancer Genome Atlas |
| TCR | T cell receptor |
| TFH | T follicular helper cell |
| TFR | T follicular regulatory cell |
| TH2 | T helper 2 cell |
| TILs | tumor infiltrating lymphocytes |
| TIM-3 | T cell membrane protein 3 |
| TKI | tyrosine kinase inhibitor |
| TMB | tumor mutational burden |
| TNBC | triple negative breast cancer |
| TNFα | tumor necrosis factor alpha |
| TOX | thymocyte selection-associated high mobility group box protein TOX |
| TPS | tumor proportion score |
| Treg | regulatory T cell |
| Tx | therapy |
| VEGF | vascular endothelial growth factor |
| VEGFR | vascular endothelial growth factor receptor |
| ZAP70 | zeta chain-associated protein kinase 70 |
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Renner, K.; Singer, K.; Koehl, G.E.; Geissler, E.K.; Peter, K.; Siska, P.J.; Kreutz, M. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2017, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.J.; Lau, P.K.H.; Unsworth, A.S.; Loi, S.; Darcy, P.K.; Kershaw, M.H.; Slaney, C.Y. Tissue-Dependent Tumor Microenvironments and Their Impact on Immunotherapy Responses. Front. Immunol. 2018, 9, 70. [Google Scholar] [CrossRef]
- Dyck, L.; Mills, K.H.G. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur. J. Immunol. 2017, 47, 765–779. [Google Scholar] [CrossRef]
- Cruz, E.; Kayser, V. Monoclonal antibody therapy of solid tumors: Clinical limitations and novel strategies to enhance treatment efficacy. Biologics 2019, 13, 33–51. [Google Scholar] [CrossRef]
- Dai, S.; Jia, R.; Zhang, X.; Fang, Q.; Huang, L. The PD-1/PD-Ls pathway and autoimmune diseases. Cell Immunol. 2014, 290, 72–79. [Google Scholar] [CrossRef]
- Vokes, E.E.; Ready, N.; Felip, E.; Horn, L.; Burgio, M.A.; Antonia, S.J.; Aren Frontera, O.; Gettinger, S.; Holgado, E.; Spigel, D.; et al. Nivolumab versus docetaxel in previously treated advanced non-small-cell lung cancer (CheckMate 017 and CheckMate 057): 3-year update and outcomes in patients with liver metastases. Ann. Oncol. 2018, 29, 959–965. [Google Scholar] [CrossRef]
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-Year Overall Survival for Patients With Advanced NonSmall-Cell Lung Cancer Treated With Pembrolizumab: Results From the Phase I KEYNOTE-001 Study. J. Clin. Oncol. 2019, 37, 2518–2527. [Google Scholar] [CrossRef]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Vuky, J.; Powles, T.; Plimack, E.R.; Hahn, N.M.; de Wit, R.; Pang, L.; et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 1483–1492. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulieres, D.; Tahara, M.; de Castro, G., Jr.; Psyrri, A.; Baste, N.; Neupane, P.; Bratland, A.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Chen, R.L.; Zhou, J.X.; Cao, Y.; Li, S.H.; Li, Y.H.; Jiang, M.; Lu, D.Y.; Li, P.; Lin, L.Z. The efficacy of PD-1/PD-L1 inhibitors in advanced squamous-cell lung cancer: A meta-analysis of 3112 patients. Immunotherapy 2019, 11, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulieres, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef]
- Goodman, A.; Pyke, R.; Kato, S.; Okamura, R.; Sokol, E.; Frampton, G.M.; Miller, V.A.; Carter, H.; Kurzrock, R. MHC-1 genotype as a predictor of response to immunotherapy. J. Clin. Oncol. 2019, 37, 149. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Massarelli, E.; William, W.; Johnson, F.; Kies, M.; Ferrarotto, R.; Guo, M.; Feng, L.; Lee, J.J.; Tran, H.; Kim, Y.U.; et al. Combining Immune Checkpoint Blockade and Tumor-Specific Vaccine for Patients With Incurable Human Papillomavirus 16-Related Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 67–73. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Schmidts, A.; Maus, M.V. Making CAR T Cells a Solid Option for Solid Tumors. Front. Immunol. 2018, 9, 2593. [Google Scholar] [CrossRef] [PubMed]
- Knochelmann, H.M.; Smith, A.S.; Dwyer, C.J.; Wyatt, M.M.; Mehrotra, S.; Paulos, C.M. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front. Immunol. 2018, 9, 1740. [Google Scholar] [CrossRef]
- Moon, E.K.; Wang, L.C.; Dolfi, D.V.; Wilson, C.B.; Ranganathan, R.; Sun, J.; Kapoor, V.; Scholler, J.; Pure, E.; Milone, M.C.; et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014, 20, 4262–4273. [Google Scholar] [CrossRef]
- Beatty, G.L.; Moon, E.K. Chimeric antigen receptor T cells are vulnerable to immunosuppressive mechanisms present within the tumor microenvironment. Oncoimmunology 2014, 3, e970027. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Nishimura, H.; Minato, N.; Nakano, T.; Honjo, T. Immunological studies on PD-1 deficient mice: Implication of PD-1 as a negative regulator for B cell responses. Int. Immunol. 1998, 10, 1563–1572. [Google Scholar] [CrossRef]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef]
- Probst, H.C.; McCoy, K.; Okazaki, T.; Honjo, T.; van den Broek, M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat. Immunol. 2005, 6, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Salmi, A.; Butler, L.D.; Chiller, J.M.; Oldstone, M.B. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J. Exp. Med. 1984, 160, 521–540. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Gassner, F.J.; Zaborsky, N.; Catakovic, K.; Rebhandl, S.; Huemer, M.; Egle, A.; Hartmann, T.N.; Greil, R.; Geisberger, R. Chronic lymphocytic leukaemia induces an exhausted T cell phenotype in the TCL1 transgenic mouse model. Br. J. Haematol. 2015, 170, 515–522. [Google Scholar] [CrossRef]
- Kim, Y.S.; Park, G.B.; Lee, H.K.; Song, H.; Choi, I.H.; Lee, W.J.; Hur, D.Y. Cross-linking of B7-H1 on EBV-transformed B cells induces apoptosis through reactive oxygen species production, JNK signaling activation, and fasL expression. J. Immunol. 2008, 181, 6158–6169. [Google Scholar] [CrossRef]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci. Signal. 2012, 5, ra46. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Catakovic, K.; Klieser, E.; Neureiter, D.; Geisberger, R. T cell exhaustion: From pathophysiological basics to tumor immunotherapy. Cell Commun. Signal. 2017, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Schildberg, F.A.; Klein, S.R.; Freeman, G.J.; Sharpe, A.H. Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. Immunity 2016, 44, 955–972. [Google Scholar] [CrossRef]
- Taylor, A.; Harker, J.A.; Chanthong, K.; Stevenson, P.G.; Zuniga, E.I.; Rudd, C.E. Glycogen Synthase Kinase 3 Inactivation Drives T-bet-Mediated Downregulation of Co-receptor PD-1 to Enhance CD8(+) Cytolytic T Cell Responses. Immunity 2016, 44, 274–286. [Google Scholar] [CrossRef]
- Krueger, J.; Rudd, C.E.; Taylor, A. Glycogen synthase 3 (GSK-3) regulation of PD-1 expression and and its therapeutic implications. Semin. Immunol. 2019, 42, 101295. [Google Scholar] [CrossRef]
- Taylor, A.; Rothstein, D.; Rudd, C.E. Small-Molecule Inhibition of PD-1 Transcription Is an Effective Alternative to Antibody Blockade in Cancer Therapy. Cancer Res. 2018, 78, 706–717. [Google Scholar] [CrossRef]
- Weichhart, T.; Hengstschlager, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Langdon, S.; Hughes, A.; Taylor, M.A.; Kuczynski, E.A.; Mele, D.A.; Delpuech, O.; Jarvis, L.; Staniszewska, A.; Cosulich, S.; Carnevalli, L.S.; et al. Combination of dual mTORC1/2 inhibition and immune-checkpoint blockade potentiates anti-tumour immunity. Oncoimmunology 2018, 7, e1458810. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [PubMed]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; Andre, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (Version 4.2020). Available online: https://www.nccn.org/professionals/physician_gls/pdf/cll.pdf (accessed on 20 March 2020).
- Long, M.; Beckwith, K.; Do, P.; Mundy, B.L.; Gordon, A.; Lehman, A.M.; Maddocks, K.J.; Cheney, C.; Jones, J.A.; Flynn, J.M.; et al. Ibrutinib treatment improves T cell number and function in CLL patients. J. Clin. Investig. 2017, 127, 3052–3064. [Google Scholar] [CrossRef]
- Dubovsky, J.A.; Beckwith, K.A.; Natarajan, G.; Woyach, J.A.; Jaglowski, S.; Zhong, Y.; Hessler, J.D.; Liu, T.M.; Chang, B.Y.; Larkin, K.M.; et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 2013, 122, 2539–2549. [Google Scholar] [CrossRef]
- Sagiv-Barfi, I.; Kohrt, H.E.; Burckhardt, L.; Czerwinski, D.K.; Levy, R. Ibrutinib enhances the antitumor immune response induced by intratumoral injection of a TLR9 ligand in mouse lymphoma. Blood 2015, 125, 2079–2086. [Google Scholar] [CrossRef]
- Chellappa, S.; Kushekhar, K.; Munthe, L.A.; Tjonnfjord, G.E.; Aandahl, E.M.; Okkenhaug, K.; Tasken, K. The PI3K p110delta Isoform Inhibitor Idelalisib Preferentially Inhibits Human Regulatory T Cell Function. J. Immunol. 2019, 202, 1397–1405. [Google Scholar] [CrossRef]
- Hock, B.D.; MacPherson, S.A.; McKenzie, J.L. Idelalisib and caffeine reduce suppression of T cell responses mediated by activated chronic lymphocytic leukemia cells. PLoS ONE 2017, 12, e0172858. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.G.; Jang, M.; Kim, Y.; Leem, G.; Kim, K.H.; Lee, H.; Kim, T.S.; Choi, S.J.; Kim, H.D.; Han, J.W.; et al. VEGF-A drives TOX-dependent T cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci. Immunol. 2019, 4, eaay0555. [Google Scholar] [CrossRef] [PubMed]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Hermann-Kleiter, N.; Baier, G. Orphan nuclear receptor NR2F6 acts as an essential gatekeeper of Th17 CD4+ T cell effector functions. Cell Commun. Signal. 2014, 12, 38. [Google Scholar] [CrossRef] [PubMed]
- Klepsch, V.; Hermann-Kleiter, N.; Do-Dinh, P.; Jakic, B.; Offermann, A.; Efremova, M.; Sopper, S.; Rieder, D.; Krogsdam, A.; Gamerith, G.; et al. Nuclear receptor NR2F6 inhibition potentiates responses to PD-L1/PD-1 cancer immune checkpoint blockade. Nat. Commun. 2018, 9, 1538. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, X.; Li, J.; Mo, L.; Zhao, H.; Zhu, Y.; Hu, Z.; Gao, J.; Tan, W. PD-1 blockade enhances the antitumor efficacy of GM-CSF surface-modified bladder cancer stem cells vaccine. Int. J. Cancer 2018, 142, 2106–2117. [Google Scholar] [CrossRef]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W.; et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 2015, 7, 283ra52. [Google Scholar] [CrossRef]
- Li, N.; Qin, H.; Li, X.; Zhou, C.; Wang, D.; Ma, W.; Lin, C.; Zhang, Y.; Wang, S.; Zhang, S. Synergistic antitumor effect of chemotactic-prostate tumor-associated antigen gene-modified tumor cell vaccine and anti-CTLA-4 mAb in murine tumor model. Immunol. Lett. 2007, 113, 90–98. [Google Scholar] [CrossRef]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef]
- Linch, S.N.; Kasiewicz, M.J.; McNamara, M.J.; Hilgart-Martiszus, I.F.; Farhad, M.; Redmond, W.L. Combination OX40 agonism/CTLA-4 blockade with HER2 vaccination reverses T-cell anergy and promotes survival in tumor-bearing mice. Proc. Natl. Acad. Sci. USA 2016, 113, E319–E327. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, M.R.; Holst, P.J.; Steffensen, M.A.; Christensen, J.P.; Thomsen, A.R. Adenoviral vaccination combined with CD40 stimulation and CTLA-4 blockage can lead to complete tumor regression in a murine melanoma model. Vaccine 2010, 28, 6757–6764. [Google Scholar] [CrossRef] [PubMed]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [PubMed]
- Wolff, F.; Leisch, M.; Greil, R.; Risch, A.; Pleyer, L. The double-edged sword of (re)expression of genes by hypomethylating agents: From viral mimicry to exploitation as priming agents for targeted immune checkpoint modulation. Cell Commun. Signal. 2017, 15, 13. [Google Scholar] [CrossRef]
- Kon, E.; Benhar, I. Immune checkpoint inhibitor combinations: Current efforts and important aspects for success. Drug Resist. Updat. 2019, 45, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Zolov, S.N.; Rietberg, S.P.; Bonifant, C.L. Programmed cell death protein 1 activation preferentially inhibits CD28.CAR-T cells. Cytotherapy 2018, 20, 1259–1266. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Boesteanu, A.C.; Binder, Z.A.; Xu, C.; Reid, R.A.; Rodriguez, J.L.; Cook, D.R.; Thokala, R.; Blouch, K.; McGettigan-Croce, B.; et al. Checkpoint Blockade Reverses Anergy in IL-13Ralpha2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol. Ther. Oncolytics 2018, 11, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef]
- Maude, S.L.; Hucks, G.E.; Self, A.E.; Talekar, M.K.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Gonzalez, V.; Nazimuddin, F.; Gupta, M.; et al. The effect of pembrolizumab in combination with CD19-targeted chimeric antigen receptor (CAR) T cells in relapsed acute lymphoblastic leukemia (ALL). J. Clin. Oncol. 2017, 35, 103. [Google Scholar] [CrossRef]
- Li, A.M.; Hucks, G.E.; Dinofia, A.M.; Seif, A.E.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Fasano, C.; McBride, B.; Gonzalez, V.; et al. Checkpoint Inhibitors Augment CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy in Relapsed B-Cell Acute Lymphoblastic Leukemia. Blood 2018, 132, 556. [Google Scholar] [CrossRef]
- Ardeshna, K.; Marzolini, M.A.V.; Osborne, W.; Al-Hajj, M.; Thomas, S.; Faulkner, J.; Pule, M.; Peddareddigari, V.G.R.; Khokhar, N.Z. Study of AUTO3, the First Bicistronic Chimeric Antigen Receptor (CAR) Targeting CD19 and CD22, Followed By Anti-PD1 Consolidation in Patients with Relapsed/Refractory (r/r) Diffuse Large B Cell Lymphoma (DLBCL): Alexander Study. Blood 2018, 132, 1679. [Google Scholar] [CrossRef]
- Suarez, E.R.; de Chang, K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Chen, N.; Morello, A.; Tano, Z.; Adusumilli, P.S. CAR T-cell intrinsic PD-1 checkpoint blockade: A two-in-one approach for solid tumor immunotherapy. Oncoimmunology 2017, 6, e1273302. [Google Scholar] [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother. 2019, 68, 365–377. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Cheng, C.; Mu, W.; Liu, X.; Li, N.; Wei, X.; Liu, X.; Xia, C.; Wang, H. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front. Med. 2017, 11, 554–562. [Google Scholar] [CrossRef]
- Zou, F.; Lu, L.; Liu, J.; Xia, B.; Zhang, W.; Hu, Q.; Liu, W.; Zhang, Y.; Lin, Y.; Jing, S.; et al. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat. Commun. 2019, 10, 4109. [Google Scholar] [CrossRef]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef]
- Zhao, W.; Jia, L.; Zhang, M.; Huang, X.; Qian, P.; Tang, Q.; Zhu, J.; Feng, Z. The killing effect of novel bi-specific Trop2/PD-L1 CAR-T cell targeted gastric cancer. Am. J. Cancer Res. 2019, 9, 1846–1856. [Google Scholar]
- Xie, Y.J.; Dougan, M.; Jailkhani, N.; Ingram, J.; Fang, T.; Kummer, L.; Momin, N.; Pishesha, N.; Rickelt, S.; Hynes, R.O.; et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc. Natl. Acad. Sci. USA 2019, 116, 7624–7631. [Google Scholar] [CrossRef]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef]
- Naidoo, J.; Page, D.B.; Li, B.T.; Connell, L.C.; Schindler, K.; Lacouture, M.E.; Postow, M.A.; Wolchok, J.D. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol. 2015, 26, 2375–2391. [Google Scholar] [CrossRef]
- Champiat, S.; Lambotte, O.; Barreau, E.; Belkhir, R.; Berdelou, A.; Carbonnel, F.; Cauquil, C.; Chanson, P.; Collins, M.; Durrbach, A.; et al. Management of immune checkpoint blockade dysimmune toxicities: A collaborative position paper. Ann. Oncol. 2016, 27, 559–574. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; Ernstoff, M.S.; Gardner, J.M.; Ginex, P.; et al. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2018, 36, 1714–1768. [Google Scholar] [CrossRef]
- Park, R.; Lopes, L.; Cristancho, C.R.; Riano, I.M.; Saeed, A. Treatment-Related Adverse Events of Combination Immune Checkpoint Inhibitors: Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10, 258. [Google Scholar] [CrossRef]
- Younes, A.; Brody, J.; Carpio, C.; Lopez-Guillermo, A.; Ben-Yehuda, D.; Ferhanoglu, B.; Nagler, A.; Ozcan, M.; Avivi, I.; Bosch, F.; et al. Safety and activity of ibrutinib in combination with nivolumab in patients with relapsed non-Hodgkin lymphoma or chronic lymphocytic leukaemia: A phase 1/2a study. Lancet Haematol. 2019, 6, e67–e78. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, T.; Li, J.; Lin, J.; Liang, W.; Huang, W.; Wan, N.; Jiang, J. Association Between Tumor Mutation Burden (TMB) and Outcomes of Cancer Patients Treated With PD-1/PD-L1 Inhibitions: A Meta-Analysis. Front. Pharmacol. 2019, 10, 673. [Google Scholar] [CrossRef] [PubMed]
- Doherty, P.C.; Zinkernagel, R.M. A biological role for the major histocompatibility antigens. Lancet 1975, 1, 1406–1409. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Huemer, F.; Rinnerthaler, G.; Westphal, T.; Hackl, H.; Hutarew, G.; Gampenrieder, S.P.; Weiss, L.; Greil, R. Impact of antibiotic treatment on immune-checkpoint blockade efficacy in advanced non-squamous non-small cell lung cancer. Oncotarget 2018, 9, 16512–16520. [Google Scholar] [CrossRef]
- Derosa, L.; Hellmann, M.D.; Spaziano, M.; Halpenny, D.; Fidelle, M.; Rizvi, H.; Long, N.; Plodkowski, A.J.; Arbour, K.C.; Chaft, J.E.; et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann. Oncol. 2018, 29, 1437–1444. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019, 4, 126908. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Robbins, P.F.; Tran, E.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Ivey, G.; Li, Y.F.; El-Gamil, M.; Lalani, A.; et al. Unique Neoantigens Arise from Somatic Mutations in Patients with Gastrointestinal Cancers. Cancer Discov. 2019, 9, 1022–1035. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Pletscher-Frankild, S.; Jensen, L.J.; Mann, M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell. Proteom. 2015, 14, 658–673. [Google Scholar] [CrossRef]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Bjerregaard, A.M.; Nielsen, M.; Jurtz, V.; Barra, C.M.; Hadrup, S.R.; Szallasi, Z.; Eklund, A.C. An Analysis of Natural T Cell Responses to Predicted Tumor Neoepitopes. Front. Immunol. 2017, 8, 1566. [Google Scholar] [CrossRef]
- Robinson, J.; Halliwell, J.A.; Hayhurst, J.D.; Flicek, P.; Parham, P.; Marsh, S.G. The IPD and IMGT/HLA database: Allele variant databases. Nucleic Acids Res. 2015, 43, D423–D431. [Google Scholar] [CrossRef]
- The problem with neoantigen prediction. Nat. Biotechnol. 2017, 35, 97. [CrossRef]
- Mamitsuka, H. Predicting peptides that bind to MHC molecules using supervised learning of hidden Markov models. Proteins 1998, 33, 460–474. [Google Scholar] [CrossRef]
- Donnes, P.; Elofsson, A. Prediction of MHC class I binding peptides, using SVMHC. BMC Bioinform. 2002, 3, 25. [Google Scholar] [CrossRef]
- Nielsen, M.; Lundegaard, C.; Worning, P.; Lauemoller, S.L.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003, 12, 1007–1017. [Google Scholar] [CrossRef]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O.; Nielsen, M. An integrative approach to CTL epitope prediction: A combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur. J. Immunol. 2005, 35, 2295–2303. [Google Scholar] [CrossRef]
- Antes, I.; Siu, S.W.; Lengauer, T. DynaPred: A structure and sequence based method for the prediction of MHC class I binding peptide sequences and conformations. Bioinformatics 2006, 22, e16–e24. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Lamberth, K.; Harndahl, M.; Justesen, S.; Roder, G.; Peters, B.; Sette, A.; Lund, O.; et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE 2007, 2, e796. [Google Scholar] [CrossRef] [PubMed]
- Lundegaard, C.; Lamberth, K.; Harndahl, M.; Buus, S.; Lund, O.; Nielsen, M. NetMHC-3.0: Accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2008, 36, W509–W512. [Google Scholar] [CrossRef] [PubMed]
- Hoof, I.; Peters, B.; Sidney, J.; Pedersen, L.E.; Sette, A.; Lund, O.; Buus, S.; Nielsen, M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 2009, 61, 1–13. [Google Scholar] [CrossRef]
- Zhang, H.; Lund, O.; Nielsen, M. The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: Application to MHC-peptide binding. Bioinformatics 2009, 25, 1293–1299. [Google Scholar] [CrossRef]
- Kim, Y.; Sidney, J.; Pinilla, C.; Sette, A.; Peters, B. Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinform. 2009, 10, 394. [Google Scholar] [CrossRef]
- Lundegaard, C.; Lund, O.; Nielsen, M. Prediction of epitopes using neural network based methods. J. Immunol. Methods 2011, 374, 26–34. [Google Scholar] [CrossRef]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Kesmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef]
- Pedersen, L.E.; Rasmussen, M.; Harndahl, M.; Nielsen, M.; Buus, S.; Jungersen, G. A combined prediction strategy increases identification of peptides bound with high affinity and stability to porcine MHC class I molecules SLA-1*04:01, SLA-2*04:01, and SLA-3*04:01. Immunogenetics 2016, 68, 157–165. [Google Scholar] [CrossRef]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef]
- Nielsen, M.; Andreatta, M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Kalaora, S.; Barnea, E.; Merhavi-Shoham, E.; Qutob, N.; Teer, J.K.; Shimony, N.; Schachter, J.; Rosenberg, S.A.; Besser, M.J.; Admon, A.; et al. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget 2016, 7, 5110–5117. [Google Scholar] [CrossRef] [PubMed]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271 e11. [Google Scholar] [CrossRef]
- Luksza, M.; Riaz, N.; Makarov, V.; Balachandran, V.P.; Hellmann, M.D.; Solovyov, A.; Rizvi, N.A.; Merghoub, T.; Levine, A.J.; Chan, T.A.; et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017, 551, 517–520. [Google Scholar] [CrossRef]
- O’Donnell, T.J.; Rubinsteyn, A.; Bonsack, M.; Riemer, A.B.; Laserson, U.; Hammerbacher, J. MHCflurry: Open-Source Class I MHC Binding Affinity Prediction. Cell Syst. 2018, 7, 129–132 e4. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.S.; Kim, E.; Lee, M.G.; Shin, E.C.; Paik, S.; Kim, S. Neopepsee: Accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann. Oncol. 2018, 29, 1030–1036. [Google Scholar] [CrossRef]
- Ling, A.; Lofgren-Burstrom, A.; Larsson, P.; Li, X.; Wikberg, M.L.; Oberg, A.; Stenling, R.; Edin, S.; Palmqvist, R. TAP1 down-regulation elicits immune escape and poor prognosis in colorectal cancer. Oncoimmunology 2017, 6, e1356143. [Google Scholar] [CrossRef]
- Einstein, M.H.; Leanza, S.; Chiu, L.G.; Schlecht, N.F.; Goldberg, G.L.; Steinberg, B.M.; Burk, R.D. Genetic variants in TAP are associated with high-grade cervical neoplasia. Clin. Cancer Res. 2009, 15, 1019–1023. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef] [PubMed]
- Schrors, B.; Lubcke, S.; Lennerz, V.; Fatho, M.; Bicker, A.; Wolfel, C.; Derigs, P.; Hankeln, T.; Schadendorf, D.; Paschen, A.; et al. HLA class I loss in metachronous metastases prevents continuous T cell recognition of mutated neoantigens in a human melanoma model. Oncotarget 2017, 8, 28312–28327. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Malu, S.; McKenzie, J.A.; Andrews, M.C.; Talukder, A.H.; Tieu, T.; Karpinets, T.; Haymaker, C.; Forget, M.A.; Williams, L.J.; et al. The RNA-binding Protein MEX3B Mediates Resistance to Cancer Immunotherapy by Downregulating HLA-A Expression. Clin. Cancer Res. 2018, 24, 3366–3376. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Sucker, A.; Zhao, F.; Pieper, N.; Heeke, C.; Maltaner, R.; Stadtler, N.; Real, B.; Bielefeld, N.; Howe, S.; Weide, B.; et al. Acquired IFNgamma resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat. Commun. 2017, 8, 15440. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef]
- Hopkins, A.C.; Yarchoan, M.; Durham, J.N.; Yusko, E.C.; Rytlewski, J.A.; Robins, H.S.; Laheru, D.A.; Le, D.T.; Lutz, E.R.; Jaffee, E.M. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight 2018, 3, 122092. [Google Scholar] [CrossRef]
- Robert, L.; Harview, C.; Emerson, R.; Wang, X.; Mok, S.; Homet, B.; Comin-Anduix, B.; Koya, R.C.; Robins, H.; Tumeh, P.C.; et al. Distinct immunological mechanisms of CTLA-4 and PD-1 blockade revealed by analyzing TCR usage in blood lymphocytes. Oncoimmunology 2014, 3, e29244. [Google Scholar] [CrossRef]
- Hogan, S.A.; Courtier, A.; Cheng, P.F.; Jaberg-Bentele, N.F.; Goldinger, S.M.; Manuel, M.; Perez, S.; Plantier, N.; Mouret, J.F.; Nguyen-Kim, T.D.L.; et al. Peripheral Blood TCR Repertoire Profiling May Facilitate Patient Stratification for Immunotherapy against Melanoma. Cancer Immunol. Res. 2019, 7, 77–85. [Google Scholar] [CrossRef]
- Ghorani, E.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Lynch, M.; Peggs, K.S.; Swanton, C.; Quezada, S.A. Differential binding affinity of mutated peptides for MHC class I is a predictor of survival in advanced lung cancer and melanoma. Ann. Oncol. 2018, 29, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Freudenmann, L.K.; Marcu, A.; Stevanovic, S. Mapping the tumour human leukocyte antigen (HLA) ligandome by mass spectrometry. Immunology 2018, 154, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Chowell, D.; Krishna, S.; Becker, P.D.; Cocita, C.; Shu, J.; Tan, X.; Greenberg, P.D.; Klavinskis, L.S.; Blattman, J.N.; Anderson, K.S. TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. Proc. Natl. Acad. Sci. USA 2015, 112, E1754–E1762. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, N.; Funt, S.A.; Blank, C.U.; Powles, T.; Rosenberg, J.E.; van der Heijden, M.S. The Cancer Immunogram as a Framework for Personalized Immunotherapy in Urothelial Cancer. Eur. Urol. 2019, 75, 435–444. [Google Scholar] [CrossRef]
- Karasaki, T.; Nagayama, K.; Kuwano, H.; Nitadori, J.I.; Sato, M.; Anraku, M.; Hosoi, A.; Matsushita, H.; Morishita, Y.; Kashiwabara, K.; et al. An Immunogram for the Cancer-Immunity Cycle: Towards Personalized Immunotherapy of Lung Cancer. J. Thorac. Oncol. 2017, 12, 791–803. [Google Scholar] [CrossRef]
- Xie, F.; Zhang, J.; Wang, J.; Reuben, A.; Xu, W.; Yi, X.; Varn, F.S.; Ye, Y.; Cheng, J.; Yu, M.; et al. Multifactorial Deep Learning Reveals Pan-Cancer Genomic Tumor Clusters with Distinct Immunogenomic Landscape and Response to Immunotherapy. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Haragan, A.; Field, J.K.; Davies, M.P.A.; Escriu, C.; Gruver, A.; Gosney, J.R. Heterogeneity of PD-L1 expression in non-small cell lung cancer: Implications for specimen sampling in predicting treatment response. Lung Cancer 2019, 134, 79–84. [Google Scholar] [CrossRef]
- Yu, H.; Boyle, T.A.; Zhou, C.; Rimm, D.L.; Hirsch, F.R. PD-L1 Expression in Lung Cancer. J. Thorac. Oncol. 2016, 11, 964–975. [Google Scholar] [CrossRef]
- Wu, C.C.; Maher, M.M.; Shepard, J.A. Complications of CT-guided percutaneous needle biopsy of the chest: Prevention and management. AJR Am. J. Roentgenol. 2011, 196, W678–W682. [Google Scholar] [CrossRef]
- Thampanitchawong, P.; Piratvisuth, T. Liver biopsy:complications and risk factors. World J. Gastroenterol. 1999, 5, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Banna, G.L.; Olivier, T.; Rundo, F.; Malapelle, U.; Fraggetta, F.; Libra, M.; Addeo, A. The Promise of Digital Biopsy for the Prediction of Tumor Molecular Features and Clinical Outcomes Associated With Immunotherapy. Front. Med. (Lausanne) 2019, 6, 172. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Dong, D.; Fang, M.; Zhu, Y.; Zang, Y.; Liu, Z.; Zhang, H.; Ying, J.; Zhao, X.; Tian, J. Can CT-based radiomics signature predict KRAS/NRAS/BRAF mutations in colorectal cancer? Eur. Radiol. 2018, 28, 2058–2067. [Google Scholar] [CrossRef]
- Huang, Q.Y.; Liu, Z. Radiomics approach to characterize microsatellite instability: A CT-based radiomic signature for the detection of the MSI-H phenotype in colorectal cancer. In Proceedings of the ECR 2019, Vienna, Austria, 27 February–3 March 2019. [Google Scholar]
- Yip, S.S.; Kim, J.; Coroller, T.P.; Parmar, C.; Velazquez, E.R.; Huynh, E.; Mak, R.H.; Aerts, H.J. Associations Between Somatic Mutations and Metabolic Imaging Phenotypes in Non-Small Cell Lung Cancer. J. Nucl. Med. 2017, 58, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, O.; Echegaray, S.; Khuong, A.; Hoang, C.D.; Shrager, J.B.; Jensen, K.C.; Berry, G.J.; Guo, H.H.; Lau, C.; Plevritis, S.K.; et al. Predictive radiogenomics modeling of EGFR mutation status in lung cancer. Sci. Rep. 2017, 7, 41674. [Google Scholar] [CrossRef]
- Herbst, R.S.; Lopes, G.; Kowalski, D.M.; Kasahara, K.; Wu, Y.-L.; De Castro, G.; Cho, B.C.; Turna, H.Z.; Cristescu, R.; Aurora-Garg, D.; et al. LBA4 Association of KRAS mutational status with response to pembrolizumab monotherapy given as first-line therapy for PD-L1-positive advanced non-squamous NSCLC in KEYNOTE-042. Ann. Oncol. 2019, 30, mdz453.001. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Lisberg, A.; Garon, E.B. The Italian Nivolumab Expanded Access Program Confirms the Limitations of Single-Agent PD-1 Inhibition in EGFR-Mutant and Never-Smoking Patients with NSCLC. J. Thorac. Oncol. 2018, 13, 1058–1059. [Google Scholar] [CrossRef]
- Lee, C.K.; Man, J.; Lord, S.; Links, M.; Gebski, V.; Mok, T.; Yang, J.C. Checkpoint Inhibitors in Metastatic EGFR-Mutated Non-Small Cell Lung Cancer-A Meta-Analysis. J. Thorac. Oncol. 2017, 12, 403–407. [Google Scholar] [CrossRef]
- Grove, O.; Berglund, A.E.; Schabath, M.B.; Aerts, H.J.; Dekker, A.; Wang, H.; Velazquez, E.R.; Lambin, P.; Gu, Y.; Balagurunathan, Y.; et al. Quantitative computed tomographic descriptors associate tumor shape complexity and intratumor heterogeneity with prognosis in lung adenocarcinoma. PLoS ONE 2015, 10, e0118261. [Google Scholar] [CrossRef]
- Aerts, H.J.; Velazquez, E.R.; Leijenaar, R.T.; Parmar, C.; Grossmann, P.; Carvalho, S.; Bussink, J.; Monshouwer, R.; Haibe-Kains, B.; Rietveld, D.; et al. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat. Commun. 2014, 5, 4006. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Hobbs, B.; Amer, A.; Li, X.; Behrens, C.; Canales, J.R.; Cuentas, E.P.; Villalobos, P.; Fried, D.; Chang, J.Y.; et al. Development of an Immune-Pathology Informed Radiomics Model for Non-Small Cell Lung Cancer. Sci. Rep. 2018, 8, 1922. [Google Scholar] [CrossRef] [PubMed]
- Patil, P.D.; Bera, K.; Vaidya, P.; Prasanna, P.; Khunger, M.; Khunger, A.; Velcheti, V.; Madabhushi, A. Correlation of radiomic features with PD-L1 expression in early stage non-small cell lung cancer (ES-NSCLC) to predict recurrence and overall survival (OS). J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Mazzaschi, G.; Quaini, F.; Milanese, G.; Madeddu, D.; Bocchialini, G.; Ampollini, L.; Gnetti, L.; Lagrasta, C.; Silva, M.; Roti, G.; et al. Differentially regulated high-throughput CT imaging features correlate to distinct tumor immune contextures portraying a radiomic signature with prognostic impact on surgically resected NSCLC. Ann. Oncol. 2019, 30, mdz072.001. [Google Scholar] [CrossRef]
- Sun, R.; Limkin, E.J.; Vakalopoulou, M.; Dercle, L.; Champiat, S.; Han, S.R.; Verlingue, L.; Brandao, D.; Lancia, A.; Ammari, S.; et al. A radiomics approach to assess tumour-infiltrating CD8 cells and response to anti-PD-1 or anti-PD-L1 immunotherapy: An imaging biomarker, retrospective multicohort study. Lancet Oncol. 2018, 19, 1180–1191. [Google Scholar] [CrossRef]
- Hettich, M.; Braun, F.; Bartholoma, M.D.; Schirmbeck, R.; Niedermann, G. High-Resolution PET Imaging with Therapeutic Antibody-based PD-1/PD-L1 Checkpoint Tracers. Theranostics 2016, 6, 1629–1640. [Google Scholar] [CrossRef]
- Xing, Y.; Chand, G.; Liu, C.; Cook, G.J.R.; O’Doherty, J.; Zhao, L.; Wong, N.C.L.; Meszaros, L.K.; Ting, H.H.; Zhao, J. Early Phase I Study of a (99m)Tc-Labeled Anti-Programmed Death Ligand-1 (PD-L1) Single-Domain Antibody in SPECT/CT Assessment of PD-L1 Expression in Non-Small Cell Lung Cancer. J. Nucl. Med. 2019, 60, 1213–1220. [Google Scholar] [CrossRef]
- Niemeijer, A.N.; Leung, D.; Huisman, M.C.; Bahce, I.; Hoekstra, O.S.; van Dongen, G.; Boellaard, R.; Du, S.; Hayes, W.; Smith, R.; et al. Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer. Nat. Commun. 2018, 9, 4664. [Google Scholar] [CrossRef]
- Massard, C.; Michiels, S.; Ferte, C.; Le Deley, M.C.; Lacroix, L.; Hollebecque, A.; Verlingue, L.; Ileana, E.; Rosellini, S.; Ammari, S.; et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017, 7, 586–595. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Champiat, S.; Dercle, L.; Ammari, S.; Massard, C.; Hollebecque, A.; Postel-Vinay, S.; Chaput, N.; Eggermont, A.; Marabelle, A.; Soria, J.C.; et al. Hyperprogressive Disease Is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1. Clin. Cancer Res. 2017, 23, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Champiat, S.; Dercle, L.; Aspeslagh, S.; Castanon, E.; Limkin, E.J.; Baldini, C.; Postel-Vinay, S.; Hollebecque, A.; Massard, C.; et al. Baseline lymphopenia should not be used as exclusion criteria in early clinical trials investigating immune checkpoint blockers (PD-1/PD-L1 inhibitors). Eur. J. Cancer 2017, 84, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Bensch, F.; van der Veen, E.L.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schroder, C.P.; Hiltermann, T.J.N.; van der Wekken, A.J.; et al. (89)Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Khorrami, M.; Prasanna, P.; Gupta, A.; Patil, P.; Velu, P.D.; Thawani, R.; Corredor, G.; Alilou, M.; Bera, K.; Fu, P.; et al. Changes in CT Radiomic Features Associated with Lymphocyte Distribution Predict Overall Survival and Response to Immunotherapy in Non-Small Cell Lung Cancer. Cancer Immunol. Res. 2019. [Google Scholar] [CrossRef]
- Trebeschi, S.; Drago, S.G.; Birkbak, N.J.; Kurilova, I.; Calin, A.M.; Pizzi, A.D.; Lalezari, F.; Lambregts, D.M.J.; Rohaan, M.; Parmar, C.; et al. Predicting Response to Cancer Immunotherapy using Non-invasive Radiomic Biomarkers. Ann. Oncol. 2019, 30, 998–1004. [Google Scholar] [CrossRef]
- Himoto, Y.; Veeraraghavan, H.; Zheng, J.; Zamarin, D.; Snyder, A.; Capanu, M.; Nougaret, S.; Vargas, H.A.; Shitano, F.; Callahan, M.; et al. Computed Tomography–Derived Radiomic Metrics Can Identify Responders to Immunotherapy in Ovarian Cancer. JCO Precis. Oncol. 2019. [Google Scholar] [CrossRef]
- Ligero, M.; Garcia-Ruiz, A.; Viaplana, C.; Raciti, M.V.; Matos, I.; Martín-Liberal, J.; Hierro, C.; Gonzalez, M.; Morales-Barrera, R.; Suárez, C.; et al. Artificial intelligence combining radiomics and clinical data for predicting response to immunotherapy. Ann. Oncol. 2019, 30, mdz253.002. [Google Scholar] [CrossRef]
- Tunali, I.; Gray, J.E.; Qi, J.; Abdalah, M.; Jeong, D.K.; Guvenis, A.; Gillies, R.J.; Schabath, M.B. Novel clinical and radiomic predictors of rapid disease progression phenotypes among lung cancer patients treated with immunotherapy: An early report. Lung Cancer 2019, 129, 75–79. [Google Scholar] [CrossRef]
- Dercle, L.; Fronheiser, M.; Lu, L.; Du, S.; Hayes, W.; Leung, D.K.; Roy, A.; Schwartz, L.H.; Zhao, B. Radiomic signatures for identification of tumors sensitive to nivolumab or docetaxel in squamous non-small cell lung cancer (sqNSCLC). Ann. Oncol. 2019, 30, v773. [Google Scholar] [CrossRef]
- Korpics, M.C.; Bhave, S.R.; Redler, G.; Pitroda, S.; Luke, J.J.; Chmura, S.J. A Validated Radiomics T Cell Score Predicts Response to Multi-site SBRT Combined with Pembrolizumab. Int. J. Radiat. Oncol. Biol. Phys. 2019, 104, 1189–1190. [Google Scholar] [CrossRef]
- Matos, I.; Martin-Liberal, J.; Garcia-Ruiz, A.; Hierro, C.; Ochoa de Olza, M.; Viaplana, C.; Azaro, A.; Vieito, M.; Brana, I.; Mur, G.; et al. Capturing Hyperprogressive disease with immune checkpoint inhibitors using RECIST 1.1 criteria. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Colen, R.R.; Fujii, T.; Bilen, M.A.; Kotrotsou, A.; Abrol, S.; Hess, K.R.; Hajjar, J.; Suarez-Almazor, M.E.; Alshawa, A.; Hong, D.S.; et al. Radiomics to predict immunotherapy-induced pneumonitis: Proof of concept. Investig. New Drugs 2018, 36, 601–607. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).