Neonatal Clonazepam Administration Induced Long-Lasting Changes in GABAA and GABAB Receptors

 ,
,  ,
,  , and
, and
Abstract
1. Introduction
2. Results
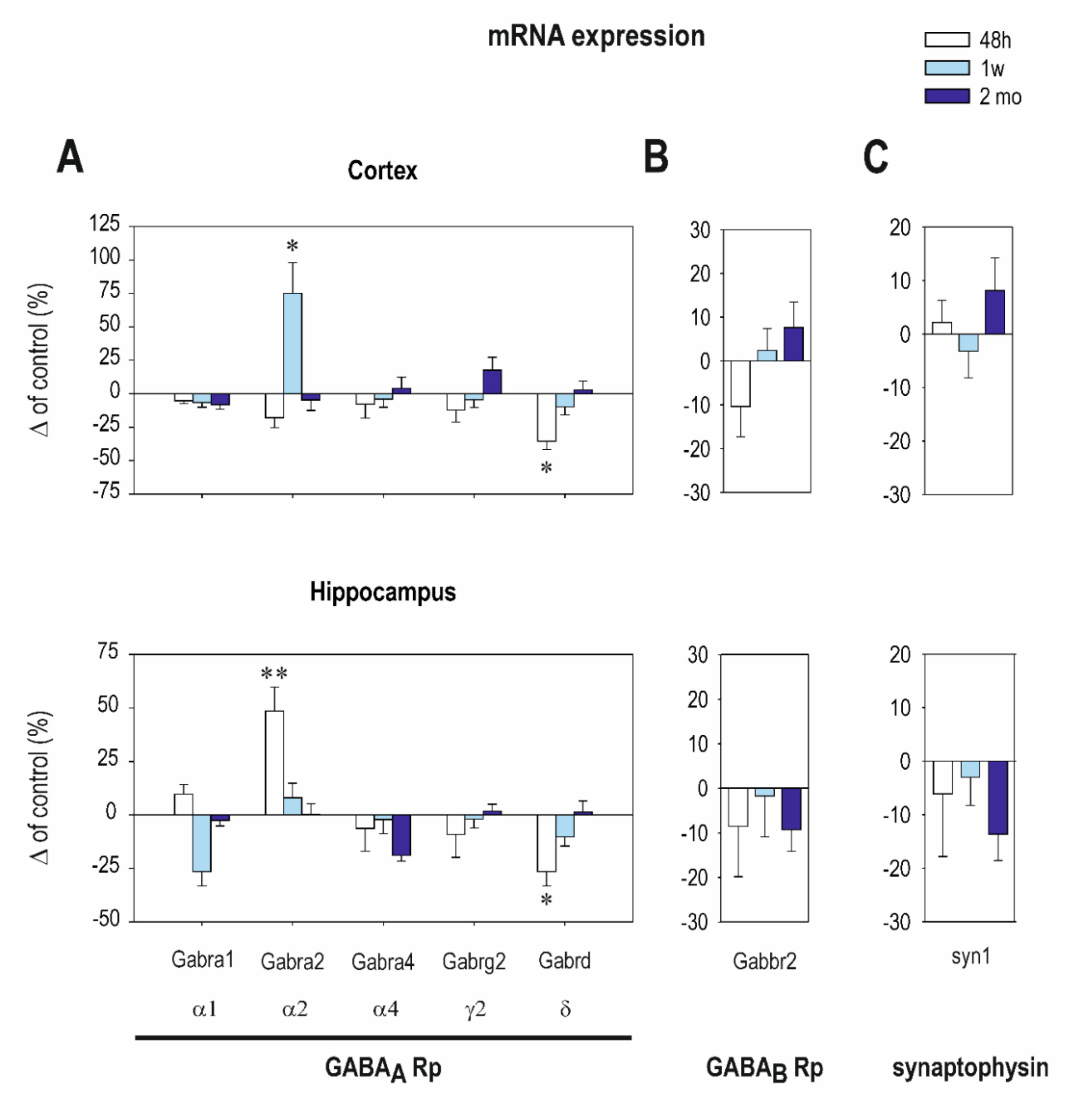
2.1. The Effect of CZP Administration on GABAA and GABAB R2 Receptor (Rp) Subunit mRNA Expression
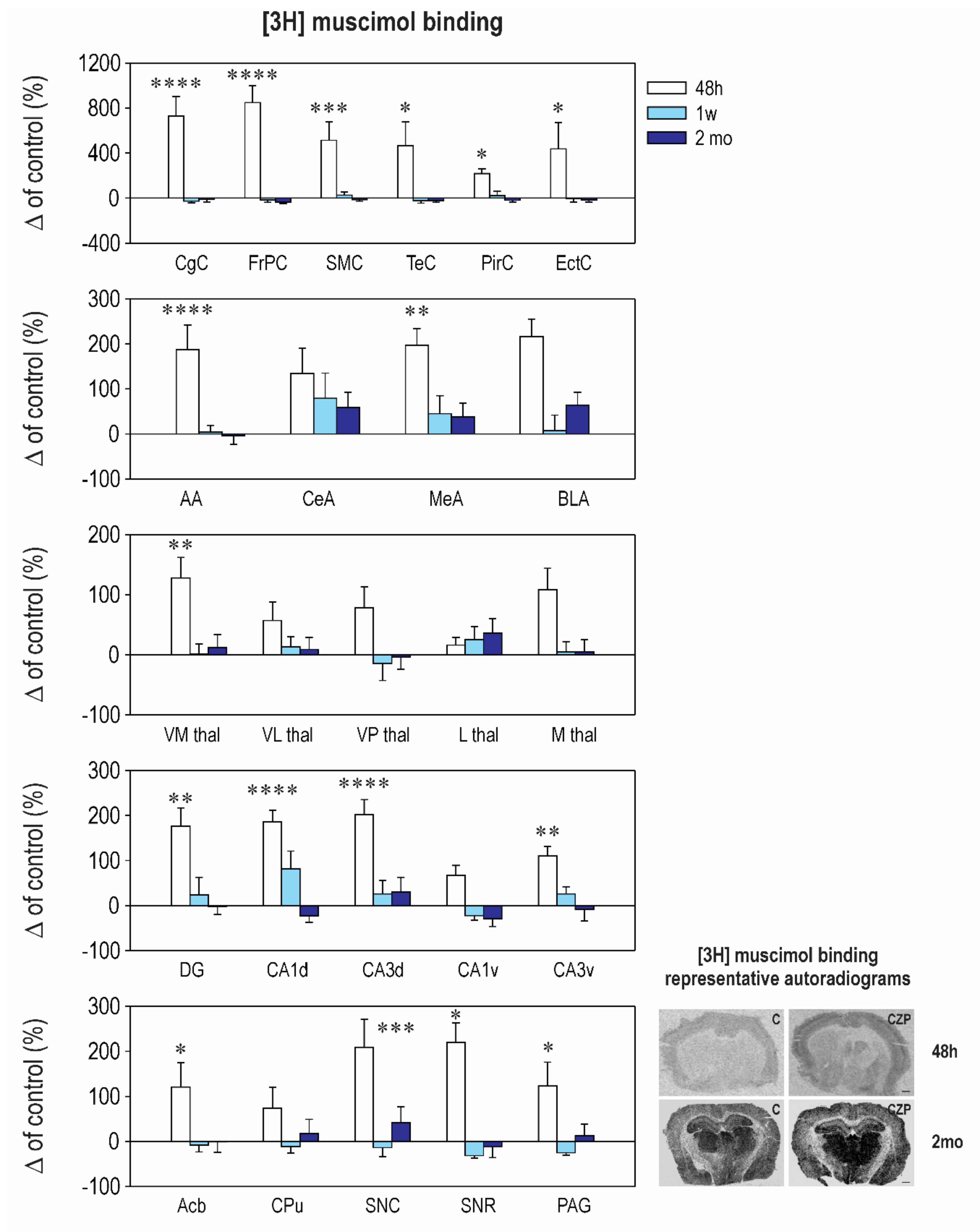
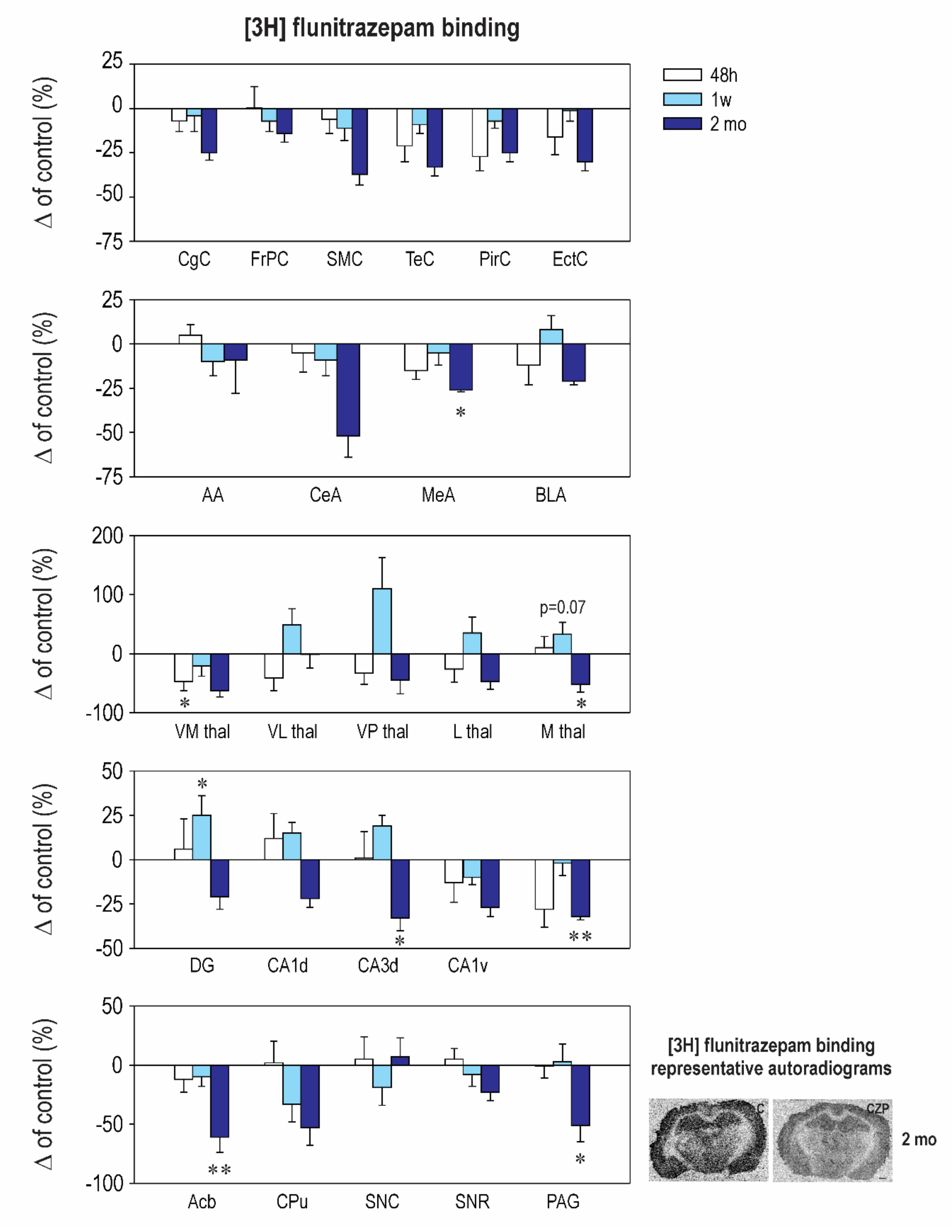
2.2. Effect of CZP Administration on [3H] Muscimol Binding and [3H] Flunitrazepam Binding
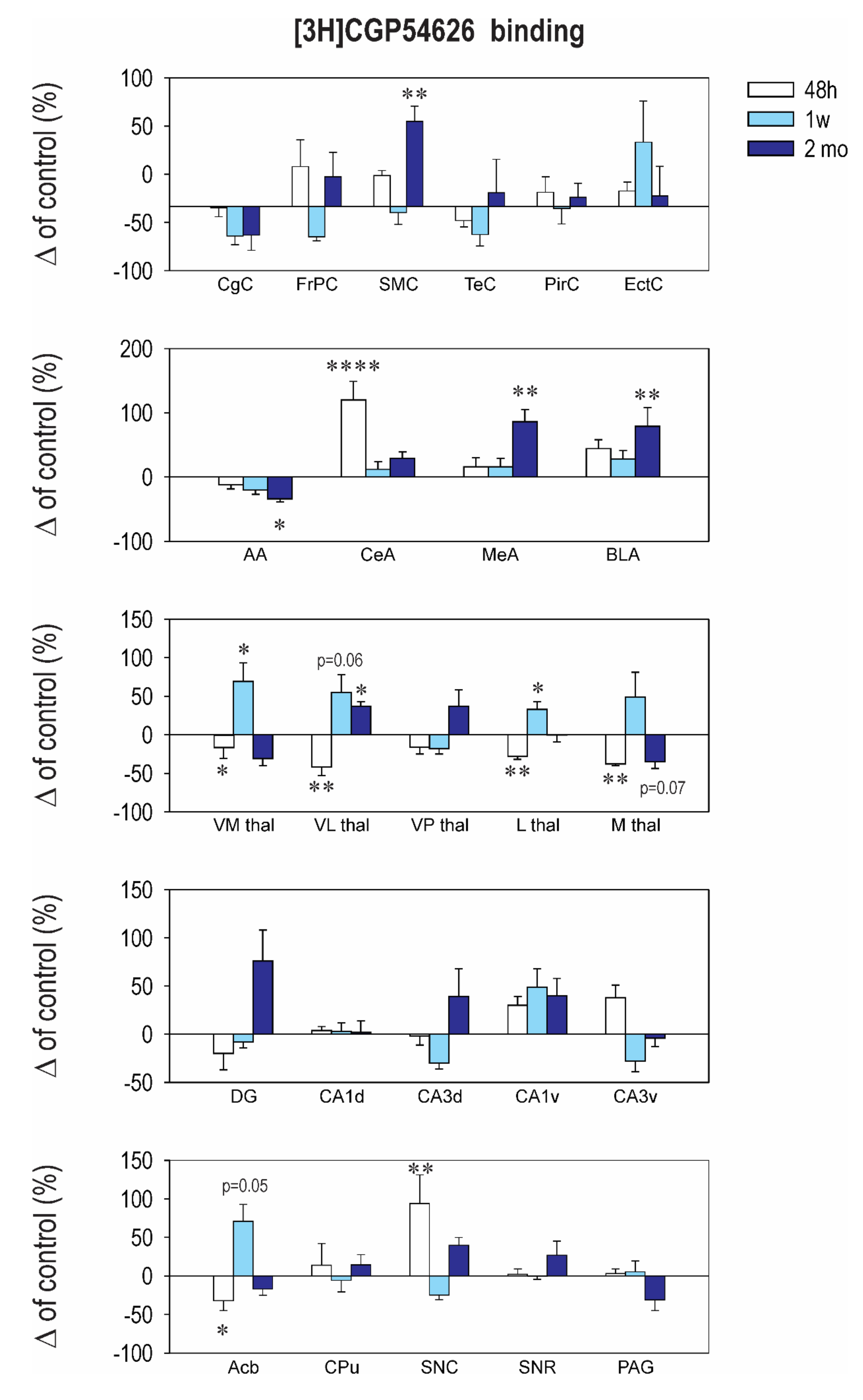
2.3. Effect of CZP Administration on [3H] CGP54626 Binding
3. Discussion
4. Materials and Methods
4.1. Pharmacological Treatment
4.2. Quantitative Real-Time RT-PCR
4.3. Receptor Binding
4.4. Statistics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DOAJ | Directory of open access journals |
| TLA | Three-letter acronym |
| LD | Linear dichroism |
Appendix

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cortex | Interval 48 h Mean ± SEM Control CZP | Interval 1 Week Mean ± SEM Control CZP | Interval 2 Months Mean ± SEM Control CZP | |||
|---|---|---|---|---|---|---|
| Gabra1 (α1) | 16.3 ± 0.7 | 15.3 ± 0.6 | 24.2 ± 1.7 | 22.7 ± 0.9 | 27.3 ± 2.4 | 26.6 ± 1.6 |
| Gabra2 (α2) | 11.7 ± 1.3 | 10.7 ± 1.1 | 6.6 ± 1.2 | ⬆ 11.1 ± 1.1 t = 2.705, df= 13, p = 0.018 | 14.5 ± 1.8 | 13.8 ± 1.3 |
| Gabra4 (α4) | 2.3 ± 0.3 | 2.2 ± 0.2 | 2.8 ± 0.3 | 2.7 ± 0.3 | 3.2 ± 0.3 | 3.3 ± 0.2 |
| Gabrg2 (γ2) | 3.4 ± 0.4 | 3.08 ± 0.32 | 3.5 ± 0.4 | 3.4 ± 0.3 | 5.5 ± 0.506 | 6.2 ± 0.3 |
| Gabrd (δ) | 0.14 ± 0.01 | ⬇ 0.06 ± 0.0 t = 2.196, df = 17, p = 0.0423 | 0.14 ± 0.02 | 0.13 ± 0.01 | 0.24 ± 0.02 | 0.25 ± 0.01 |
| Synaptophysin | 3.03 ± 0.27 | 3.400 ± 0.1 | 3.8 ± 0.4 | 3.8 ± 0.4 | 3.7 ± 0.27 | 4.0 ± 0.1 |
| Gabbr2 | 2.1 ± 0.1 | 1.8 ± 0.1 | 1.9 ± 0.2 | 2.02 ± 0.2 | 2.1 ± 0.2 | 2.3 ± 0.1 |
| Hippocampus | ||||||
| Gabra1 (α1) | 7.8 ± 0.9 | 8.6 ± 0.9 | 10.0 ± 1.1 | 9.6 ± 0.6 | 12.2 ± 1.0 | 11.2 ± 0.9 |
| Gabra2 (α2) | 17.2 ± 1.3 | ⬆ 26.1 ± 2.6 t = 2.956, df = 15, p = 0.0098 | 29.7 ± 4.0 | 27.4 ± 3.9 | 36.3 ± 4.7 | 37.8 ± 5.7 |
| Gabra4 (α4) | 2.1 ± 0.5 | 1.7 ± 0.4 | 3.2 ± 0.8 | 3.2 ± 0.8 | 9.2 ± 1.42 | 4.267 ± 1.1 |
| Gabrg2 (γ2) | 9.9 ± 1.1 | 7.8 ± 0.6 | 13.5 ± 1.6 | 12.6 ± 1.6 | 19.0 ± 2.2 | 19.50 ± 2.3 |
| Gabrd (δ) | 0.1 ± 0.02 | ⬇ 0.064 ± 0.01 t = 2.129, df = 16, p = 0.0435 | 0.2 ± 0.03 | 0.13 ± 0.03 | 0.3 ± 0.06 | 0.28 ± 0.06 |
| Synaptophysin | 45.0 ± 9.7 | 31.4 ± 5.1 | 69.0 ± 15.2 | 65.0 ± 16.9 | 103.2 ± 3.0 | 86.8 ± 6.2 |
| Gabbr2 | 3.3 ± 0.2 | 3.04 ± 0.4 | 3.9 ± 0.2 | 3.9 ± 0.4 | 4.7 ± 0.1 | 4.2 ± 0.2 |
| Interval 48 h Mean ± SEM | Interval 1 week Mean ± SEM | Interval 2 months Mean ± SEM | ||||
|---|---|---|---|---|---|---|
| Control | CZP | Control | CZP | Control | CZP | |
| Cingulate Cx. | 50.1 ± 8.6 | ⬆ 416.0 ± 86.8 t = 6.921, df = 35, p < 0.0001 | 120.0 ± 35.3 | 88.3 ± 18.8 | 67.8 ± 14.8 | 60.0 ± 14.1 |
| Frontoparital Cx. | 55.8 ± 10.4 | ⬆ 529.0 ± 84.3 t = 8.006, df = 35, p < 0.0001 | 152.0 ± 49.6 | 127.3 ± 32.9 | 93.8 ± 15.7 | 59.8 ± 12.3 |
| Sensorimotor Cx. | 52.8 ± 12.0 | ⬆ 324.0 ± 88.0 t = 4.289, df = 35, p = 0.0004 | 151.1 ± 49.0 | 189.0 ± 42.4 | 72.1 ± 14.6 | 62.4 ± 9.9 |
| Temporal Cx. | 50.0 ± 10.5 | ⬆ 283.2 ± 105.0 t = 2.901, df = 35, p = 0.0191 | 211.7 ± 75.3 | 165.9 ± 48.7 | 76.5 ± 23.7 | 59.4 ± 11.3 |
| Piriform Cx. | 34.2 ± 5.5 | ⬆ 109.0 ± 14.2 t = 3.036, df = 35, p = 0.0135 | 66.4 ± 18.5 | 81.8 ± 26.5 | 52.3 ± 12 | 42.2 ± 8.3 |
| Enthorinal Cx. | 34.5 ± 2.8 | ⬆ 104 ± 7.4 t = 3.036, df = 35, p = 0.0134 | 51.7 ± 19.6 | 37 ± 6.1 | 52.3 ± 16.1 | 43.2 ± 10.0 |
| Anterior AMG | 28.8 ± 4.6 | ⬆ 82.8 ± 15.8 t = 4.756, df = 36, p < 0.0001 | 33.2 ± 5.3 | 34.5 ± 5.0 | 43.6 ± 9.1 | 42.1 ± 8.1 |
| Central AMG | 25.2 ± 2.5 | 79.0 ± 19.0 | 32.7 ± 6.6 | 58.5 ± 18.2 | 27.0 ± 8.4 | 43.0 ± 8.8 |
| Medial AMG | 34.0 ± 3.1 | ⬆ 101.0 ± 12.0 t = 3.688, df = 35, p = 0.0023 | 49.2 ± 10.3 | 71.3 ± 19.0 | 35.5 ± 5.8 | 49.1 ± 10.8 |
| Basolateral AMG | 40.1 ± 11.0 | 126.8 ± 15.0 | 110.3 ± 37.6 | 119.6 ± 37.6 | 36.3 ± 7.5 | 59.4 ± 10 |
| N. Accumbens | 25.7 ± 3.9 | ⬆ 56.8 ± 13.0 t = 2.791, df = 35, p = 0.0252 | 22.0 ± 2.7 | 20.2 ± 3.3 | 30.1 ± 12.0 | 30.0 ± 7.0 |
| CPU | 31.0 ± 5.3 | 53.8 ± 14.0 | 20.4 ± 2.5 | 18.2 ± 3.0 | 29.3 ± 11.0 | 34.7 ± 9.0 |
| Ventromedial TH | 28.0 ± 4.1 | ⬆ 63.8 ± 9.0 t = 3.09, df = 35, p = 0.0117 | 37.4 ± 8.8 | 37.8 ± 6.0 | 44.5 ± 8.5 | 49.7 ± 9.6 |
| Ventrolateral TH | 24.1 ± 4.2 | 37.8 ± 7.0 | 27.0 ± 5.7 | 30.6 ± 4.5 | 46.3 ± 6.6 | 50.5 ± 9.4 |
| Ventralpost. TH | 34.7 ± 4.7 | 61.6 ± 12.0 | 137.4 ± 56.1 | 118.3 ± 39.0 | 63.6 ± 4.2 | 61.4 ± 13 |
| Lateral TH | 32.4 ± 5.9 | 37.6 ± 4.0 | 27.8 ± 4.9 | 34.8 ± 6.2 | 43.5 ± 10.6 | 59.0 ± 10.0 |
| Medial TH | 19.7 ± 2.8 | 41.0 ± 7.1 | 27.5 ± 4.8 | 29.0 ± 4.7 | 46.1 ± 7.9 | 48.4 ± 9.4 |
| Dentate Gyrus | 31.8 ± 2.8 | ⬆ 106.0 ± 15.9 t = 3.671, df = 35, p = 0.0024 | 47.1 ± 7.0 | 58.2 ± 18 | 49.6 ± 11.0 | 48.7 ± 8.7 |
| CA1 Dorsal | 29.4 ± 2.5 | ⬆ 84.1 ± 7.7 t = 5.424, df = 35, p < 0.0001 | 23.8 ± 2.6 | 43.3 ± 9.3 | 68.3 ± 2.9 | 52.7 ± 9.7 |
| CA2 Dorsal | 29.2 ± 5.1 | ⬆ 79.0 ± 14.0 t = 3.941, df = 35, p = 0.0011 | 26.2 ± 5.2 | 35.8 ± 6.8 | 36.1 ± 10.6 | 39.1 ± 9.3 |
| CA3 Dorsal | 31.2 ± 4.3 | ⬆ 94.5 ± 10.0 t = 5.175, df = 35, p < 0.0001 | 31.0 ± 5.9 | 39.0 ± 9.3 | 30.0 ± 9.0 | 39.0 ± 9.9 |
| CA1 Ventral | 44.1 ± 12.3 | 73.6 ± 9.6 | 40.5 ± 15.5 | 31.7 ± 4.0 | 55.8 ± 10.7 | 39.7 ± 9.7 |
| CA2 Ventral | 34.8 ± 8.4 | ⬆ 83.6 ± 10.0 t = 4.228, df = 35, p = 0.0005 | 24.8 ± 5.5 | 32.8 ± 3.2 | 56.8 ± 6.7 | 43.1 ± 11.2 |
| CA3 Ventral | 35.5 ± 7.3 | ⬆ 74.8 ± 7.6 t = 3.175, df = 35, p = 0.0093 | 25.5 ± 6.1 | 32.2 ± 4.1 | 45.1 ± 12.6 | 41.5 ± 11.9 |
| SN Compacta | 26.8 ± 4.0 | ⬆ 83.0 ± 16.5 t = 4.591, df = 35, p = 0.0002 | 35.7 ± 6.1 | 30.8 ± 7.2 | 24.3 ± 3.3 | 34.5 ± 8.5 |
| SN Reticulata | 31.5 ± 4.9 | ⬆ 101.0 ± 13.0 t = 2.883, df = 35, p = 0.0199 | 88.2 ± 23.5 | 60.6 ± 15.4 | 64.0 ± 20.3 | 56.1 ± 15.4 |
| PAG | 27.7 ± 3.3 | ⬆ 61.6 ± 14.0 t = 2.764, df = 35, p = 0.0269 | 41.2 ± 6.7 | 30.8 ± 4.9 | 35.6 ± 10.7 | 40.4 ± 8.7 |
| Interval 48 h Mean ± SEM | Interval 1 Week Mean ± SEM | Interval 2 Months Mean ± SEM | ||||
|---|---|---|---|---|---|---|
| Structure | Control | CZP | Control | CZP | Control | CZP |
| Cingulate Cx. | 388.0 ± 25.5 | 361.0 ± 17.2 | 387.4 ± 15.5 | 373.0 ± 36.0 | 370.2 ± 35.1 | 276.0 ± 15.0 |
| Frontoparital Cx. | 380.8 ± 23.9 | 382.4 ± 45.2 | 396.6 ± 26.9 | 368.9 ± 22.0 | 306.0 ± 34.0 | 263.1 ± 15.0 |
| Sensorimotor Cx. | 363.9 ± 48.2 | 342.1 ± 28.0 | 410.4 ± 37.6 | 364.1 ± 30.6 | 314.8 ± 25.2 | 198.4 ±19.0 |
| Temporal Cx. | 389.8 ± 55.2 | 270.3 ± 25.5 | 389.7 ± 19.0 | 355.5 ± 19.8 | 374.3 ± 49.6 | 250.7 ± 18.0 |
| Piriform Cx. | 336.0 ± 42.9 | 245.7 ± 25.5 | 281.0 ± 24.6 | 260.4 ± 11.0 | 282.3 ± 18.1 | 212.7 ± 14.0 |
| Enthorinal Cx. | 367.6 ± 54.0 | 271.3 ± 7.8 | 305.9 ± 22.0 | 301.6 ± 19.7 | 318.8 ± 52.2 | 223.0 ± 15.8 |
| Anterior AMG | 215.9 ± 21.8 | 226.1 ± 12.0 | 233.6 ± 7.8 | 211.1 ± 19.3 | 126.8 ± 44.4 | 115.0 ± 24.5 |
| Central AMG | 200.9 ± 40.0 | 190.9 ± 21.0 | 210.5 ± 7.2 | 210.6 ± 20.0 | 160.7 ± 39.0 | 77.2 ± 20.0 |
| Medial AMG | 317.3 ± 32.8 | 269.3 ± 15.9 | 295.0 ± 33.4 | 280.4 ± 19.0 | 304.0 ± 30.3 | ⬇ 224.7 ± 4.2 t = 2.572, df = 37, p = 0.0422 |
| Basolateral AMG | 343.6 ± 38.0 | 301.1 ± 37.1 | 318.0 ± 30.3 | 343.0 ± 24.0 | 319.3 ± 43.9 | 252.9 ± 5.5 |
| N. Accumbens | 223.3 ± 29.4 | 197.6 ± 24.0 | 216.7 ± 22.4 | 196.0 ± 17.4 | 233.8 ± 33.1 | ⬇ 91.3 ± 3.0 t = 3.637, df = 37, p = 0.0025 |
| CPU | 145.1 ± 30.1 | 148.7 ± 25.6 | 121.6 ± 26.1 | 81.1 ± 17.9 | 109.3 ± 21.4 | 51.1 ± 16.1 |
| Ventromedial TH | 230.1 ± 5.3 | ⬇ 122.9 ± 37.2 t = 2.859, df = 37, p = 0.0207 | 222.4 ± 17.2 | 175.1 ± 37.2 | 92 ± 37.0 | 33.9 ± 9.0 |
| Ventrolateral TH | 146.4 ± 28.6 | 86.1 ± 31.6 | 102.4 ± 27.4 | 153.0 ± 28.0 | 50.3 ± 19.1 | 49.8 ± 11.5 |
| Ventralpost. TH | 116.8 ± 26.7 | 78.2 ± 21.8 | 57.2 ± 12.9 | 120.5 ± 30.4 | 24.0 ± 11.4 | 13.1 ± 5.4 |
| Lateral TH | 157.0 ± 31.6 | 115.9 ± 33.9 | 106.6 ± 30.3 | 143.8 ± 28.5 | 74.0 ± 19.6 | 39.0 ± 9.7 |
| Medial TH | 193.3 ± 37.1 | 212.1 ± 36.5 | 158.1 ± 34.0 | 235.7 ± 19.7 | 178.0 ± 29.8 | 85.0 ± 23.4 |
| Dentate Gyrus | 313.4 ± 26.8 | 333.3 ± 53.6 | 311.1 ± 19.0 | 390.1 ± 33.6 | 337.8 ± 56.7 | 267.3 ± 23.2 |
| CA1 Dorsal | 261.5 ± 32.9 | 292.4 ± 35.7 | 288.9 ± 24.2 | 331.6 ± 17 | 249.7 ± 34.8 | 195.4 ± 16.4 |
| CA2 Dorsal | 228.9 ± 33.0 | 231.0 ± 41.4 | 230.6 ± 25.3 | 272.9 ± 15.1 | 206.8 ± 29.2 | 120.1 ± 23.2 |
| CA3 Dorsal | 294.8 ± 25.4 | 297.0 ± 45.5 | 261.6 ± 26.5 | 311.3 ± 14.9 | 267.8 ± 20.9 | ⬇ 179.0 ± 18.3 t = 3.795, df = 37, p = 0.0016 |
| CA1 Ventral | 373.1 ± 63.0 | 251.5 ± 10.9 | 289.1 ± 12.2 | 261.6 ± 12.9 | 280.0 ± 31.8 | 205.0 ± 14.8 |
| CA2 Ventral | 316.9 ± 60.8 | 248.9 ± 19.4 | 294.0 ± 20.5 | 280.9 ± 12.9 | 304.2 ± 41.4 | 218.0 ± 15.1 |
| CA3 Ventral | 356.9 ± 72.9 | 256.3 ± 35.0 | 294.4 ± 24.6 | 289.8 ± 19.8 | 320.8 ± 31.2 | 218.6 ± 7.3 |
| SN Compacta | 168.3 ± 49.4 | 177.3 ± 31.8 | 206.7 ± 11.9 | 168.1 ± 31.0 | 132.3 ± 49.2 | 141.0 ± 20.7 |
| SN Reticulata | 228.0 ± 60.4 | 238.4 ± 21.6 | 252.0 ± 21.6 | 232.1 ± 26.3 | 249.8 ± 27.6 | 192.0 ± 16.7 |
| PAG | 223.8 ± 35.9 | 220.9 ± 22.8 | 227.0 ± 22.7 | 233.3 ± 33.8 | 197.0 ± 22.0 | ⬇ 97.0 ± 28.1 t = 2.522, df = 37, p = 0.0492 |
| Structure | Control 48 h | CZP 48 h | Control 1 week | CZP 1 week | Control 50 days | CZP 50 days |
|---|---|---|---|---|---|---|
| Cingulate Cx. | 274.1 ± 25.1 | 271.0 ± 19.0 | 311.1 ± 18.9 | 239.3 ± 20.9 | 404.7 ± 75.1 | 314.9 ± 49.3 |
| Frontoparital Cx. | 266.4 ± 9.3 | 349.8 ± 55.0 | 377.1 ± 39.6 | 287.6 ± 11.6 | 206.0 ± 26.3 | 253.6 ± 39.3 |
| Sensorimotor Cx. | 227.4 ± 15.0 | 283.0 ± 10.0 | 242.6 ± 14.7 | 230.6 ± 21.4 | 141.5 ± 16.5 | ⬆ 235.0 ± 17.7 t = 2.416, df = 37, p = 0.0017 |
| Temporal Cx. | 292.3 ± 27.4 | 260.1 ± 15.5 | 327.9 ± 38.6 | 256.0 ± 25.2 | 195.0 ± 26.9 | 215.7 ± 50.5 |
| Piriform Cx. | 210.1 ± 21.2 | 233.1 ± 25.6 | 237.1 ± 28.3 | 233.3 ± 27.5 | 269.0 ± 48.7 | 287.7 ± 30.6 |
| Enthorinal Cx. | 233.5 ± 24.7 | 262.3 ± 15.7 | 276.6 ± 24.0 | 414.9 ± 88.4 | 427.5 ± 87.3 | 462.6 ± 97.1 |
| Anterior AMG | 260.6 ± 20.0 | 228.7 ± 16.9 | 315.7 ± 31.2 | 253.9 ± 21.6 | 229.3 ± 49.3 | ⬇152.3 ± 10.7 t = 2.72, df = 37, p = 0.0293 |
| Central AMG | 231.1 ± 12.8 | ⬆ 507.7 ± 66.0 t = 5.706, df = 37, p < 0.0001 | 198.4 ± 19.4 | 221.5 ± 23.9 | 210.7 ± 37.8 | 271.9 ± 21.3 |
| Medial AMG | 439.9 ± 47.3 | 511.4 ± 63.3 | 298.1 ± 23.2 | 347.3 ± 40.0 | 365.5 ± 92.5 | ⬆ 678.6 ± 70.0 t = 3.679, df = 39, p = 0.0023 |
| Basolateral AMG | 251.9 ± 14.9 | 362.1 ± 34.0 | 218.7 ± 23.5 | 279.9 ± 27.0 | 268.0 ± 48.3 | ⬆ 479.1 ± 77 t = 3.422, df = 36, p = 0.0047 |
| N. Accumbens | 1028.0 ± 76.0 | ⬇ 697.6 ± 134.0 t = 2.857, df = 36, p = 0.021 | 384.6 ± 33.4 | 657.0 ± 85.0 | 369.5 ± 78.6 | 306.7 ± 29.6 |
| CPU | 673.1 ± 65.7 | 767.1 ± 191.0 | 454.3 ± 52.1 | 428.4 ± 68.0 | 174.8 ± 17.8 | 200.3 ± 22.8 |
| Ventromedial TH | 1151.0 ± 56.5 | 892.5 ± 68.7 | 734.3 ± 113 | ⬆ 1238.0 ± 175.0 t = 2.88, df = 36, p = 0.0199 | 601.0 ± 131 | 415.1 ± 51.4 |
| Ventrolateral TH | 1085.0 ± 66.5 | ⬇ 632.0 ± 120.0 t = 2.961, df = 36, p = 0.0161 | 735.6 ± 79.0 | ⬆ 1139.0 ± 166.0 t = 2.961, df = 36, p = 0.0293 | 606.5 ± 94.2 | 380.7 ± 37 |
| Ventralpost. TH | 1898.0 ± 321.0 | 1592.0 ± 178.0 | 1409.0 ± 77.8 | 1161.0 ± 99.0 | 249.7 ± 36.9 | ⬆ 342.4 ± 52.6 t = 3.738, df = 35, p = 0.0020 |
| Lateral TH | 2187.0 ± 125.0 | ⬇ 1566 ± 83.0 t = 3.608, df = 36, p = 0.0028 | 1532.0 ± 162.2 | ⬆ 2033.0 ± 156.0 t = 3.006, df = 39, p = 0.0143 | 718.7 ± 66.2 | 718.3 ± 63.8 |
| Medial TH | 1585.0 ± 119.0 | 1081 ± 16.5 | 1558.0 ± 190.3 | 2318.0 ± 501.0 | 1076 ± 179.6 | 694.0 ± 95 |
| Dentate Gyrus | 552.3 ± 66.9 | 441.9 ± 96.0 | 272.3 ± 17.3 | 250.8 ± 15.6 | 200.8 ± 24.5 | 353.1 ± 64 |
| CA1 Dorsal | 257.0 ± 18.8 | 267.0 ± 9.8 | 205.6 ± 19.6 | 211.8 ± 19.3 | 154.2 ± 10.3 | 138.7 ± 6.1 |
| CA2 Dorsal | 546.9 ± 61 | 611.6 ± 123.0 | 256.3 ± 13.0 | 281.8 ± 27.5 | 391.3 ± 87.2 | 324.9 ± 56.5 |
| CA3 Dorsal | 1087.0 ± 53.1 | 1060.0 ± 93.0 | 500.4 ± 76.0 | 351.8 ± 29.0 | 361.2 ± 34.5 | 500.9 ± 104 |
| CA1 Ventral | 257.7 ± 21.9 | 335.1 ± 22.0 | 352.1 ± 46.2 | 523.0 ± 69.0 | 503.8 ± 83.8 | 708.4 ± 92 |
| CA2 Ventral | 264.3 ± 35.5 | 312.3 ± 21.0 | 381.3 ± 40.0 | 442.0 ± 53.9 | 385.0 ± 68.2 | 292.7 ± 49.6 |
| CA3 Ventral | 693.0 ± 72.0 | 954.1 ± 88.0 | 714.3 ± 102.0 | 511.0 ± 78.0 | 772.7 ± 35.2 | 742.9 ± 71 |
| SN Compacta | 276.1 ± 15.0 | ⬆ 536.7 ± 102.0 t = 4.026, df = 36, p = 0.0008 | 352.4 ± 31.4 | 265.8 ± 22.0 | 113.5 ± 4.4 | 158.7 ± 11 |
| SN Reticulata | 264.4 ± 20.8 | 269.1 ± 17.3 | 290.0 ± 22.0 | 288.8 ± 12.0 | 124.5 ± 6.5 | 158.6 ± 22 |
| PAG | 967.6 ± 60.0 | 999.7 ± 54.0 | 564.1 ± 69.2 | 594.0 ± 76.0 | 154.0 ± 9.5 | 107.0 ± 22 |
References
- Wu, C.; Sun, D. GABA receptors in brain development, function, and injury. Metab. Brain Dis. 2015, 30, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Fritschy, J.M.; Panzanelli, P. GABAA receptors and plasticity of inhibitory neurotransmission in the central nervous system. Eur. J. Neurosci. 2014, 39, 1845–1865. [Google Scholar] [CrossRef] [PubMed]
- Terunuma, M. Diversity of structure and function of GABAB receptors: A complexity of GABAB-mediated signaling. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2018, 94, 390–411. [Google Scholar] [CrossRef] [PubMed]
- Fritschy, J.M. Significance of GABA(A) receptor heterogeneity: Clues from developing neurons. Adv. Pharmacol. 2015, 73, 13–39. [Google Scholar]
- Wang, D.D.; Kriegstein, A.R. Defining the role of GABA in cortical development. J. Physiol. 2009, 1873–1879. [Google Scholar] [CrossRef]
- Rennie, J.M.; Boylan, G.B. Neonatal seizures and their treatment. Curr. Opin. Neurol. 2003, 16, 177–181. [Google Scholar] [CrossRef]
- Gai, N.; Grimm, V.E. The effect of prenatal exposure to diazepam on aspects of postnatal development and behavior in rats. Psychopharmacology (Berl) 1982, 78, 225–229. [Google Scholar] [CrossRef]
- Tucker, J.C. Benzodiazepines and the developing rat:a critical review. Neurosci. Biobehav. Rev. 1985, 9, 101–111. [Google Scholar] [CrossRef]
- Kellogg, C.K. Benzodiazepines: Influence on the developing brain. Prog. Brain Res. 1988, 73, 207–228. [Google Scholar]
- Ikonomidoou, C.; Turski, L. Antiepileptic drugs and brain development. Epilepsy Res. 2010, 88, 11–22. [Google Scholar] [CrossRef]
- Sundbakk, L.M.; Wood, M.; Gran, J.M.; Nordeng, H. Impact of prenatal exposure to benzodiazepines and z-hypnotics on behavioral problems at 5 years of age: A study from the Norwegian Mother and Child Cohort Study. PLoS ONE 2019, 14, e0217830. [Google Scholar] [CrossRef] [PubMed]
- Kubová, H.; Mareš, P. Time course of the anticonvulsant action of clonazepam in the developing rats. Arch. Int. Pharmacodyn. 1989, 298, 15–24. [Google Scholar] [PubMed]
- Kubová, H.; Mareš, P.; Vorlíček, J. Stable anticonvulsant action of benzodiazepines during development in rats. J. Pharm. Pharmacol. 1993, 45, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Mikulecká, A.; Mareš, P.; Kubová, H. Rebound increase in seizure susceptibility but not isolation-induced calls after single administration of clonazepam and Ro 19-8022 in infant rats. Epilepsy Behav. 2011, 20, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Mikulecká, A.; Šubrt, M.; Stuchlík, A.; Kubová, H. Consequences of early postnatal benzodiazepines exposure in rats. I. Cognitive-like behavior. Front. Behav. Neurosci. 2014, 8, 101, eCollection 2014. [Google Scholar] [CrossRef]
- Mikulecká, A.; Šubrt, M.; Pařízková, M.; Mareš, P.; Kubová, H. Consequences of early postnatal benzodiazepines exposure in rats. II. Social behavior. Front. Behav. Neurosci. 2014, 8, 169, eCollection 2014. [Google Scholar] [CrossRef]
- File, S.E. Behavioral changes persisting in to adulthood after neonatal benzodiazepine administration in the rat. Neurobehav. Toxicol. Teratol. 1986, 8, 453–461. [Google Scholar]
- File, S.E. Effects of neonatal administration of diazepam and lorazepam on performance of adolescent rats in tests of anxiety, aggression, learning and convulsions. Neurobehav. Toxicol. Teratol. 1986, 8, 301–306. [Google Scholar]
- File, S.E. The effects of neonatal administration of clonazepam on passive avoidance and on social, aggressive and exploratory behavior of adolescent male rats. Neurobehav. Toxicol. Teratol. 1986, 8, 447–452. [Google Scholar]
- File, S.E. Diazepam and caffeine administration during the first week of life: Changes in neonatal and adolescent behavior. Neurotoxicol. Teratol. 1987, 9, 9–16. [Google Scholar] [CrossRef]
- Kubová, H.; Mareš, P. Partial agonist of benzodiazepine receptors Ro 19-8022 elicits withdrawal symptoms after short-term administration in immature rats. Physiol. Res. 2012, 61, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Kubová, H.; Bendová, Z.; Moravcová, S.; Pačesová, D.; Rocha, L.; Mareš, P. Neonatal clonazepam administration induced long-lasting changes in glutamate receptors. Front. Mol. Neurosci. 2018, 11, 382. [Google Scholar] [CrossRef] [PubMed]
- Whissell, P.D.; Rosenzweig, S.; Lecker, I.; Wang, D.S.; Wojtowicz, J.M.; Orser, B.A. γ-aminobutyric acid type A receptors that contain the δ subunit promote memory and neurogenesis in the dentate gyrus. Ann. Neurol. 2013, 74, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Whissell, P.D.; Lecker, I.; Wang, D.S.; Yu, J.; Orser, B.A. Altered expression of δGABAA receptors in health and disease. Neuropharmacology 2015, 88, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Laurie, D.J.; Wisden, W.; Seeburg, P.H. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J. Neurosci. 1992, 12, 4151–4172. [Google Scholar] [CrossRef]
- Martini, C.; Rigacci, T.; Lucacchini, A. [3H]muscimol binding site on purified benzodiazepine receptor. J. Neurochem. 1983, 41, 1183–1185. [Google Scholar] [CrossRef]
- Benkherouf, A.Y.; Taina, K.R.; Meera, P.; Aalto, A.J.; Li, X.G.; Soini, S.L.; Wallner, M.; Uusi-Oukari, M.J. Extrasynaptic δ-GABAA receptors are high-affinity muscimol receptors. J. Neurochem. 2019, 149, 41–53. [Google Scholar] [CrossRef]
- Brett, R.R.; Pratt, J.A. Changes in benzodiazepine-GABA receptor coupling in an accumbens-habenula circuit after chronic diazepam treatment. Br. J. Pharmacol. 1995, 116, 2375–2384. [Google Scholar] [CrossRef]
- Andersen, S.L.; Navalta, C.P. Altering the course of neurodevelopment: A framework for understanding the enduring effects of psychotropic drugs. Int. J. Dev. Neurosci. 2004, 22, 423–440. [Google Scholar] [CrossRef]
- Dobbing, J.; Smart, J.L. Vulnerability of developing brain and behaviour. Br. Med. Bull. 1974, 30, 164–168. [Google Scholar] [CrossRef]
- Lohmann, C.; Kessels, H.W. The developmental stages of synaptic plasticity. J. Physiol. 2014, 592, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Forcelli, P.A.; Janssen, M.J.; Vicini, S.; Gale, K. Neonatal exposure to antiepileptic drugs disrupts striatal synaptic development. Ann. Neurol. 2012, 72, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Uusi-Oukari, M.; Korpi, E.R. Regulation of GABA(A) receptor subunit expression by pharmacological agents. Pharmacol. Rev. 2010, 62, 97–135. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Rosenberg, H.C.; Chiu, T.H.; Zhao, T.J. Subunit- and brain region-specific reduction of GABAA receptor subunit mRNAs during chronic treatment of rats with diazepam. J. Mol. Neurosci. 1994, 5, 105–120. [Google Scholar] [CrossRef]
- Holt, R.A.; Bateson, A.N.; Martin, I.L. Chronic treatment with diazepam or abecarnil differently affects the expression of GABAA receptor subunit mRNAs in the rat cortex. Neuropharmacology 1996, 35, 1457–1463. [Google Scholar] [CrossRef]
- Tietz, E.I.; Huang, X.; Chen, S.; Ferencak, W.F., 3rd. Temporal and regional regulation of alpha1, beta2 and beta3, but not alpha2, alpha4, alpha5, alpha6, beta1 or gamma2 GABA(A) receptor subunit messenger RNAs following one-week oral flurazepam administration. Neuroscience 1999, 91, 327–341. [Google Scholar] [CrossRef]
- Tietz, E.I.; Huang, X.; Weng, X.; Rosenberg, H.C.; Chiu, T.H. Expression of alpha 1, alpha 5, and gamma 2 GABAA receptor subunit mRNAs measured in situ in rat hippocampus and cortex following chronic flurazepam administration. J. Mol. Neurosci. 1993, 4, 277–292. [Google Scholar] [CrossRef]
- Chen, S.; Huang, X.; Zeng, X.J.; Sieghart, W.; Tietz, E.I. Benzodiazepine-mediated regulation of alpha1, alpha2, beta1-3 and gamma2 GABA(A) receptor subunit proteins in the rat brain hippocampus and cortex. Neuroscience 1999, 93, 33–44. [Google Scholar] [CrossRef]
- Owens, D.F.; Kriegstein, A.R. Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci. 2002, 3, 715–727. [Google Scholar] [CrossRef]
- Sato, T.N.; Neale, J.H. Type I and type II gamma-aminobutyric acid/benzodiazepine receptors: Purification and analysis of novel receptor complex from neonatal cortex. J. Neurochem. 1989, 52, 1114–1122. [Google Scholar] [CrossRef]
- Zhang, J.H.; Sato, M.; Tohyama, M. Different postnatal development profiles of neurons containing distinct GABAA receptor beta subunit mRNAs (beta 1, beta 2, and beta 3) in the rat forebrain. J. Comp. Neurol. 1991, 308, 586–613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Sato, M.; Tohyama, M. Different postnatal ontogenic profiles of neurons containing beta (beta 1, beta 2 and beta 3) subunit mRNAs of GABAA receptor in the rat thalamus. Brain Res. Dev. Brain Res. 1991, 58, 289–292. [Google Scholar] [CrossRef]
- Zhang, J.H.; Sato, M.; Araki, T.; Tohyama, M. Postnatal ontogenesis of neurons containing GABAA alpha 1 subunit mRNA in the rat forebrain. Brain Res. Mol. Brain Res. 1992, 16, 193–203. [Google Scholar] [CrossRef]
- Hornung, J.P.; Fritschy, J.M. Developmental profile of GABAA-receptors in the marmoset monkey: Expression of distinct subtypes in pre- and postnatal brain. J. Comp. Neurol. 1996, 367, 413–430. [Google Scholar] [CrossRef]
- Stell, B.M.; Brickley, S.G.; Tang, C.Y.; Farrant, M.; Mody, I. Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by delta subunit-containing GABAA receptors. Proc. Natl. Acad. Sci. USA 2003, 100, 14439–14444. [Google Scholar] [CrossRef]
- Glykys, J.; Peng, Z.; Chandra, D.; Homanics, G.E.; Houser, C.R.; Mody, I. A new naturally occurring GABA(A) receptor subunit partnership with high sensitivity to ethanol. Nat. Neurosci. 2007, 10, 40–48. [Google Scholar] [CrossRef]
- Holter, N.I.; Zylla, M.M.; Zuber, N.; Bruehl, C.; Draguhn, A. Tonic GABAergic control of mouse dentate granule cells during postnatal development. Eur. J. Neurosci. 2010, 32, 1300–1309. [Google Scholar] [CrossRef]
- Korpi, E.R.; Mihalek, R.M.; Sinkkonen, S.T.; Hauer, B.; Hevers, W.; Homanics, G.E.; Sieghart, W.; Lüddens, H. Altered receptor subtypes in the forebrain of GABA(A) receptor delta subunit-deficient mice: Recruitment of gamma 2 subunits. Neuroscience 2002, 109, 733–743. [Google Scholar] [CrossRef]
- Allison, C.; Pratt, J.A. Neuroadaptive processes in GABAergic and glutamatergic systems in benzodiazepine dependence. Pharmacol. Ther. 2003, 98, 171–195. [Google Scholar] [CrossRef]
- Tietz, E.I.; Rosenberg, H.C.; Chiu, T.H. Autoradiographic localization of benzodiazepine receptor downregulation. J. Pharmacol. Exp. Ther. 1986, 236, 284–292. [Google Scholar]
- Percic, D.; Svob Strac, D.; Jazvincak Jemberek, M.; Vlainic, J. Allosteric uncoupling and up-regulation of benzodiazepine and GABA recognition sites following chronic diazepam treatment of HEK 293 cells stably transfected with α1β2γ2S subunits of GABAA receptors. Naunyn. Schmiedebergs Arch. Pharmacol. 2007, 375, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Fyhn, M.; Molden, S.; Witter, M.P.; Moser, E.I.; Moser, M.B. Spatial representation in the entorhinal cortex. Science 2004, 305, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- White, N.M. Some highlights of research on the effects of caudate nucleus lesions over the past 200 years. Behav. Brain Res. 2009, 199, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Panksepp, J. The basic emotional circuits of mammalian brains: Do animals have affective lives? Neurosci. Biobehav. Rev. 2011, 35, 1791–1804. [Google Scholar] [CrossRef]
- Benarroch, E.E. Periaqueductal gray: An interface for behavioral control. Neurology 2012, 78, 210–217. [Google Scholar] [CrossRef]
- Fanselow, M.S.; Dong, H.W. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 2010, 65, 7–19. [Google Scholar] [CrossRef]
- Schroeder, H.; Humbert, A.C.; Desor, D.; Nehlig, A. Long-term consequences of neonatal exposure to diazepam on cerebral glucose utilization, learning, memory and anxiety. Brain Res. 1997, 766, 142–152. [Google Scholar] [CrossRef]
- Chalifoux, J.R.; Carter, A.G. GABAB receptor modulation of synaptic function. Curr. Opin. Neurobiol. 2011, 21, 339–344. [Google Scholar] [CrossRef]
- Gaiarsa, J.L.; Porcher, C. Emerging neurotrophic role of GABAB receptors in neuronal circuit development. Front. Cell Neurosci. 2013, 7, 206. [Google Scholar] [CrossRef]
- Prosser, H.M.; Gill, C.H.; Hirst, W.D.; Grau, E.; Robbins, M.; Calver, A.; Soffin, E.M.; Farmer, C.E.; Lanneau, C.; Gray, J.; et al. Epileptogenesis and enhanced prepulse inhibition in GABA(B1)-deficient mice. Mol. Cell Neurosci. 2001, 17, 1059–1070. [Google Scholar] [CrossRef]
- Haller, C.; Casanova, E.; Müller, M.; Vacher, C.M.; Vigot, R.; Doll, T.; Barbieri, S.; Gassmann, M.; Bettler, B. Floxed allele for conditional inactivation of the GABAB(1) gene. Genesis 2004, 40, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Bony, G.; Szczurkowska, J.; Tamagno, I.; Shelly, M.; Contestabile, A.; Cancedda, L. Non-hyperpolarizing GABAB receptor activation regulates neuronal migration and neurite growth and specification by cAMP/LKB1. Nat. Commun. 2013, 4, 1800. [Google Scholar] [CrossRef] [PubMed]
- Heaney, C.F.; Kinney, J.W. Role of GABA(B) receptors in learning and memory and neurological disorders. Neurosci. Biobehav. Rev. 2016, 63, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef]
- Benarroch, E.E. GABAB receptors: Structure, functions, and clinical implications. Neurology 2012, 78, 578–584. [Google Scholar] [CrossRef]
- Bittigau, P.; Sifringer, M.; Genz, K.; Reith, E.; Pospischil, D.; Govindarajalu, S.; Dzietko, M.; Pesditschek, S.; Mai, I.; Dikranian, K.; et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc. Natl. Acad. Sci. USA 2002, 99, 15089–15094. [Google Scholar] [CrossRef]
- Stefovska, V.G.; Uckeremann, O.; Czuczwar, M.; Smitka, M.; Czuczwar, P.; Kis, J.; Kaindl, A.M.; Turski, L.; Turski, W.A.; Ikonomidou, C. Sedative and anticonvulsant drugs suppress postnatal neurogenesis. Ann. Neurol. 2008, 64, 434–445. [Google Scholar] [CrossRef]
- Conklin, P.; Heggeness, F.W. Maturation of temperature homeostasis in the rat. Am. J. Physiol. 1971, 220, 333–336. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)). Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Rocha, L.; Alonso-Vanegas, M.; Martínez-Juárez, I.E.; Orozco-Suárez, S.; Escalante-Santiago, D.; Feria-Romero, I.A.; Zavala-Tecuapetla, C.; Cisneros-Franco, J.M.; Buentello-García, R.M.; Cienfuegos, J. GABAergic alterations in neocortex of patients with pharmacoresistant temporal lobe epilepsy can explain the comorbidity of anxiety and depression: The potential impact of clinical factors. Front. Cell Neurosci. 2015, 8, 442. [Google Scholar] [CrossRef]




| Ref. No | Gene Symbol | Gene Name | Gene Aliases |
|---|---|---|---|
| Rn00690933_m1 | Ppia | peptidylprolyl isomerase A, cyclophilin A | CYCA, CyP-A |
| Rn00788315_m1 | Gabra1 | gamma-aminobutyric acid (GABA) A receptor, alpha 1 | - |
| Rn01413643_m1 | Gabra2 | gamma-aminobutyric acid (GABA) A receptor, alpha 2 | - |
| Rn00589846_m1 | Gabra4 | gamma-aminobutyric acid (GABA) A receptor, alpha 4 | - |
| Rn01464079_m1 | Gabrg2 | gamma-aminobutyric acid (GABA) A receptor, gamma 2 | - |
| Rn00568740_m1 | Gabrd | gamma-aminobutyric acid (GABA) A receptor, delta | GABAA-RD |
| Rn00561986_m1 | Syp | synaptophysin | Syp1 |
| Rn00582550_m1 | Gabbr2 | gamma-aminobutyric acid (GABA) B receptor 2 | Gpr51 |
| Binding | Ligand (nM) and S.A. | Buffer pH 7.4 | Incubation | Exposition (RT) | Non-Labeled Ligand |
|---|---|---|---|---|---|
| GABAA | [3H] Muscimol (20 nM); 20 Ci/mmol | Tris citrate (50 mM) | 45 min at 4 °C | 8 weeks | GABA (10 µM) |
| GABAB | [3H] CGP54626 (4 nM); 30 Ci/mmol | Tris HCl (50 mM) and CaCl2 (10 mM) | 90 min at 22 °C | 12 weeks | CGP 55845 (100 µM) |
| BDZ | [3H] Flunirazepam (2 nM); 85.2 Ci/mmol | Tris HCl (170 mM) | 45 min at 4 °C | 3 weeks | Clonazepam (1µM) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubová, H.; Bendová, Z.; Moravcová, S.; Pačesová, D.; Rocha, L.; Mareš, P. Neonatal Clonazepam Administration Induced Long-Lasting Changes in GABAA and GABAB Receptors. Int. J. Mol. Sci. 2020, 21, 3184. https://doi.org/10.3390/ijms21093184
Kubová H, Bendová Z, Moravcová S, Pačesová D, Rocha L, Mareš P. Neonatal Clonazepam Administration Induced Long-Lasting Changes in GABAA and GABAB Receptors. International Journal of Molecular Sciences. 2020; 21(9):3184. https://doi.org/10.3390/ijms21093184
Chicago/Turabian StyleKubová, Hana, Zdeňka Bendová, Simona Moravcová, Dominika Pačesová, Luisa Rocha, and Pavel Mareš. 2020. "Neonatal Clonazepam Administration Induced Long-Lasting Changes in GABAA and GABAB Receptors" International Journal of Molecular Sciences 21, no. 9: 3184. https://doi.org/10.3390/ijms21093184
APA StyleKubová, H., Bendová, Z., Moravcová, S., Pačesová, D., Rocha, L., & Mareš, P. (2020). Neonatal Clonazepam Administration Induced Long-Lasting Changes in GABAA and GABAB Receptors. International Journal of Molecular Sciences, 21(9), 3184. https://doi.org/10.3390/ijms21093184