Extracellular Matrix Derived from High Metastatic Human Breast Cancer Triggers Epithelial-Mesenchymal Transition in Epithelial Breast Cancer Cells through αvβ3 Integrin

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Differences in the Composition among Matrices Produced by MCF-7 and MDA-MB-231 Cells
2.2. Interaction with MDA-ECM Induced EMT-Associated Changes in MCF-7 Cells
2.3. Interaction with MDA-ECM Increased MCF-7 Cells Migratory Capacity
2.4. Interaction with MDA-ECM Triggered Integrin and TGF-β Signaling Pathways in MCF-7 Cells
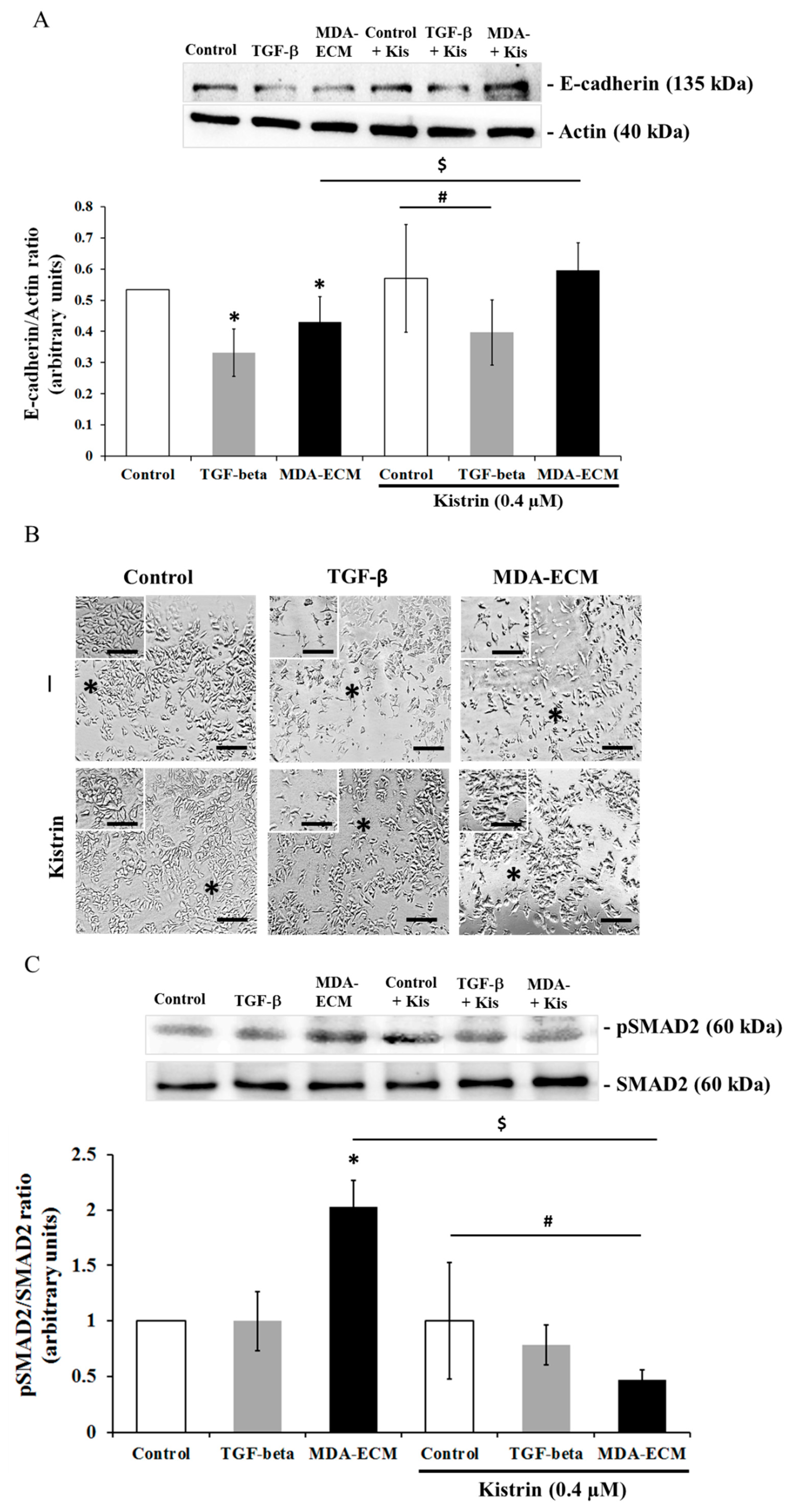
2.5. Involvement of αvβ3 Integrin in the EMT of MCF-7 Cells Cultured on MDA-ECM
3. Discussion
4. Material and Methods
4.1. Reagents
4.2. Antibodies
4.3. Cell Cultures
4.4. Isolation of Immobilized Cell-Derived Matrices
4.5. Stimulation of MCF-7 Cells to Epithelial-Mesenchymal Transition (EMT)
4.6. Analysis of ECM Composition by Indirect ELISA
4.7. Immunofluorescence of E-cadherin and Confocal Microscopy
4.8. Migration Assay
4.9. Nuclear Extract
4.10. SDS-PAGE and Western Blot
4.11. qRT-PCR Analysis
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MCF-ECM | Extracellular matrix-derived from the MCF-7 cell line |
| MDA-ECM | Extracellular matrix-derived from the MDA-MB-231 cell line |
References
- Bertucci, F.; Birnbaum, D. Reasons for breast cancer heterogeneity. J. Biol. 2008, 7, 6. [Google Scholar] [CrossRef]
- Pritchard, K.I. Endocrine therapy: Is the first generation of targeted drugs the last? J. Int. Med. 2013, 274, 144–152. [Google Scholar] [CrossRef]
- Lynce, F.; Blackburn, M.J.; Cai, L.; Wang, H.; Rubinstein, L.; Harris, P.; Isaacs, C.; Pohlmann, P.R. Characteristics and outcomes of breast cancer patients, enrolled in the National Cancer Institute Cancer Therapy Evaluation Program sponsored phase I clinical trials. Breast Cancer Res. Treat. 2018, 168, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Kusuma, N.; Denoyer, D.; Eble, J.A.; Redvers, R.P.; Parker, B.S.; Pelzer, R.; Anderson, R.L.; Pouliot, N. Integrin-dependent response to laminin-511 regulates breast tumor cell invasion and metastasis. Int. J. Cancer 2012, 130, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C. Epithelial-mesenchymal transition. J. Cell Sci. 2005, 118, 4325–4326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Neilson, E.G. Epithelial mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2009, 112, 1776–1784. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGFbeta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Mamuya, F.A.; Duncan, M.K. aV integrins and TGF-β-induced EMT: A circle of regulation. J. Cell Mol. Med. 2012, 164, 45–55. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, 344. [Google Scholar] [CrossRef] [Green Version]
- Schlaepfer, D.D.; Mitra, S.K. Multiple connections link FAK to cell motility and invasion. Curr. Opin. Genet. Dev. 2004, 14, 92–101. [Google Scholar] [CrossRef]
- Anthis, N.J.; Campbell, I.D. The tail of integrin activation. Trends Biochem. Sci. 2011, 36, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, D.; Nagaharu, K.; Shimojo, N.; Hanamura, N.; Yamashita, M.; Kozuka, Y.; Imanaka-Yoshida, K.; Yoshida, T. Binding of αvβ1 and αvβ6 integrins to tenascin-C induces epithelial-mesenchymal transition-like change of breast cancer cells. Oncogenesis 2013, 2, e65. [Google Scholar] [CrossRef] [Green Version]
- Nagaharu, K.; Zhang, X.; Yoshida, T.; Katoh, D.; Hanamura, N.; Kozuka, Y.; Ogawa, T.; Shiraishi, T.; Imanaka-Yoshida, K. Tenascin C induces epithelial-mesenchymal transition-like change accompanied by SRC activation and focal adhesion kinase phosphorylation in human breast cancer cells. Am. J. Pathol. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Tayeh, H.; Weidenfeld, K.; Zhilin-Roth, A.; Schif-Zuck, S.; Thaler, S.; Cotarelo, C.; Tan, T.Z.; Thiery, J.P.; Green, J.E.; Klorin, G.; et al. ‘Normalizing’ the malignant phenotype of luminal breast cancer cells via alpha(v)beta(3)-integrin. Cell Death Dis. 2016, 7, e2491. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Ishii, S.; Ikeda, T.; Masamura, S.; Doi, M.; Kitajima, M. The relationship between bone metastasis from human breast cancer and integrin alpha(v)beta3 expression. Anticancer Res. 2005, 25, 79–83. [Google Scholar] [PubMed]
- Flamini, M.I.; Uzair, I.D.; Pennacchio, G.E.; Neira, F.J.; Mondaca, J.M.; Cuello-Carrión, F.D.; Jahn, G.A.; Simoncini, T.; Sanchez, A.M. Thyroid Hormone Controls Breast Cancer Cell Movement via Integrin αV/β3/SRC/FAK/PI3-Kinases. Horm. Cancer 2017, 8, 16–27. [Google Scholar] [CrossRef]
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer 2011, 11, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Cichon, M.A.; Radisky, D.C. Extracellular matrix as a contextual determinant of transforming growth factor-β signaling in epithelial-mesenchymal transition and in cancer. Cell Adhes. Migr. 2014, 8, 588–594. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- Chia, J.; Kusuma, N.; Anderson, R.; Parker, B.; Bidwell, B.; Zamurs, L.; Nice, E.; Pouliot, N. Evidence for a role of tumor-derived laminin-511 in the metastatic progression of breast cancer. Am. J. Pathol. 2007, 170, 2135–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudland, P.S.; Platt-Higgins, A.; El-Tanani, M.; De Silva Rudland, S.; Barraclough, R.; Winstanley, J.H.; Howitt, R.; West, C.R. Prognostic significance of the metastasis-associated protein osteopontin in human breast cancer. Cancer Res. 2002, 62, 3417–3427. [Google Scholar] [PubMed]
- Hancox, R.A.; Allen, M.D.; Holliday, D.L.; Edwards, D.R.; Pennington, C.J.; Guttery, D.S.; Shaw, J.A.; Walker, R.A.; Pringle, J.H.; Jones, J.L. Tumour associated tenascin-C isoforms promote breast cancer cell invasion and growth by matrix metalloproteinase-dependent and independent mechanisms. Breast Cancer Res. 2009, 11, R24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massagué, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.M.; Onodera, Y.; Bissell, M.J.; Park, C.C. Breast cancer cells in three-dimensional culture display an enhanced radioresponse after coordinate targeting of integrin alpha5beta1 and fibronectin. Cancer Res. 2010, 70, 5238–5248. [Google Scholar] [CrossRef] [Green Version]
- Kenny, H.A.; Chiang, C.Y.; White, E.A.; Schryver, E.M.; Habis, M.; Romero, I.L.; Ladanyi, A.; Penicka, C.V.; George, J.; Matlin, K.; et al. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. J. Clin. Investig. 2014, 124, 4614–4628. [Google Scholar] [CrossRef] [Green Version]
- Eke, I.; Storch, K.; Krause, M.; Cordes, N. Cetuximab attenuates its cytotoxic and radiosensitizing potential by inducing fibronectin biosynthesis. Cancer Res. 2013, 73, 5869–5879. [Google Scholar] [CrossRef] [Green Version]
- Sisci, D.; Aquila, S.; Middea, E.; Gentile, M.; Maggiolini, M.; Mastroianni, F.; Montanaro, D.; Andò, S. Fibronectin and type IV collagen activate ERalpha AF-1 by c-Src pathway: Effect on breast cancer cell motility. Oncogene 2004, 23, 8920–8930. [Google Scholar] [CrossRef] [Green Version]
- Benton, G.; Crooke, G.E.; George, J. Laminin-1 induces E-cadherin expression in 3-dimensional cultured breast cancer cells by inhibiting DNA methyltransferase 1 and reversing promoter methylation status. FASEB J. 2009, 23, 3884–3895. [Google Scholar] [CrossRef]
- Pal, S.; Moulik, S.; Dutta, A.; Chatterjee, A. Extracellular matrix protein laminin induces matrix metalloproteinase-9 in human breast cancer cell line MCF-7. Cancer Microenviron. 2014, 7, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero, J.; Tobar, N.; Cáceres, M.; Espinoza, L.; Escobar, P.; Dotor, J.; Smith, P.C.; Martínez, J. Soluble factors derived from tumor mammary cell lines induce a stromal mammary adiposereversion in human and mice adipose cells. Possible role of TGF-beta1 and TNF-alpha. Breast Cancer Res. Treat. 2010, 119, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Sánchez, N.A.; Vadillo, E.; Balandrán, J.C.; Monroy-García, A.; Pelayo, R.; Fuentes-Pananá, E.M. Evidence of lateral transmission of aggressive features between different types of breast cancer cells. Int. J. Oncol. 2017, 51, 1482–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwajin, S.; Aree, M. Epithelial-mesenchymal Transition and Cell Invasion. Toxicol. Res. 2010, 26, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Comşa, S.; Cîmpean, A.M.; Raica, M. The Story of MCF-7 Breast Cancer Cell Line: 40 years of Experience in Research. Anticancer Res. 2015, 35, 3147–3154. [Google Scholar]
- Huttenlocher, A.; Horwitz, A.R. Integrins in cell migration. Cold Spring Harb. Perspect. Biol. 2011, 3, a005074. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signaling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Helal-Neto, E.; Brandão-Costa, R.M.; Saldanha-Gama, R.; Ribeiro-Pereira, C.; Midlej, V.; Benchimol, M.; Morandi, V.; Barja-Fidalgo, C. Priming Endothelial Cells With a Melanoma-Derived Extracellular Matrix Triggers the Activation of αvβ3/VEGFR2 Axis. J. Cell. Physiol. 2016, 231, 2464–2473. [Google Scholar] [CrossRef]
- Marelli, U.K.; Rechenmacher, F.; Sobahi, T.R.; Mas-Moruno, C.; Kessler, H. Tumor targeting via integrin ligands. Front. Oncol. 2013, 30, 222. [Google Scholar] [CrossRef] [Green Version]
- Taherian, A.; Li, X.; Liu, Y.; Haas, T.A. Differences in integrin expression and signaling within human breast cancer cells. BMC Cancer 2011, 11, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juliano, D.; Wang, Y.; Marcinkiewicz, C.; Rosenthal, L.A.; Stewart, G.J.; Niewiarowski, S. Disintegrin interaction with alpha V beta 3 integrin on human umbilical vein endothelial cells: Expression of ligand-induced binding site on beta 3 subunit. Exp. Cell Res. 1996, 225, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Whittaker, C.A.; Carr, S.A.; Tanabe, K.K.; Hynes, R.O. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer 2014, 14, 514–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foroni, L.; Vasuri, F.; Valente, S.; Gualandi, C.; Focarete, M.L.; Caprara, G.; Scandola, M.; D’Errico-Grigioni, A.; Pasquinelli, G. The role of 3D microenvironmental organization in MCF-7 epithelial-mesenchymal transition after 7 culture days. Exp. Cell Res. 2013, 319, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- BisselL, M.J.; Weaver, V.M.; Lelièvre, S.A.; Wang, F.; Petersen, O.W.; Schmeichel, K.L. Tissue structure, nuclear organization, and gene expression in normal and malignant breast. Cancer Res. 1999, 59, 1757–1763. [Google Scholar] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Kalembeyi, I.; Inada, H.; Nishiura, R.; Imanaka-Yoshida, K.; Sakakura, T.; Yoshida, T. Tenascin-C upregulates matrix metalloproteinase-9 in breast cancer cells: Direct and synergistic effects with transforming growth fator beta 1. Int. J. Cancer 2003, 105, 53–60. [Google Scholar] [CrossRef]
- Ilunga, K.; Nishiura, R.; Inada, H.; El-Karef, A.; Imanaka-Yoshida, K.; Sakakura, T.; Yoshida, T. Co-stimulation of human breast cancer cells with transforming growth factor-beta and tenascin-C enhances matrix metalloproteinase-9 expression and cancer cell invasion. Int. J. Exp. Pathol. 2004, 85, 373–379. [Google Scholar] [CrossRef]
- Neubauer, H.; Ruoff, A.; Paessler, N.; Solomayer, E.; Wallwiener, D.; Fehm, T. A laminin-rich basement membrane matrix influences estrogen receptor beta expression and morphology of MDA-MB-231 breast cancer cells. Oncol. Rep. 2009, 21, 475–481. [Google Scholar]
- Polyak, K. Heterogeneity in breast cancer. J. Clin. Investig. 2011, 121, 3786–3788. [Google Scholar] [CrossRef] [Green Version]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midwood, K.S.; Orend, G. The role of tenascin-C in tissue injury and tumorigenesis. J. Cell Commun. Signal. 2009, 3, 287–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadar, A.; Tõkés, A.M.; Kulka, J.; Robert, L. Extracellular matrix components in breast carcinomas. Semin. Cancer Biol. 2002, 12, 243–257. [Google Scholar] [CrossRef]
- Saitoh, Y.; Kuratsu, J.; Takeshima, H.; Yamamoto, S.; Ushio, Y. Expression of osteopontin in human glioma. Its correlation with the malignancy. Lab. Investig. 1995, 72, 55–63. [Google Scholar]
- Su, L.; Mukherjee, A.B.; Mukherjee, B.B. Expression of antisense osteopontin RNA inhibits tumor promoter-induced neoplastic transformation of mouse JB6 epidermal cells. Oncogene 1995, 10, 2163–2169. [Google Scholar]
- Niu, Y.; Zhang, L.; Bi, X.; Yuan, S.; Chen, P. Evaluation of vitronectin Expression in prostate cancer and the clinical significance of the association of vitronectin expression with prostate specific antigen in detecting prostate cancer. Urol. J. 2016, 13, 2527–2532. [Google Scholar]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide3-kinase pathway in cancer. Nat.Rev.Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [Green Version]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Hébert, M.C.; Zhang, Y.E. TGF-β receptor-activated p38 MAP kinase mediates Smad-independent TGF-β responses. EMBO J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.K.; Matchett, K.B.; McEnhill, P.M.; Dakir, E.H.; McMullin, M.F.; El-Tanani, Y.; Patterson, L.; Faheem, A.; Rudland, P.S.; McCarron, P.A.; et al. Protein deregulation associated with breast cancer metastasis. Cytokine Growth Factor Rev. 2015, 26, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, Y.; Menke, A. Signaling pathways involved in collagen-induced disruption of the E-cadherin complex during epithelial-mesenchymal transition. Cells Tissues Organs 2007, 185, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Varner, J. Integrins: Roles in cancer development and as treatment targets. Br. J. Cancer 2004, 90, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Akatsuka, T.; Imanaka-Yoshida, K. Tenascin-C and integrins in cancer. Cell Adhes. Migr. 2015, 9, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, B.S.; Sarnella, A.; Capasso, D.; Comegna, D.; Del Gatto, A.; Gramanzini, M.; Albanese, S.; Saviano, M.; Zaccaro, L.; Zannetti, A. Therapeutic Potential of a Novel αvβ3 Antagonist to Hamper the Aggressiveness of Mesenchymal Triple Negative Breast Cancer Sub-Type. Cancers (Basel) 2019, 11, E139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cammareri, P.; Rose, A.M.; Vincent, D.F.; Wang, J.; Nagano, A.; Libertini, S.; Ridgway, R.A.; Athineos, D.; Coates, P.J.; McHugh, A.; et al. Inactivation of TGFbeta receptors in stem cells drives cutaneous squamous cell carcinoma. Nat. Commun. 2016, 7, 12493. [Google Scholar] [CrossRef]
- Zhang, H.; Fredericks, T.; Xiong, G.; Qi, Y.; Rychahou, P.G.; Li, J.D.; Pihlajaniemi, T.; Xu, W.; Xu, R. Membrane associated collagen XIII promotes cancer metastasis and enhances anoikis resistance. Breast Cancer Res. 2018, 20, 116. [Google Scholar] [CrossRef]
- Tan, F.; Huang, Y.; Pei, Q.; Liu, H.; Pei, H.; Zhu, H. Matrix stiffness mediates stemness characteristics via activating the Yes-associated protein in colorectal cancer cells. J. Cell Biochem. 2018. [Google Scholar] [CrossRef]
- Celia-Terrassa, T.; Kang, Y. Distinctive properties of metastasis-initiating cells. Genes Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef] [Green Version]
- Kröger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2016, 116, 7353–7362. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado Brandão-Costa, R.; Helal-Neto, E.; Maia Vieira, A.; Barcellos-de-Souza, P.; Morgado-Diaz, J.; Barja-Fidalgo, C. Extracellular Matrix Derived from High Metastatic Human Breast Cancer Triggers Epithelial-Mesenchymal Transition in Epithelial Breast Cancer Cells through αvβ3 Integrin. Int. J. Mol. Sci. 2020, 21, 2995. https://doi.org/10.3390/ijms21082995
Machado Brandão-Costa R, Helal-Neto E, Maia Vieira A, Barcellos-de-Souza P, Morgado-Diaz J, Barja-Fidalgo C. Extracellular Matrix Derived from High Metastatic Human Breast Cancer Triggers Epithelial-Mesenchymal Transition in Epithelial Breast Cancer Cells through αvβ3 Integrin. International Journal of Molecular Sciences. 2020; 21(8):2995. https://doi.org/10.3390/ijms21082995
Chicago/Turabian StyleMachado Brandão-Costa, Renata, Edward Helal-Neto, Andreza Maia Vieira, Pedro Barcellos-de-Souza, Jose Morgado-Diaz, and Christina Barja-Fidalgo. 2020. "Extracellular Matrix Derived from High Metastatic Human Breast Cancer Triggers Epithelial-Mesenchymal Transition in Epithelial Breast Cancer Cells through αvβ3 Integrin" International Journal of Molecular Sciences 21, no. 8: 2995. https://doi.org/10.3390/ijms21082995