The 15q11.2 BP1-BP2 Microdeletion (Burnside–Butler) Syndrome: In Silico Analyses of the Four Coding Genes Reveal Functional Associations with Neurodevelopmental Disorders



Abstract
:1. Introduction
2. Results
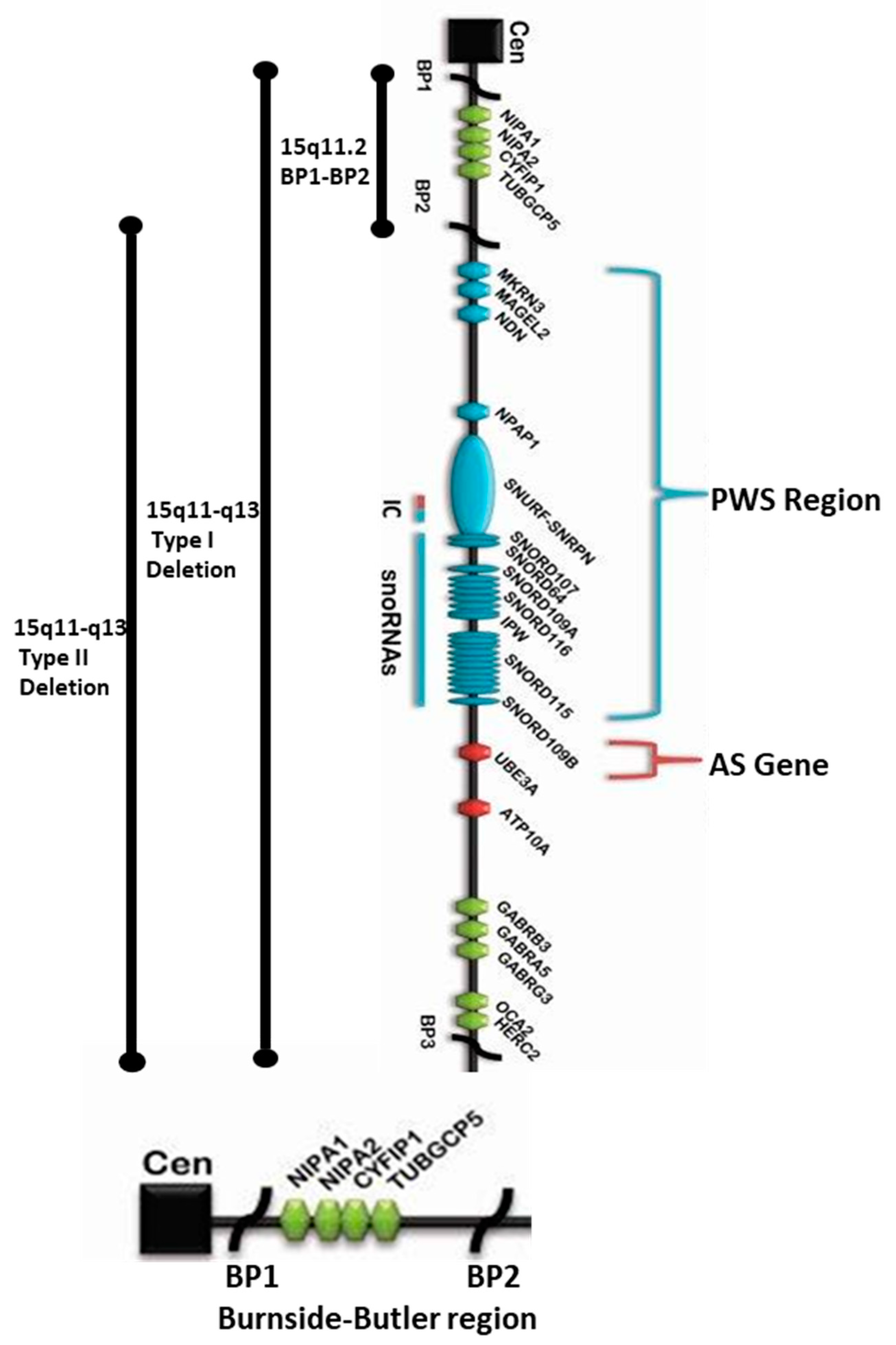
2.1. Overview of the Four Genes in the 15q11.2 BP1-BP2 Region
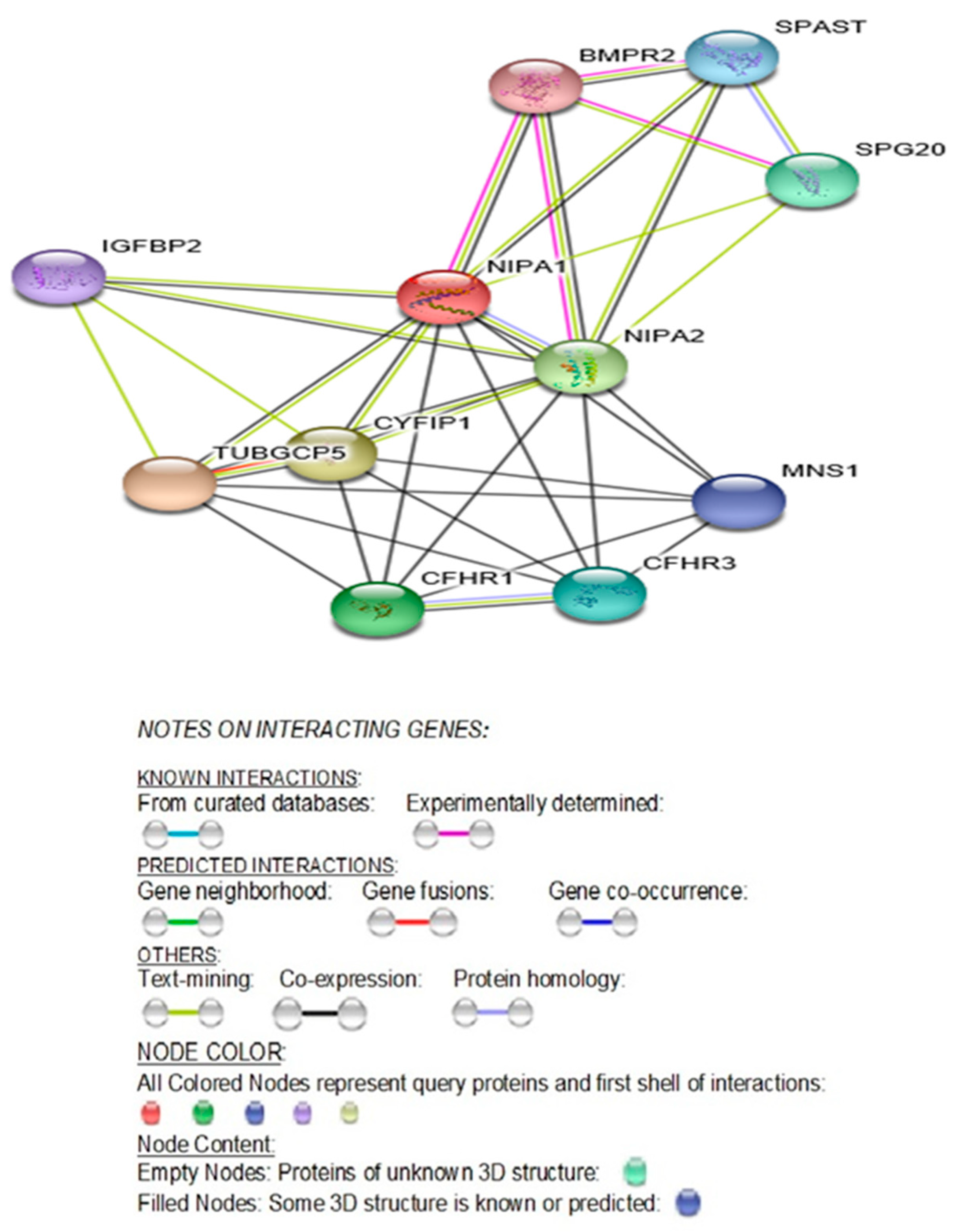
2.2. NIPA1 (Non-Imprinted in Prader–Willi/Angelman Syndrome Region Protein 1) Gene
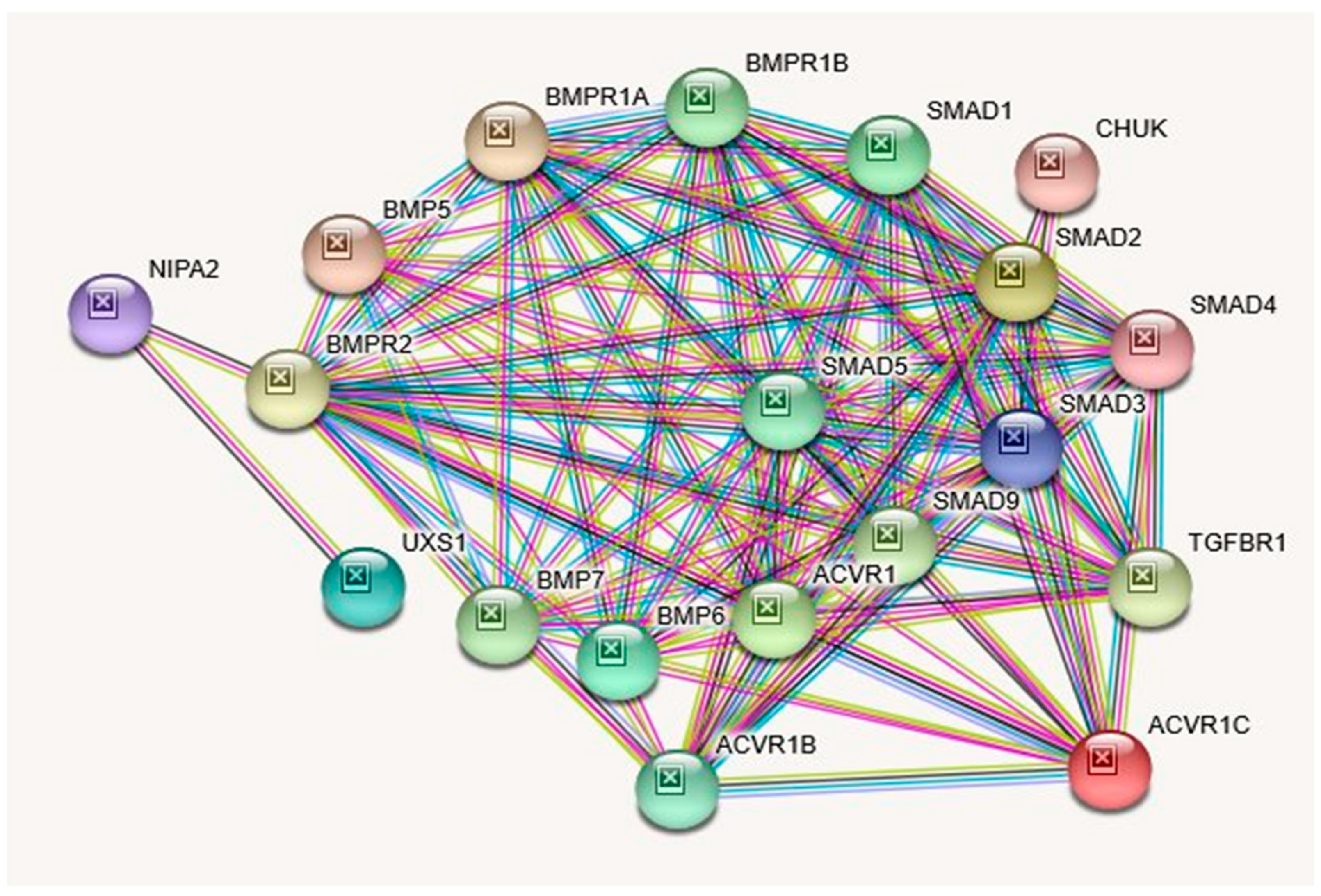
2.3. NIPA2 (Non-Imprinted in Prader–Willi/Angelman Syndrome Region Protein 2) Gene
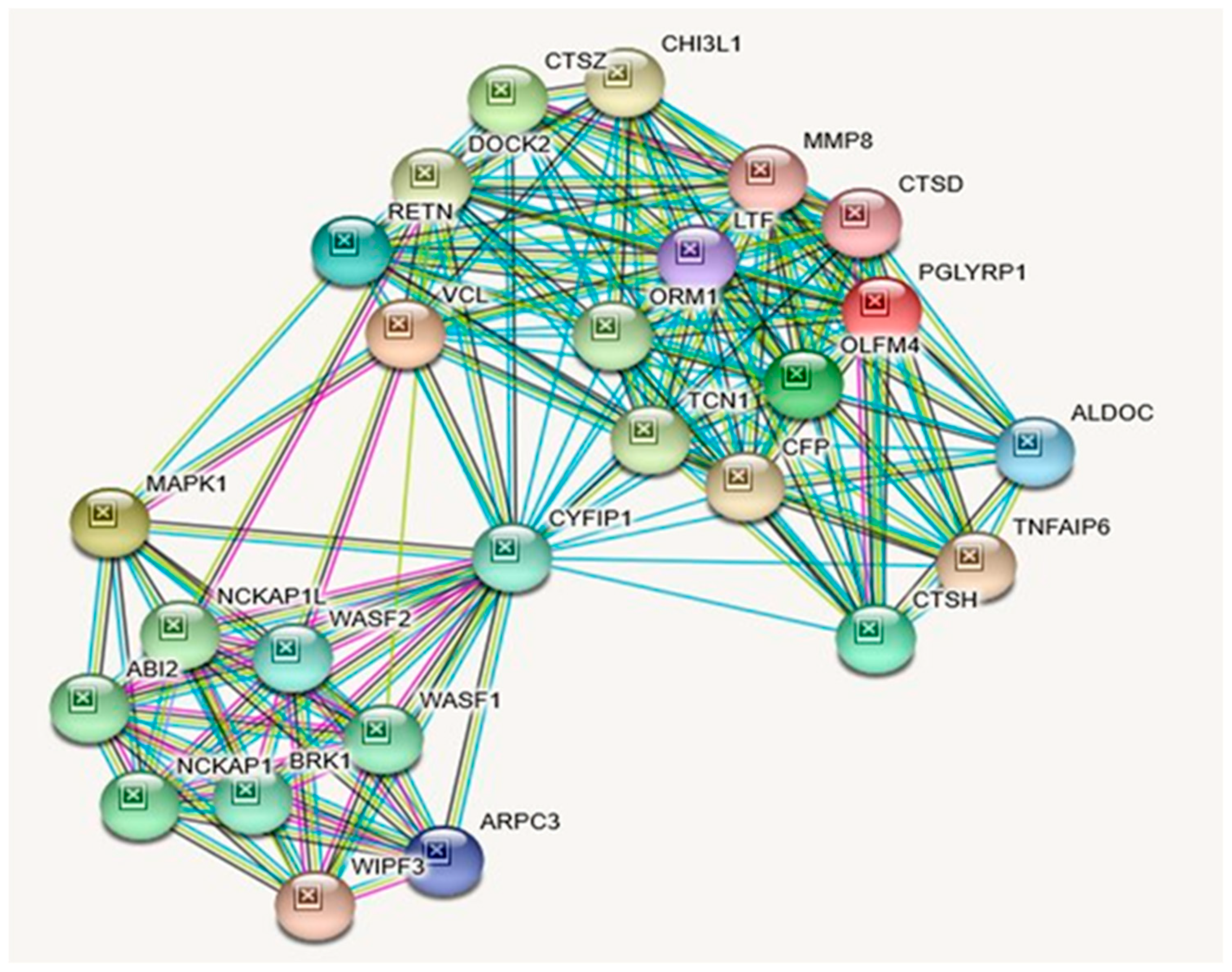
2.4. CYFIP1 (Cytoplasmic FMR1 Interacting Protein 1) Gene
2.5. TUBGCP5 (Tubulin Gamma Complex Associated Protein 5) Gene
3. Discussion
3.1. Overlapping Association of PWS and AS Specific Genes with Both Syndromes at Varying Degrees
3.2. 15q11.2 B-P1-BP2 Microdeletion (Burnside–Butler) Syndrome: Characterization of Genes Within the BP1-BP2 Region, Meta-Analysis, and Parent-of-origin Effects Reveal Neurodevelopmental Associated Phenotypes
3.3. All Four BP1-BP2 Region Genes Are Significantly Associated with Autism Spectrum Disorder
3.4. 15q11.2 B–P1–BP2 Microdeletion (Burnside–Butler) Syndrome: Frequency
3.5. Summary
4. Materials and Methods
t in q
- tf stands for term frequency—the more times a search term appears in a document, the higher the score
- idf stands for inverse document frequency—matches on rarer terms count more than matches on common terms
- coord is the coordination factor—if there are multiple terms in a query, the more terms that match, the higher the score
- lengthNorm—matches on a smaller field score higher than matches on a larger field
- index-time boost—if a boost was specified for a document at index time, scores for searches that match that document will be boosted.
- query clause boost—a user may explicitly boost the contribution of one part of a query over another.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Butler, M.G. Clinical and genetic aspects of the 15q11.2 BP1-BP2 microdeletion disorder. J. Intellect. Disabil. Res. 2017, 61, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Bittel, D.C.; Butler, M.G. Prader-Willi syndrome: Clinical genetics, cytogenetics and molecular biology. Expert Rev. Mol. Med. 2005, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Lee, P.D.K.; Whitman, B.Y. Management of Prader-Willi Syndrome, 3rd ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Cox, D.M.; Butler, M.G. The 15q11.2 BP1-BP2 microdeletion syndrome: A review. Int. J. Mol. Sci. 2015, 16, 4068–4082. [Google Scholar] [PubMed]
- Writing Committee for the ENIGMA-CNV Working Group; Van Der Meer, D.; Sønderby, I.E.; Kaufmann, T.; Walters, G.B.; Abdellaoui, A.; Ames, D.; Amunts, K.; Andersson, M.; Armstrong, N.J.; et al. Association of copy number variation of the 15q11.2 BP1-BP2 region with cortical and subcortical morphology and cognition. JAMA Psychiatry 2019, 30, 1–11. [Google Scholar]
- Ho, K.S.; Wassman, E.R.; Baxter, A.L.; Hensel, C.H.; Martin, M.M.; Prasad, A.; Twede, H.; Vanzo, R.J.; Butler, M.G. Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders using an ultra-high-resolution chromosomal microarray optimized for neurodevelopmental disorders. Int. J. Mol. Sci. 2016, 17, 2070. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.G. Magnesium supplement and the 15q11.2 BP1-BP2 microdeletion (Burnside-Butler) syndrome: A potential treatment? Int. J. Mol. Sci. 2019, 20, 2914. [Google Scholar] [CrossRef] [Green Version]
- Das, D.K.; Tapias, V.; D’’Aiuto, L.; Chowdari, K.V.; Francis, L.; Zhi, Y.; Ghosh, A.; Surti, U.; Tischfield, J.; Sheldon, M.; et al. Genetic and morphological features of human iPSC-derived neurons with chromosome 15q11.2 (BP1-BP2) deletions. Mol. Neuropsychiatry 2015, 1, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.-H.; Locke, D.; Greally, J.M.; Knoll, J.H.M.; Ohta, T.; Dunai, J.; Yavor, A.; Eichler, E.E.; Nicholls, R.D. Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am. J. Hum. Genet. 2003, 73, 898–925. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zhang, Y.; Zhang, P.; Sang, T.; Zhang, F.; Ji, T.; Huang, Q.; Xie, H.; Du, R.; Cai, B.; et al. NIPA2 located in 15q11.2 is mutated in patients with childhood absence epilepsy. Hum. Genet. 2012, 131, 1217–1224. [Google Scholar]
- Jiang, Y.; Zhang, Y.; Zhang, P.; Zhang, F.; Xie, H.; Chan, P.; Wu, X. NIPA2 mutations are correlative with childhood absence epilepsy in the Han Chinese population. Hum. Genet. 2014, 133, 675–676. [Google Scholar] [CrossRef]
- Jerkovich, A.M.; Butler, M.G. Further phenotypic expansion of 15q11.2 BP1-BP2 microdeletion (Burnside-Butler) syndrome. J. Pediatric Genet. 2014, 3, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Jerkovich, A.M.; Butler, M.G. 15q11.2 Microdeletion (BP1-BP2) and developmental delay, behaviour issues, epilepsy and congenital heart disease: A series of 52 patients. Eur. J. Med. Genet. 2015, 58, 140–147. [Google Scholar]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteomics 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goytain, A.; Hines, R.; El-Husseini, A.; Quamme, G.A. NIPA1(SPG6), the basis for autosomal dominant form of hereditary spastic paraplegia, encodes a functional Mg2+ transporter. J. Biol. Chem. 2007, 282, 8060–8068. [Google Scholar] [CrossRef] [Green Version]
- Rainier, S.; Chai, J.-H.; Tokarz, D.; Nicholls, R.D.; Fink, J.K. NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6). Am. J. Hum. Genet. 2003, 73, 967–971. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Matthies, D.S.; Botzolakis, E.J.; Macdonald, R.L.; Blakely, R.D.; Hedera, P. Hereditary spastic paraplegia-associated mutations in the NIPA1 gene and its Caenorhabditis elegans homolog trigger neural degeneration in vitro and in vivo through a gain-of-function mechanism. J. Neurosci. 2008, 28, 13938–13951. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-P.; Lin, S.-P.; Lee, C.; Chern, S.-R.; Wu, P.-S.; Chen, Y.-N.; Chen, S.-W.; Wang, W. Familial transmission of recurrent 15q11.2 (BP1-BP2) microdeletion encompassing NIPA1, NIPA2, CYFIP1, and TUBGCP5 associated with phenotypic variability in developmental, speech, and motor delay. Taiwan J. Obstet. Gynecol. 2017, 56, 93–97. [Google Scholar] [CrossRef]
- Leblond, C.S.; Heinrich, J.; Delorme, R.; Proepper, C.; Betancur, C.; Huguet, G.; Konyukh, M.; Chaste, P.; Ey, E.; Råstam, M.; et al. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012, 8, E1002521. [Google Scholar] [CrossRef] [Green Version]
- Klebe, S.; Durr, A.; Bouslam, N.; Grid, D.; Paternotte, C.; Depienne, C.; Hanein, S.; Bouhouche, A.; Elleuch, N.; Azzedine, H.; et al. Spastic Paraplegia 5: Locus refinement, candidate gene analysis and clinical description. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144B, 854–861. [Google Scholar] [CrossRef]
- Botzolakis, E.J.; Zhao, J.; Gurba, K.N.; Macdonald, R.L.; Hedera, P. The effect of HSP-causing mutations in SPG3A and NIPA1 on the assembly, trafficking, and interaction between atlastin-1 and NIPA1. Mol. Cell Neurosci. 2011, 46, 122–135. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.G. Single gene and syndromic causes of obesity: Illustrative examples. In Progress in Molecular Biology and Translational Science; Tao, Y., Ed.; Elsevier Inc: Chennai, India, 2016; Volume 140, pp. 1–45. [Google Scholar]
- Butler, M.G.; Hartin, S.N.; Hossain, W.A.; Manzardo, A.M.; Kimonis, V.E.; Dykens, E.; Gold, J.A.; Kim, S.-J.; Weisensel, N.; Tamura, R.; et al. Molecular genetic classification in Prader-Willi syndrome: A multisite cohort study. J. Med. Genet. 2019, 56, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Song, C.; Guo, H.; Xu, P.; Huang, W.; Zhou, Y.; Sun, J.; Li, C.-X.; Du, Y.; Li, X.; et al. Distinct novel mutations affecting the same base in the NIPA1 gene cause autosomal dominant hereditary spastic paraplegia in two Chinese families. Hum. Mutat. 2005, 25, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Chen, Y.-J.; Wang, M.-W.; Lin, X.-H.; Dong, E.-L.; Chen, W.-J.; Wang, N.; Lin, X. Genetic and clinical profile of Chinese patients with autosomal dominant spastic paraplegia. Mol. Diagn. Ther. 2019, 23, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.S.; Damiano, J.A.; Mullen, S.A.; Bellows, S.; Scheffer, I.E.; Berkovic, S.F. Does variation in NIPA2 contribute to genetic generalized epilepsy? Hum. Genet. 2014, 133, 673–674. [Google Scholar] [CrossRef] [Green Version]
- Cowan, C.M.; Jiang, X.; Hsu, T.; Soo, C.; Zhang, B.; Wang, J.Z.; Kuroda, S.; Wu, B.; Zhang, Z.; Zhang, X.; et al. Synergistic effects of Nell-1 and BMP-2 on the osteogenic differentiation of myoblasts. J. Bone Miner. Res. 2007, 22, 918–930. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Shaw, W.R.; Tsang, H.T.H.; Reid, E.; O’Kane, C.J. Drosophila spichthyin inhibits BMP signaling and regulates synaptic growth and axonal microtubules. Nat. Neursci. 2007, 10, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.J.; Mendelsohn, M.; Jessel, T.M. Neuronal patterning by BMPs: A requirement for GDF7 in the generation of a discrete class of commissural interneurons in the mouse spinal cord. Genes Dev. 1998, 12, 3394–3407. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-H.; Ivkovic, S.; Murray, R.C.; Jaramillo, S.; Lyons, K.S.; Johnson, J.E.; Calof, A.L. Autoregulation of neurogenesis by GDF11. Neuron 2003, 37, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, I.; Taniguchi, J.; Hata, K.; Saeki, N.; Yamashita, T. BMP inhibition enhances axonal growth and functional recovery after spinal cord injury. J. Neurochem. 2008, 105, 1471–1479. [Google Scholar] [CrossRef]
- Sahni, V.; Mukhopadhyay, A.; Tysseling, V.; Hebert, A.; Birch, D.; Mcguire, T.L.; Kessler, J.A. BMPR1a and BMPR1b signaling exert opposing effects on gliosis after spinal cord injury. J. Neurosci. 2010, 30, 1839–1855. [Google Scholar] [CrossRef] [Green Version]
- Bayat, V.; Jaiswal, M.; Bellen, H.J. The BMP signaling pathway at the Drosophila neuromuscular junction and its links to neurodegenerative diseases. Curr. Opin. Neurobiol. 2011, 21, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackstone, C. Cellular pathways of hereditary spastic paraplegia. Annu. Rev. Neurosci. 2012, 35, 25–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein commmunities and disease networks. Nature 2017, 25, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.W.; Serrano, M.A.; Loddo, S.; Robinson, C.; Alesi, V.; Dallapiccola, B.; Novelli, A.; Butler, M.G. Parent-of-origin effects in 15q11.2 BP1-BP2 microdeletion (Burnside-Butler) syndrome. Int. J. Mol. Sci. 2019, 20, 1459. [Google Scholar] [CrossRef] [Green Version]
- Bogdan, S.; Grewe, O.; Strunk, M.; Mertens, A.; Klämbt, C. Sra-1 interacts with Kette and Wasp and is required for neuronal and bristle development in Drosophila. Development 2004, 131, 3981–3989. [Google Scholar] [CrossRef] [Green Version]
- Napoli, I.; Mercaldo, V.; Boyl, P.P.; Eleuteri, B.; Zalfa, F.; De Rubeis, S.; Di Marino, D.; Mohr, E.; Massimi, M.; Falconi, M.; et al. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 2008, 134, 1042–1054. [Google Scholar] [CrossRef] [Green Version]
- Doe, C.M.; Relkovic, D.; Garfield, A.S.; Dalley, J.W.; Theobald, D.E.; Humby, T.; Wilkinson, L.S.; Isles, A.R. Loss of the imprinted snoRNA mbii-52 leads to increased 5htr2c pre-RNA editing and altered 5HT2CR-mediated behaviour. Hum. Mol. Genet. 2009, 18, 2140–2148. [Google Scholar] [CrossRef]
- Stelzer, Y.; Sagi, I.; Yanuka, O.; Eiges, R.; Benvenisty, N. The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader-Willi syndrome. Nat. Genet. 2014, 46, 551–557. [Google Scholar] [CrossRef]
- Garfield, A.; Davies, J.; Burke, L.; Furby, H.; Wilkinson, L.; Heisler, L.; Isles, A. Increased alternate splicing of Htr2c in a mouse model for Prader-Willi syndrome leads disruption of 5HT2C receptor mediated appetite. Mol. Brain 2016, 9, 95. [Google Scholar] [CrossRef] [Green Version]
- Clayton-Smith, J.; Laan, L. Angelman syndrome: A review of the clinical and genetic aspects. J. Med. Genet. 2003, 40, 87–95. [Google Scholar] [CrossRef]
- Galiveti, C.R.; Raabe, C.A.; Konthur, Z.; Rozhdestvensky, T.S. Differential regulation of non-protein coding RNAs from Prader-Willi syndrome locus. Sci. Rep. 2014, 4, 6445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnside, R.D.; Pasion, R.; Mikhail, F.M. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: A susceptibility region for neurological dysfunction including developmental and language delay. Hum. Genet. 2011, 130, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Rafi, S.K.; Manzardo, A.M. High-resolution chromosome ideogram representation of currently recognized genes for autism spectrum disorders. Int. J. Mol. Sci. 2015, 16, 6464–6495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafi, S.; Fernández-Jaén, A.; Alvarez, S.; Nadeau, O.W.; Butler, M.G. High functioning autism with missense mutations in Synaptotagmin-like protein 4 (SYTL4) and Transmembrane protein 187 (TMEM187) genes: SYTL4- protein modeling, protein-protein interaction, expression profiling and microRNA studies. Int. J. Mol. Sci. 2019, 20, 3358. [Google Scholar] [CrossRef] [Green Version]
- Ontology, G.; Genet, N. The Gene Ontology Consortium. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar]
- Rappaport, N.; Twik, M.; Plaschkes, I.; Nudel, R.; Iny Stein, T.; Levitt, J.; Lancet, D. MalaCards: An amalgamated human disease compendium with diverse clinical and genetic annotation and structured search. Nucleic Acids Res. 2017, 45, D877–D887. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Kaplan, S. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE | Description | Categories | Score | ||||||
|---|---|---|---|---|---|---|---|---|---|
| NIPA1 | Magnesium transporter NIPA1; Acts as a Mg (2+) transporter. Can also transport other divalent cations such as Fe(2+), Sr(2+), Ba(2+), Mn(2+) and Co(2+) but to a much less extent than Mg(2+) (By similarity); Belongs to the NIPA family. | Gene Fusion | Co-occurrence | Co-expression | Experiments | Databases | Text mining | [Homology] | |
| TUBGCP5 | Gamma-tubulin complex component 5; Gamma-tubulin complex is necessary for microtubule nucleation at the centrosome. | YES | YES | 0.995 | |||||
| CYFIP1 | Cytoplasmic FMR1-interacting protein 1; Component of the CYFIP1-EIF4E-FMR1 complex which binds to the mRNA cap and mediates translational repression. In the CYFIP1-EIF4E-FMR1 complex this subunit is an adapter between EIF4E and FMR1. Promotes the translation repression activity of FMR1 in brain probably by mediating its association with EIF4E and mRNA (By similarity). Regulates formation of membrane ruffles and lamellipodia. Plays a role in axon outgrowth. Binds to F-actin but not to RNA. Part of the WAVE complex that regulates actin filament reorganization. | YES | YES | 0.967 | |||||
| NIPA2 | Magnesium transporter NIPA2; Acts as a selective Mg (2+) transporter; Belongs to the NIPA family. | YES | YES | 0.941 | |||||
| CFHR1 | Complement factor H-related protein 1; Involved in complement regulation. | YES | 0.937 | ||||||
| SPG20 | Spartin; May be implicated in endosomal trafficking, or microtubule dynamics, or both. Participates in cytokinesis. | YES | 0.87 | ||||||
| CFHR3 | Complement factor H-related protein 3; Might be involved in complement regulation. | YES | 0.832 | ||||||
| SPAST | Spastin; ATP-dependent microtubule severing protein that recognizes and cuts polyglutamylated microtubules. | YES | 0.808 | ||||||
| MNS1 | Meiosis-specific nuclear structural protein 1; May play a role in the control of meiotic division and germ cell differentiation through regulation of pairing and recombination during meiosis. | YES | 0.769 | ||||||
| IGFBP2 | Insulin-like growth factor-binding protein 2; Inhibits IGF-mediated growth and developmental rates. | YES | 0.746 | ||||||
| BMPR2 | Bone morphogenetic protein receptor type 2 | YES | 0.736 | ||||||
| GO-term | Description | Count in Gene Set | False Discovery Rate |
|---|---|---|---|
| Biological Process (GO) | |||
| GO:0061387 | Regulation of extent of cell growth | 3 of 96 | 0.0147 |
| GO:1903830 | Magnesium ion transmembrane transport | 2 of 17 | 0.0179 |
| GO:0050770 | Regulation of axonogenesis | 3 of 162 | 0.0179 |
| GO:0001558 | Regulation of cell growth | 4 of 402 | 0.0179 |
| GO:0120035 | Regulation of plasma membrane-bounded cell projection organization | 4 of 600 | 0.0243 |
| GO:0045773 | Positive regulation of axon extension | 2 of 39 | 0.0243 |
| GO:0090287 | Regulation of cellular response to growth factor stimulus | 3 of 254 | 0.025 |
| GO:0048638 | Regulation of developmental growth | 3 of 302 | 0.0317 |
| GO:0031346 | Positive regulation of cell projection organization | 3 of 343 | 0.0375 |
| GO:0007052 | Mitotic spindle organization | 2 of 70 | 0.0375 |
| GO:0030510 | Regulation of BMP signaling pathway | 2 of 86 | 0.0412 |
| GO:0120034 | Positive regulation of plasma membrane-bounded cell projection assembly | 2 of 93 | 0.0456 |
| Molecular Function (GO) | |||
| GO:0015095 | Magnesium ion transmembrane transporter activity | 2 of 17 | 0.0042 |
| GO-term | Description | Count in Gene Set | False Discovery Rate |
|---|---|---|---|
| Biological Process (GO) | |||
| GO:0030509 | BMP signaling pathway | 10 of 92 | 3.30 × 10−15 |
| GO:0010862 | Positive regulation of pathway restricted SMAD protein phosphorylation | 9 of 49 | 3.30 × 10−15 |
| GO:0061448 | Connective tissue development | 11 of 194 | 7.18 × 10−15 |
| GO:0030501 | Positive regulation of bone mineralization | 8 of 35 | 1.05 × 10−14 |
| GO:0051216 | Cartilage development | 10 of 147 | 3.31 × 10−15 |
| Molecular Function (GO) | |||
| GO:0005160 | Transforming growth factor beta receptor binding | 7 of 50 | 1.37 × 10−11 |
| GO:0070700 | BMP receptor binding | 5 of 9 | 9.71 × 10−11 |
| GO:0019199 | Transmembrane receptor protein kinase activity | 6 of 78 | 5.79 × 10−9 |
| GO:0008083 | Growth factor activity | 7 of 160 | 5.79 × 10−9 |
| GO:0005125 | Cytokine activity | 7 of 216 | 3.33 × 10−8 |
| Cellular Component (GO) | |||
| GO:0043025 | Neuronal cell body | 6 of 460 | 0.00089 |
| GO:1990712 | HFE-transferrin receptor complex | 2 of 8 | 0.0026 |
| GO:0005615 | Extracellular space | 7 of 1134 | 0.0031 |
| GO:0030425 | Dendrite | 5 of 531 | 0.0048 |
| GO:0043235 | Receptor complex | 4 of 305 | 0.0052 |
| KEGG Pathways | |||
| HSA04350 | TGF-beta signaling pathway | 9 of 83 | 5.28 × 10−15 |
| HSA04390 | Hippo signaling pathway | 9 of 152 | 4.68 × 10−13 |
| HSA04060 | Cytokine–cytokine receptor interaction | 6 of 263 | 3.07 × 10−6 |
| HSA04360 | Axon guidance | 5 of 173 | 8.95 × 10−6 |
| Reactome Pathways | |||
| HSA201451 | Signaling by BMP | 5 of 27 | 9.97 × 10−9 |
| HAS9006936 | Signaling by TGF-beta family members | 6 of 100 | 3.10 × 10−8 |
| HSA2129379 | Molecules associated with elastic fibers | 4 of 37 | 2.03 × 10−6 |
| HSA1474244 | Extracellular matrix organization | 5 of 298 | 0.00017 |
| HSA8866652 | Synthesis of active ubiquitin: roles of E1 and E2 enzymes | 2 of 30 | 0.0055 |
| Number | MalaCards ID | Name of Associated Disease | MIFTS | Solr Relevance Score |
|---|---|---|---|---|
| 1 | SPS127 | Spastic Paraplegia 6, Autosomal Dominant | 40 | 7.269 |
| 2 | SPS041 | Spastic Paraplegia 6 | 26 | 6.526 |
| 3 | PRP016 | Paraplegia | 54 | 5.9 |
| 4 | HRD010 | Hereditary Spastic Paraplegia | 67 | 5.563 |
| 5 | ANG001 | Angelman Syndrome | 65 | 3.787 |
| 6 | ATS013 | Autosomal Recessive Congenital Ichthyosis | 65 | 3.647 |
| 7 | SPS215 | Spastic Paraplegia 3, Autosomal Dominant | 56 | 3.104 |
| 8 | SPS147 | Spastic Paraplegia 4, Autosomal Dominant | 50 | 3.027 |
| 9 | SPS148 | Spastic Paraplegia 31, Autosomal Dominant | 41 | 3.027 |
| 10 | CMP101 | Complex Hereditary Spastic Paraplegia | 23 | 3.027 |
| 11 | PRH002 | Pure Hereditary Spastic Paraplegia | 22 | 3.027 |
| 12 | PRD006 | Prader–Willi Syndrome | 60 | 2.978 |
| 13 | SPS107 | Spastic Paraplegia 18, Autosomal Recessive | 34 | 2.978 |
| 14 | SPS099 | Spastic Paraplegia 42, Autosomal Dominant | 34 | 2.978 |
| 15 | SPS021 | Spastic Paraplegia 10 | 35 | 1.081 |
| 16 | SCH015 | Schizophrenia | 76 | 0.187 |
| 17 | ATS364 | Autism | 68 | 0.171 |
| 18 | SPS057 | Spasticity | 38 | 0.171 |
| 19 | AMY091 | Amyotrophic Lateral Sclerosis 1 | 88 | 0.132 |
| 20 | LTR001 | Lateral Sclerosis | 56 | 0.132 |
| 21 | SPS012 | Spastic Paraplegia 3a | 26 | 0.132 |
| 22 | EPL164 | Epilepsy | 73 | 0.108 |
| 23 | HYP595 | Hypertension- Essential | 87 | 0.076 |
| 24 | BDY004 | Body Mass Index Quantitative Trait Locus 11 | 78 | 0.076 |
| 25 | DWN001 | Down Syndrome | 70 | 0.076 |
| 26 | ATS007 | Autism Spectrum Disorder | 69 | 0.076 |
| 27 | THR014 | Thrombocytopenia | 67 | 0.076 |
| 28 | DYS154 | Dystonia | 65 | 0.076 |
| 29 | PRP019 | Peripheral Nervous System Disease | 64 | 0.076 |
| 30 | PRC016 | Pre-Eclampsia | 63 | 0.076 |
| GENE | Description |
|---|---|
| NIPA1 | Non-Imprinted in Prader-Willi/Angelman Syndrome Region Protein 1; Acts as a Mg (2+) transporter. Can also transport other divalent cations such as Fe (2+), Sr (2+), Ba (2+), Mn (2+) and Co (2+) but to a much less extent than Mg (2+) Belongs to the NIPA family. |
| TGFB1 | Transforming growth factor beta 1; Multifunctional protein that controls proliferation, differentiation and other functions in many cell types. |
| BMP4 | Bone morphogenetic protein- 4; Induces cartilage and bone formation. Also act in mesoderm induction, tooth development, limb formation and fracture repair. |
| BMP6 | Bone morphogenetic protein 6; Induces cartilage and bone formation. |
| BMP8B | Bone morphogenetic protein 8B; Induces cartilage and bone formation. Plays a role in calcium regulation and bone homeostasis; Belongs to the TGF-beta family. |
| MUSK | Muscle, skeletal receptor tyrosine-protein kinase; Receptor tyrosine kinase which plays a central role in the formation and the maintenance of the neuromuscular junction (NMJ), the synapse between the motor neuron and the skeletal muscle. |
| BMPR2 | Bone morphogenetic protein receptor type 2; On ligand binding, forms a receptor complex consisting of two type II and two type I transmembrane serine/threonine kinases., |
| BMP2 | Bone morphogenetic protein 2; Induces cartilage and bone formation. Stimulates the differentiation of myoblasts into osteoblasts via the EIF2AK3-EIF2A- ATF4 pathway |
| BMP7 | Bone morphogenetic protein 7; Induces cartilage and bone formation. May be the osteoinductive factor responsible for the phenomenon of epithelial osteogenesis. Plays a role in calcium regulation and bone homeostasis. |
| BMPR1B | Bone morphogenetic protein receptor type-1B; On ligand binding, forms a receptor complex consisting of two type II and two type I transmembrane serine/threonine kinases. |
| GDF2 | Growth/differentiation factor 2; Potent circulating inhibitor of angiogenesis. Signals through the type I activin receptor ACVRL1. Signaling through SMAD1 in endothelial cells requires TGF-beta coreceptor endoglin. |
| GO-term | Description | Count in Gene Set | False Discovery Rate |
|---|---|---|---|
| Biological Process (GO) | |||
| GO:0007178 | Transmembrane receptor protein serine/threonine kinase signaling pathway | 16 of 189 | 1.49 × 10−23 |
| GO:0030509 | BMP signaling pathway | 11 of 92 | 7.85 × 10−17 |
| GO:0007167 | Enzyme-linked receptor protein signaling pathway | 17 of 698 | 7.85 × 10−17 |
| GO:0010862 | Positive regulation of pathway restricted SMAD protein phosphorylation | 9 of 49 | 3.68 × 10−15 |
| GO:0090100 | Positive regulation of transmembrane receptor protein serine/threonine kinase signaling pathway | 10 of 102 | 1.24 × 10−14 |
| Molecular Function (GO) | |||
| GO:0046332 | SMAD binding | 9 of 73 | 6.30 × 10−14 |
| GO:0004675 | Transmembrane receptor protein serine/threonine kinase activity | 7 of 17 | 6.30 × 10−14 |
| GO:0005072 | Transforming growth factor beta receptor, cytoplasmic mediator activity | 6 of 10 | 9.18 × 10−13 |
| GO:0019199 | Transmembrane receptor protein kinase activity | 8 of 78 | 3.47 × 10−12 |
| GO:0030618 | Transforming growth factor beta receptor, pathway-specific cytoplasmic mediator activity | 5 of 5 | 1.79 × 10−11 |
| Cellular Component (GO) | |||
| GO:0071141 | SMAD protein complex | 6 of 7 | 5.77 × 10−13 |
| GO:0098802 | Plasma membrane receptor complex | 7 of 158 | 8.00 × 10−8 |
| GO:0043235 | Receptor complex | 8 of 305 | 1.50 × 10−7 |
| GO:1902554 | Serine/threonine protein kinase complex | 5 of 69 | 1.33 × 10−6 |
| GO:0048179 | Activin receptor complex | 3 of 3 | 1.33 × 10−6 |
| KEGG Pathways | |||
| hsa04350 | TGF-beta signaling pathway | 16 of 83 | 2.41 × 10−30 |
| hsa04550 | Signaling pathways regulating pluripotency of stem cells | 12 of 138 | 2.17 × 10−18 |
| hsa04390 | Hippo signaling pathway | 11 of 152 | 4.78 × 10−16 |
| hsa04060 | Cytokine-cytokine receptor interaction | 7 of 263 | 4.15E × 10−7 |
| Reactome Pathways | |||
| HSA9006936 | Signaling by TGF-beta family members | 12 of 100 | 2.36 × 10−19 |
| HSA201451 | Signaling by BMP | 7 of 27 | 5.78 × 10−13 |
| HSA1502540 | Signaling by Activin | 5 of 13 | 6.95 × 10−10 |
| HSA181150 | Signaling by NODAL | 5 of 20 | 3.21 × 10−9 |
| HSA3315487 | SMAD2/3 MH2 Domain Mutants in Cancer | 4 of 6 | 1.01 × 10−8 |
| Number | MalaCards ID | Name of Associated Diseases | MIFTS | Solr Relevance Score |
|---|---|---|---|---|
| 1 | PD006 | Prader–Willi Syndrome | 60 | 5.045 |
| 2 | CHL002 | Childhood Absence Epilepsy | 60 | 4.53 |
| 3 | ANG001 | Angelman Syndrome | 65 | 4.495 |
| 4 | CHL058 | Childhood Electroclinical Syndrome | 21 | 3.568 |
| 5 | ATS013 | Autosomal Recessive Congenital Ichthyosis | 65 | 2.523 |
| 6 | SCH015 | Schizophrenia | 76 | 0.215 |
| 7 | ATS364 | Autism | 68 | 0.196 |
| 8 | EPL164 | Epilepsy | 73 | 0.124 |
| 9 | HYP595 | Hypertension, Essential | 87 | 0.088 |
| 10 | OST002 | Osteoporosis | 79 | 0.088 |
| 11 | BDY004 | Body Mass Index Quantitative Trait Locus 11 | 78 | 0.088 |
| 12 | DWN001 | Down Syndrome | 70 | 0.088 |
| 13 | ATS007 | Autism Spectrum Disorder | 69 | 0.088 |
| 14 | THR014 | Thrombocytopenia | 67 | 0.088 |
| 15 | MCR010 | Microcephaly | 56 | 0.088 |
| 16 | BNM029 | Bone Mineral Density Quantitative Trait Locus 15 | 51 | 0.088 |
| 17 | HYD064 | Hydrocephalus, Congenital, 1 | 47 | 0.088 |
| 18 | BNM022 | Bone Mineral Density Quantitative Trait Locus 8 | 43 | 0.088 |
| 19 | CHR523 | Chromosome 15q11.2 Deletion Syndrome | 31 | 0.088 |
| 20 | IMM162 | Immunoglobulin E Concentration, Serum | 29 | 0.088 |
| GENE | Description |
|---|---|
| NIPA2 | Magnesium transporter NIPA2; Acts as a selective Mg(2+) transporter; Belongs to the NIPA family. |
| ACVR1C | Activin receptor type-1C; Serine/threonine protein kinase which forms a receptor complex on ligand binding. |
| SMAD2 | Mothers against decapentaplegic homolog 2; also known as SMAD family member 2; Receptor-regulated SMAD (R-SMAD) that is an intracellular signal transducer and transcriptional modulator activated by TGF-beta (transforming growth factor) and activin type 1 receptor kinases. |
| ACVR1 | Activin receptor type 1; On ligand binding, forms a receptor complex consisting of two type II and two type I transmembrane serine/threonine kinases. |
| BMP6 | Bone morphogenetic protein 6; Induces cartilage and bone formation; Bone morphogenetic proteins. |
| UXS1 | UDP-glucuronic acid decarboxylase 1; Catalyzes the NAD-dependent decarboxylation of UDP- glucuronic acid to UDP-xylose. Necessary for the biosynthesis of the core tetrasaccharide in glycosaminoglycan biosynthesis. |
| SMAD3 | Mothers against decapentaplegic homolog 3; also known as SMAD family member 3; Receptor-regulated SMAD (R-SMAD) that is an intracellular signal transducer and transcriptional modulator activated by TGF-beta (transforming growth factor) and activin type 1 receptor kinases. |
| SMAD4 | Mothers against decapentaplegic homolog 4; also known as SMAD family member 4; In muscle physiology, plays a central role in the balance between atrophy and hypertrophy. |
| CHUK | Inhibitor of nuclear factor kappa-B kinase subunit alpha; Serine kinase that plays an essential role in the NF- kappa-B signaling pathway. |
| BMP5 | Bone morphogenetic protein 5; Induces cartilage and bone formation; Bone morphogenetic proteins. |
| BMPR1A | Bone morphogenetic protein receptor type-1A; On ligand binding, forms a receptor complex consisting of two type II and two type I transmembrane serine/threonine kinases. |
| BMPR2 | Bone morphogenetic protein receptor type- 2; On ligand binding, forms a receptor complex consisting of two type II and two type I transmembrane serine/threonine kinases. |
| TGFBR1 | TGF-beta receptor type-1; Transmembrane serine/threonine kinase forming with the TGF-beta type II serine/threonine kinase receptor. Regulating a plethora of physiological and pathological processes including cell cycle arrest in epithelial and hematopoietic cells, control of mesenchymal cell proliferation and differentiation, wound healing, extracellular matrix production, and immunosuppression. |
| SMAD9 | Mothers against decapentaplegic homolog 9; also known as SMAD family member 9; Transcriptional modulator activated by BMP (bone morphogenetic proteins) type 1 receptor kinase. |
| BMP7 | Bone morphogenetic protein 7; Induces cartilage and bone formation. May be the osteoinductive factor responsible for the phenomenon of epithelial osteogenesis. Plays a role in calcium regulation and bone homeostasis; Bone morphogenetic proteins. |
| BMPR1B | Bone morphogenetic protein receptor type-1B; On ligand binding, forms a receptor complex consisting of two type II and two type I transmembrane serine/threonine kinases. |
| SMAD1 | Mothers against decapentaplegic homolog 1; also known as SMAD family member 1; Transcriptional modulator activated by BMP (bone morphogenetic proteins) type 1 receptor kinase. |
| SMAD5 | Mothers against decapentaplegic homolog 5; also known as SMAD family member 5; Transcriptional modulator activated by BMP (bone morphogenetic proteins) type 1 receptor kinase. SMAD5 is a receptor-regulated SMAD (R-SMAD). |
| ACVR1B | Activin receptor type-1B; Transmembrane serine/threonine kinase activin type-1 receptor forming an activin receptor complex with activin receptor type-2 (ACVR2A or ACVR2B). Transduces the activin signal from the cell surface to the cytoplasm and is thus regulating many physiological and pathological processes including neuronal differentiation and neuronal survival, hair follicle development and cycling. |
| GO-term | Description | Count in Gene Set | False Discovery Rate |
|---|---|---|---|
| Biological Process (GO) | |||
| GO:0002252 | Immune effector process | 24 of 927 | 9.03 × 10−27 |
| GO:0016192 | Vesicle-mediated transport | 25 of 1699 | 5.71 × 10−23 |
| GO:0043312 | Neutrophil degranulation | 19 of 485 | 1.14 × 10−22 |
| GO:0051179 | Localization | 26 of 5233 | 7.42 × 10−14 |
| GO:0038096 | Fc-gamma receptor signaling pathway involved in phagocytosis | 8 of 73 | 4.66 × 10−12 |
| Molecular Function (GO) | |||
| GO:0044877 | Protein-containing complex binding | 8 of 968 | 0.00062 |
| GO:0008092 | Cytoskeletal protein binding | 8 of 882 | 0.00062 |
| GO:0004252 | Serine-type endopeptidase activity | 5 of 180 | 0.00062 |
| GO:0004175 | Endopeptidase activity | 6 of 399 | 0.00062 |
| GO:0003779 | Actin binding | 6 of 413 | 0.00062 |
| Cellular Component (GO) | |||
| GO:0035580 | Specific granule lumen | 14 of 62 | 9.87 × 10−26 |
| GO:0034774 | Secretory granule lumen | 17 of 323 | 2.31 × 10−22 |
| GO:1904724 | Tertiary granule lumen | 12 of 55 | 3.63 × 10−22 |
| GO:0030141 | Secretory granule | 19 of 828 | 1.51 × 10−19 |
| GO:0070820 | Tertiary granule | 13 of 164 | 4.33 × 10−19 |
| KEGG Pathways | |||
| hsa04810 | Regulation of actin cytoskeleton | 10 of 205 | 1.08 × 10−11 |
| hsa05131 | Shigellosis | 5 of 63 | 1.55 × 10−6 |
| hsa04666 | Fc gamma R-mediated phagocytosis | 5 of 89 | 5.32 × 10−6 |
| hsa04520 | Adherens junction | 4 of 71 | 7.99 × 10−5 |
| Reactome Pathways | |||
| HSA168249 | Innate Immune System | 26 of 1012 | 9.01 × 10−32 |
| HSA6798695 | Neutrophil degranulation | 19 of 471 | 8.77 × 10−24 |
| HSA5663213 | RHO GTPases Activate WASPs and WAVEs | 10 of 35 | 3.26 × 10−19 |
| HSA2029482 | Regulation of actin dynamics for phagocytic cup formation | 10 of 60 | 3.18 × 10−17 |
| HSA4420097 | VEGFA-VEGFR2 Pathway | 7 of 95 | 1.15 × 10−9 |
| Number | MalaCards ID | Name of Associated Diseases | MIFTS | Solr |
|---|---|---|---|---|
| Relevance | ||||
| Score | ||||
| 1 | FRG001 | Fragile X Syndrome | 69 | 5.035 |
| 2 | ATS364 | Autism | 68 | 4.578 |
| 3 | SCH015 | Schizophrenia | 76 | 4.481 |
| 4 | PRD006 | Prader–Willi Syndrome | 60 | 3.561 |
| 5 | CHR523 | Chromosome 15q11.2 Deletion Syndrome | 31 | 3.561 |
| 6 | PRV006 | Pervasive Developmental Disorder | 58 | 2.518 |
| 7 | ATS007 | Autism Spectrum Disorder | 69 | 0.377 |
| 8 | ALC028 | Alacrima, Achalasia, and Mental Retardation Syndrome | 65 | 0.188 |
| 9 | HYP595 | Hypertension, Essential | 87 | 0.084 |
| 10 | BDY004 | Body Mass Index Quantitative Trait Locus 11 | 78 | 0.084 |
| 11 | EPL164 | Epilepsy | 73 | 0.084 |
| 12 | DWN001 | Down Syndrome | 70 | 0.084 |
| 13 | LKM002 | Leukemia | 69 | 0.084 |
| 14 | LKM062 | Leukemia, Acute Lymphoblastic | 68 | 0.084 |
| 15 | NSP012 | Nasopharyngeal Carcinoma | 67 | 0.084 |
| 16 | THR014 | Thrombocytopenia | 67 | 0.084 |
| 17 | TRN020 | Turner Syndrome | 66 | 0.084 |
| 18 | ANG001 | Angelman Syndrome | 65 | 0.084 |
| 19 | ETN001 | Eating Disorder | 61 | 0.084 |
| 20 | SQM006 | Squamous Cell Carcinoma | 60 | 0.084 |
| GENE | Description |
|---|---|
| CYFIP1 | Cytoplasmic FMR1-interacting protein 1; Component of the CYFIP1-EIF4E-FMR1 complex which binds to the mRNA cap and mediates translational repression. In the CYFIP1-EIF4E-FMR1 complex this subunit is an adapter between EIF4E and FMR1. Promotes the translation repression activity of FMR1 in brain probably by mediating its association with EIF4E and mRNA (By similarity). Regulates formation of membrane ruffles and lamellipodia. Plays a role in axon outgrowth. Binds to F-actin but not to RNA. Part of the WAVE complex that regulates actin filament reorganization. |
| PGLYRP1 | Peptidoglycan recognition protein 1; Pattern receptor that binds to murein peptidoglycans (PGN) of Gram-positive bacteria. Has bactericidal activity towards Gram-positive bacteria. |
| VCL | Vinculin; Actin filament (F-actin)-binding protein involved in cell-matrix adhesion and cell-cell adhesion. Regulates cell- surface E-cadherin expression and potentiates mechanosensing by the E-cadherin complex. May also play important roles in cell morphology and locomotion; Belongs to the vinculin/alpha-catenin family. |
| MAPK1 | Mitogen-activated protein kinase 1; Serine/threonine kinase which acts as an essential component of the MAP kinase signal transduction pathway. MAPK1/ERK2 and MAPK3/ERK1 are the 2 MAPKs which play an important role in the MAPK/ERK cascade. Depending on the cellular context, the MAPK/ERK cascade mediates diverse biological functions such as cell growth, adhesion, survival and differentiation through the regulation of transcription, translation, cytoskeletal rearrangements. |
| CTSZ | Cathepsin Z; Exhibits carboxy-monopeptidase as well as carboxy- dipeptidase activity. |
| OLFM4 | Olfactomedin-4; May promote proliferation of pancreatic cancer cells by favoring the transition from the S to G2/M phase. In myeloid leukemic cell lines, inhibits cell growth and induces cell differentiation and apoptosis. |
| CTSH | Pro-cathepsin H; Important for the overall degradation of proteins in lysosomes; Belongs to the peptidase C1 family. |
| RETN | Resistin; Hormone that seems to suppress insulin ability to stimulate glucose uptake into adipose cells (By similarity). Potentially links obesity to diabetes (By similarity). Promotes chemotaxis in myeloid cells. |
| ALDOC | Aldolase, fructose-bisphosphate C. |
| ARPC3 | Actin-related protein 2/3 complex subunit 3; Functions as component of the Arp2/3 complex which is involved in regulation of actin polymerization and together with an activating nucleation-promoting factor (NPF) mediates the formation of branched actin networks. |
| LTF | Lactotransferrin; Lactoferroxins A, B and C have opioid antagonist activity. |
| CTSD | Cathepsin D; Acid protease active in intracellular protein breakdown. Involved in the pathogenesis of several diseases such as breast cancer and possibly Alzheimer disease; Cathepsins. |
| MMP8 | Neutrophil collagenase; Can degrade fibrillar type I, II, and III collagens; Belongs to the peptidase M10A family. |
| WIPF3 | WAS/WASL-interacting protein family member 3; May be a regulator of cytoskeletal organization. May have a role in spermatogenesis (By similarity); Belongs to the verprolin family. |
| TNFAIP6 | Tumor necrosis factor-inducible gene 6 protein; Possibly involved in cell-cell and cell-matrix interactions during inflammation and tumorigenesis. |
| CFP | Properdin; A positive regulator of the alternate pathway of complement. It binds to and stabilizes the C3- and C5-convertase enzyme complexes. |
| CHI3L1 | Chitinase-3-like protein 1; Carbohydrate-binding lectin with a preference for chitin. Has no chitinase activity. May play a role in tissue remodeling and in the capacity of cells to respond to and cope with changes in their environment. |
| DOCK2 | Dedicator of cytokinesis protein 2; Involved in cytoskeletal rearrangements required for lymphocyte migration in response of chemokines. |
| TCN1 | Transcobalamin-1; Binds vitamin B12 with femtomolar affinity and protects it from the acidic environment of the stomach; Belongs to the eukaryotic cobalamin transport proteins family. |
| ORM1 | Alpha-1-acid glycoprotein 1; Functions as transport protein in the blood stream. Binds various ligands in the interior of its beta-barrel domain. Appears to function in modulating the activity of the immune system during the acute-phase reaction. |
| NCKAP1L | Nck-associated protein 1-like; Essential hematopoietic-specific regulator of the actin cytoskeleton (Probable). Controls lymphocyte development, activation, proliferation and homeostasis, erythrocyte membrane stability, as well as phagocytosis and migration by neutrophils and macrophages. Component of the WAVE2 complex which signals downstream of RAC to stimulate F- actin polymerization. |
| ABI2 | Abl interactor 2; May act in regulation of cell growth and transformation by interacting with nonreceptor tyrosine kinases ABL1 and/or ABL2. Part of the WAVE complex that regulates lamellipodia formation. The WAVE complex regulates actin filament reorganization via its interaction with the Arp2/3 complex. |
| NCKAP1 | Nck-associated protein 1; Part of the WAVE complex that regulates lamellipodia formation. The WAVE complex regulates actin filament reorganization via its interaction with the Arp2/3 complex. As component of the WAVE1 complex, required for BDNF-NTRK2 endocytic trafficking and signaling from early endosomes. |
| WASF1 | Wiskott-Aldrich syndrome protein family member 1; Downstream effector molecule involved in the transmission of signals from tyrosine kinase receptors and small GTPases to the actin cytoskeleton. Promotes formation of actin filaments. Part of the WAVE complex that regulates lamellipodia formation. |
| BRK1 | Protein BRICK1; Involved in regulation of actin and microtubule organization. Part of a WAVE complex that activates the Arp2/3 complex. As component of the WAVE1 complex, required for BDNF- NTRK2 endocytic trafficking and signaling from early endosomes. |
| WASF2 | Wiskott-Aldrich syndrome protein family member 2; Downstream effector molecule involved in the transmission of signals from tyrosine kinase receptors and small GTPases to the actin cytoskeleton. Promotes formation of actin filaments. Part of the WAVE complex that regulates lamellipodia formation. The WAVE complex regulates actin filament reorganization via its interaction with the Arp2/3 complex; Wiskott-Aldrich Syndrome protein family. |
| GO-term | Description | Count in Gene Set | False Discovery Rate |
|---|---|---|---|
| Biological Process (GO) | |||
| GO:0000086 | G2/M transition of mitotic cell cycle | 20 of 123 | 4.48 × 10−36 |
| GO:1903047 | Mitotic cell cycle process | 25 of 564 | 2.99 × 10−35 |
| GO:0010389 | Regulation of G2/M transition of mitotic cell cycle | 20 of 149 | 6.29 × 10−35 |
| GO:0070925 | Organelle assembly | 25 of 666 | 6.97 × 10−34 |
| Molecular Function (GO) | |||
| GO:0043015 | Gamma-tubulin binding | 6 of 28 | 3.66 × 10−10 |
| GO:0015631 | Tubulin binding | 8 of 344 | 5.61 × 10−7 |
| GO:0005200 | Structural constituent of cytoskeleton | 4 of 106 | 0.0004 |
| GO:0008017 | Microtubule binding | 4 of 253 | 0.0065 |
| GO:0030291 | Protein serine/threonine kinase inhibitor activity | 2 of 33 | 0.0155 |
| Cellular Component (GO) | |||
| GO:0005813 | Centrosome | 23 of 468 | 3.98 × 10−32 |
| GO:0005815 | Microtubule organizing center | 24 of 683 | 4.49 × 10−31 |
| GO:0015630 | Microtubule cytoskeleton | 25 of 1118 | 1.64 × 10−28 |
| GO:0044430 | Cytoskeletal part | 25 of 1547 | 3.73 × 10−25 |
| GO:0044450 | Microtubule organizing center part | 14 of 167 | 5.27 × 10−21 |
| KEGG Pathways | |||
| hsa04114 | Oocyte meiosis | 4 of 116 | 0.0013 |
| hsa04110 | Cell cycle | 3 of 123 | 0.0218 |
| hsa05203 | Viral carcinogenesis | 3 of 183 | 0.0447 |
| hsa05169 | Epstein–Barr virus infection | 3 of 194 | 0.0447 |
| Reactome Pathways | |||
| HSA380270 | Recruitment of mitotic centrosome proteins and complexes | 26 of 79 | 5.07 × 10−59 |
| HSA380320 | Recruitment of NuMA to mitotic centrosomes | 26 of 91 | 4.11 × 10−58 |
| HSA380259 | Loss of Nlp from mitotic centrosomes | 20 of 68 | 4.97 × 10−42 |
| HSA8854518 | AURKA Activation by TPX2 | 20 of 71 | 8.45 × 10−42 |
| HSA2565942 | Regulation of PLK1 Activity at G2/M Transition | 20 of 85 | 1.69 × 10−40 |
| Number | MalaCards ID | Name of Associated Diseases | MIFTS | Solr Relevance Score |
|---|---|---|---|---|
| 1 | PRD006 | Prader–Willi Syndrome | 60 | 4.293 |
| 2 | SCH015 | Schizophrenia | 76 | 0.23 |
| 3 | ATS364 | Autism | 68 | 0.21 |
| 4 | MCR010 | Microcephaly | 56 | 0.188 |
| 5 | HYP595 | Hypertension, Essential | 87 | 0.094 |
| 6 | BDY004 | Body Mass Index Quantitative Trait Locus 11 | 78 | 0.094 |
| 7 | EPL164 | Epilepsy | 73 | 0.094 |
| 8 | DWN001 | Down Syndrome | 70 | 0.094 |
| 9 | ATS007 | Autism Spectrum Disorder | 69 | 0.094 |
| 10 | THR014 | Thrombocytopenia | 67 | 0.094 |
| 11 | HYD064 | Hydrocephalus, Congenital, 1 | 47 | 0.094 |
| 12 | PRM031 | Primary Autosomal Recessive Microcephaly | 47 | 0.094 |
| 13 | PRM212 | Primary Microcephaly | 42 | 0.094 |
| GENE | Description |
|---|---|
| TUBGCP5 | Gamma-tubulin complex component 5; Gamma-tubulin complex is necessary for microtubule nucleation at the centrosome. |
| HAUS4 | HAUS augmin-like complex subunit 4; Contributes to mitotic spindle assembly, maintenance of centrosome integrity and completion of cytokinesis. |
| CEP41 | Centrosomal protein of 41 kDa; Required during ciliogenesis for tubulin glutamylation in cilium. |
| HAUS3 | HAUS augmin-like complex subunit 3; Contributes to mitotic spindle assembly, maintenance of centrosome integrity and completion of cytokinesis. |
| TUBGCP6 | Gamma-tubulin complex component 6; Gamma-tubulin complex is necessary for microtubule nucleation at the centrosome. |
| HAUS8 | HAUS augmin-like complex subunit 8; Contributes to mitotic spindle assembly, maintenance of centrosome integrity and completion of cytokinesis. |
| CEP135 | Centrosomal protein of 135 kDa; Centrosomal protein involved in centriole biogenesis. Acts as a scaffolding protein during early centriole biogenesis. Required for the targeting of centriole satellite proteins to centrosomes such as of PCM1, SSX2IP and CEP290 and recruitment of WRAP73 to centrioles. Also required for centriole-centriole cohesion during interphase. |
| DCTN3 | Dynactin subunit 3; Together with dynein may be involved in spindle assembly and cytokinesis. |
| HAUS2 | HAUS augmin-like complex subunit 2; Contributes to mitotic spindle assembly, maintenance of centrosome integrity and completion of cytokinesis. |
| TUBGCP4 | Gamma-tubulin complex component 4; Gamma-tubulin complex is necessary for microtubule nucleation at the centrosome. |
| TUBGCP3 | Gamma-tubulin complex component 3; Gamma-tubulin complex is necessary for microtubule nucleation at the centrosome. |
| CEP76 | Centrosomal protein of 76 kDa; Centrosomal protein involved in regulation of centriole duplication. Required to limit centriole duplication to once per cell cycle by preventing centriole reduplication; |
| CLASP1 | CLIP-associating protein 1; Microtubule plus-end tracking protein that promotes the stabilization of dynamic microtubules. Involved in the nucleation of noncentrosomal microtubules originating from the trans-Golgi network (TGN). Required for the polarization of the cytoplasmic microtubule arrays in migrating cells towards the leading edge of the cell. |
| YWHAE | 14-3-3 protein epsilon; Adapter protein implicated in the regulation of a large spectrum of both general and specialized signaling pathways. |
| CEP72 | Centrosomal protein of 72 kDa; Involved in the recruitment of key centrosomal proteins to the centrosome. Provides centrosomal microtubule-nucleation activity on the gamma-tubulin ring complexes (gamma-TuRCs) and has critical roles in forming a focused bipolar spindle, which is needed for proper tension generation between sister chromatids. Involved in centriole duplication. |
| CEP70 | Centrosomal protein of 70 kDa; Plays a role in the organization of both preexisting and nascent microtubules in interphase cells. During mitosis, required for the organization and orientation of the mitotic spindle. |
| PRKAR2B | cAMP-dependent protein kinase type II-beta regulatory subunit; Regulatory subunit of the cAMP-dependent protein kinases involved in cAMP signaling in cells. |
| PLK4 | Serine/threonine-protein kinase PLK4; Serine/threonine-protein kinase that plays a central role in centriole duplication. Able to trigger procentriole formation on the surface of the parental centriole cylinder. |
| NINL | Ninein-like protein; Involved in the microtubule organization in interphase cells. Overexpression induces the fragmentation of the Golgi and causes lysosomes to disperse toward the cell periphery; it also interferes with mitotic spindle assembly. |
| CEP164 | Centrosomal protein of 164 kDa; Plays a role in microtubule organization and/or maintenance for the formation of primary cilia (PC), a microtubule-based structure that protrudes from the surface of epithelial cells. Plays a critical role in G2/M checkpoint and nuclear divisions. A key player in the DNA damage-activated ATR/ATM signaling cascade. |
| MZT2B | Mitotic spindle organizing protein 2B. |
| HAUS1 | HAUS augmin-like complex subunit 1; Contributes to mitotic spindle assembly, maintenance of centrosome integrity and completion of cytokinesis. |
| PLK1 | Serine/threonine-protein kinase PLK1; Serine/threonine-protein kinase that performs several important functions throughout M phase of the cell cycle, including the regulation of centrosome maturation and spindle assembly, the removal of cohesins from chromosome arms, the inactivation of anaphase-promoting complex/cyclosome (APC/C) inhibitors, and the regulation of mitotic exit and cytokinesis. |
| YWHAG | 14-3-3 protein gamma; Adapter protein implicated in the regulation of a large spectrum of both general and specialized signaling pathways. |
| PRKACA | cAMP-dependent protein kinase catalytic subunit alpha; Phosphorylates a large number of substrates in the cytoplasm and the nucleus. Regulates the abundance of compartmentalized pools of its regulatory subunits through phosphorylation of PJA2 which binds and ubiquitinates these subunits, leading to their subsequent proteolysis. Required for glucose- mediated adipogenic differentiation increase and osteogenic differentiation inhibition from osteoblasts. |
| MZT1 | Mitotic-spindle organizing protein 1; Required for gamma-tubulin complex recruitment to the centrosome. |
| NIPA1 | NIPA2 | CYFIP1 | TUBGCP5 |
|---|---|---|---|
| NIPA Magnesium Transporter 1 | NIPA Magnesium Transporter 2 | Cytoplasmic FMR1 Interacting Protein 1 | Tubulin Gamma Complex Associated Protein 5 |
| Protein Coding Gene | Protein Coding Gene | Protein Coding Gene | Protein Coding Gene |
| Cellular Compartmental Distribution with confidence number: | Cellular Compartmental Distribution with confidence number: | Cellular Compartmental Distribution with confidence number: | Cellular Compartmental Distribution with confidence number: |
| plasma membrane (5) * | plasma membrane (4) * | extracellular (4) * | cytoskeleton (5) * |
| endosome (4) * | endosome (3) * | cytosol (4) * | cytosol (5) * |
| Golgi apparatus (1) * | cytoskeleton (1) * | nucleus (2) * | |
| mitochondrion (1) * | |||
| nucleus (1) * | |||
| Among its Related Pathways are: | Among its Related Pathways are: | Among its Related Pathways are: | Among its Related Pathways are: |
| miscellaneous transport and binding events and transport of glucose and other sugars, bile salts and organic acids, metal ions and amine compounds. | miscellaneous transport and binding events and transport of glucose and other sugars, bile salts and organic acids, metal ions and amine compounds. | Regulation of actin dynamics for phagocytic cup formation and signaling by Rho GTPases. | Nanog in Mammalian ESC Pluripotency and G-Beta Gamma Signaling. |
| Cardinal Diseases Associated with NIPA1: | Cardinal Diseases Associated with NIPA2: | Cardinal Diseases Associated with CYFIP1: | Cardinal Disease Associated with TUBGCP5: |
| Spastic Paraplegia 6, Autosomal Dominant & Spastic Paraplegia 6. | Angelman Syndrome and Prader–Willi Syndrome. | Fragile X Syndrome and Autism. | Prader–Willi Syndrome. |
| Number | Neurodevelopmental Disorders/Diseases | NIPA1 | NIPA2 | CYFIP1 | TUBGCP5 |
|---|---|---|---|---|---|
| 1 | Prader–Willi Syndrome | YES | YES | YES | YES |
| 2 | Angelman Syndrome | YES | YES | YES | NO |
| 3 | 15q11.2 Deletion Syndrome with Attention Deficit Hyperactive Disorder & Learning Disability | YES | YES | YES | NO |
| 4 | Autism Spectrum Disorder | YES | YES | YES | YES |
| 5 | Schizophrenia | YES | YES | YES | YES |
| 6 | Epilepsy | YES | YES | YES | YES |
| 7 | Down Syndrome | YES | YES | YES | YES |
| 8 | Microcephaly | YES | YES | NO | YES |
| 9 | Developmental Disorder | NO | NO | YES | NO |
| 10 | Peripheral Nervous System Disease | YES | NO | NO | NO |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rafi, S.K.; Butler, M.G. The 15q11.2 BP1-BP2 Microdeletion (Burnside–Butler) Syndrome: In Silico Analyses of the Four Coding Genes Reveal Functional Associations with Neurodevelopmental Disorders. Int. J. Mol. Sci. 2020, 21, 3296. https://doi.org/10.3390/ijms21093296
Rafi SK, Butler MG. The 15q11.2 BP1-BP2 Microdeletion (Burnside–Butler) Syndrome: In Silico Analyses of the Four Coding Genes Reveal Functional Associations with Neurodevelopmental Disorders. International Journal of Molecular Sciences. 2020; 21(9):3296. https://doi.org/10.3390/ijms21093296
Chicago/Turabian StyleRafi, Syed K., and Merlin G. Butler. 2020. "The 15q11.2 BP1-BP2 Microdeletion (Burnside–Butler) Syndrome: In Silico Analyses of the Four Coding Genes Reveal Functional Associations with Neurodevelopmental Disorders" International Journal of Molecular Sciences 21, no. 9: 3296. https://doi.org/10.3390/ijms21093296
APA StyleRafi, S. K., & Butler, M. G. (2020). The 15q11.2 BP1-BP2 Microdeletion (Burnside–Butler) Syndrome: In Silico Analyses of the Four Coding Genes Reveal Functional Associations with Neurodevelopmental Disorders. International Journal of Molecular Sciences, 21(9), 3296. https://doi.org/10.3390/ijms21093296