The Emerging Role of the FGF/FGFR Pathway in Gastrointestinal Stromal Tumor

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. FGF/FGFR Family
3. FGF/FGFR Events in GIST
4. FGFR Mutations and Gene Fusions
5. FGFR Ligand Overexpression
6. FGFR Events and Imatinib Resistance in KIT/PDGFRA Mutant GIST
7. Clinical Perspectives
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Demetri, G.D.; von Mehren, M.; Blanke, C.D.; van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. New Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Nemunaitis, J.; Bauer, S.; Blay, J.Y.; Choucair, K.; Gelderblom, H.; George, S.; Schöffski, P.; von Mehren, M.; Zalcberg, J.; Achour, H.; et al. Intrigue: Phase III study of ripretinib versus sunitinib in advanced gastrointestinal stromal tumor after imatinib. Future Oncol. 2019, 16, 4251–4264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebreyohannes, Y.K.; Wozniak, A.; Zhai, M.E.; Wellens, J.; Cornillie, J.; Vanleeuw, U.; Evans, E.; Gardino, A.K.; Lengauer, C.; Debiec-Rychter, M.; et al. Robust Activity of Avapritinib, Potent and Highly Selective Inhibitor of Mutated KIT, in Patient-derived Xenograft Models of Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2019, 25, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, M.; Wang, Z.-F.; Sarlomo-Rikala, M.; Osuch, C.; Rutkowski, P.; Lasota, J. Succinate Dehydrogenase-Deficient GISTs. Am. J. Surg. Pathol. 2011, 35, 1712–1721. [Google Scholar] [CrossRef] [Green Version]
- Boikos, S.A.; Pappo, A.S.; Killian, J.K.; LaQuaglia, M.P.; Weldon, C.B.; George, S.; Trent, J.C.; von Mehren, M.; Wright, J.A.; Schiffman, J.D.; et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors. Jama Oncol. 2016, 2, 922. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, M.A.; Astolfi, A.; Urbini, M.; Nannini, M.; Paterini, P.; Indio, V.; Saponara, M.; Formica, S.; Ceccarelli, C.; Casadio, R.; et al. Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. Eur. J. Hum. Genet. 2014, 22, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, M.A.; Lolli, C.; Nannini, M.; Astolfi, A.; Indio, V.; Saponara, M.; Urbini, M.; la Rovere, S.; Gill, A.; Goldstein, D.; et al. Good survival outcome of metastatic SDH-deficient gastrointestinal stromal tumors harboring SDHA mutations. Genet. Med. 2015, 17, 391–395. [Google Scholar] [CrossRef]
- Killian, J.K.; Kim, S.Y.; Miettinen, M.; Smith, C.; Merino, M.; Tsokos, M.; Quezado, M.; Smith, W.I.; Jahromi, M.S.; Xekouki, P.; et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013, 3, 648–657. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, M.A.; Astolfi, A.; di Battista, M.; Heinrich, M.C.; Paterini, P.; Scotlandi, K.; Santini, D.; Catena, F.; Manara, M.C.; Nannini, M.; et al. Insulin-like growth factor 1 receptor expression in wild-type GISTs: A potential novel therapeutic target. Int. J. Cancer 2009, 125, 2991–2994. [Google Scholar] [CrossRef] [PubMed]
- Beadling, C.; Patterson, J.; Justusson, E.; Nelson, D.; Pantaleo, M.A.; Hornick, J.L.; Chacón, M.; Corless, C.L.; Heinrich, M.C. Gene expression of the IGF pathway family distinguishes subsets of gastrointestinal stromal tumors wild type for KIT and PDGFRA. Cancer Med. 2013, 2, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, M.A.; Astolfi, A.; Nannini, M.; Ceccarelli, C.; Formica, S.; Santini, D.; Heinrich, M.C.; Corless, C.; Dei Tos, A.P.; Paterini, P.; et al. Differential expression of neural markers in KIT and PDGFRA wild-type gastrointestinal stromal tumours. Histopathology 2011, 59, 1071–1080. [Google Scholar] [CrossRef]
- Miettinen, M.; Fetsch, J.F.; Sobin, L.H.; Lasota, J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: A clinicopathologic and molecular genetic study of 45 cases. Am. J. Surg. Pathol. 2006, 30, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Daniels, M.; Lurkin, I.; Pauli, R.; Erbstößer, E.; Hildebrandt, U.; Hellwig, K.; Zschille, U.; Lüders, P.; Krüger, G.; Knolle, J.; et al. Spectrum of KIT/PDGFRA/BRAF mutations and Phosphatidylinositol-3-Kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett. 2011, 312, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Gasparotto, D.; Rossi, S.; Polano, M.; Tamborini, E.; Lorenzetto, E.; Sbaraglia, M.; Mondello, A.; Massani, M.; Lamon, S.; Bracci, R.; et al. Quadruple-Negative GIST Is a Sentinel for Unrecognized Neurofibromatosis Type 1 Syndrome. Clin. Cancer Res. 2017, 23, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantaleo, M.A.; Nannini, M.; Corless, C.L.; Heinrich, M.C. Quadruple wild-type (WT) GIST: Defining the subset of GIST that lacks abnormalities of KIT, PDGFRA, SDH, or RAS signaling pathways. Cancer Med. 2015, 4, 101–103. [Google Scholar] [CrossRef]
- Nannini, M.; Astolfi, A.; Urbini, M.; Indio, V.; Santini, D.; Heinrich, M.C.; Corless, C.L.; Ceccarelli, C.; Saponara, M.; Mandrioli, A.; et al. Integrated genomic study of quadruple-WT GIST (KIT/PDGFRA/SDH/RAS pathway wild-type GIST). BMC Cancer 2014, 14, 685. [Google Scholar] [CrossRef] [Green Version]
- Nannini, M.; Urbini, M.; Astolfi, A.; Biasco, G.; Pantaleo, M.A. The progressive fragmentation of the KIT/PDGFRA wild-type (WT) gastrointestinal stromal tumors (GIST). J. Transl. Med. 2017, 15, 113. [Google Scholar] [CrossRef]
- Brenca, M.; Rossi, S.; Polano, M.; Gasparotto, D.; Zanatta, L.; Racanelli, D.; Valori, L.; Lamon, S.; Dei Tos, A.P.; Maestro, R. Transcriptome sequencing identifies ETV6-NTRK3 as a gene fusion involved in GIST. J. Pathol. 2016, 238, 543–549. [Google Scholar] [CrossRef]
- Shi, E.; Chmielecki, J.; Tang, C.M.; Wang, K.; Heinrich, M.C.; Kang, G.; Corless, C.L.; Hong, D.; Fero, K.E.; Murphy, J.D.; et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J. Transl. Med. 2016, 14, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantaleo, M.A.; Urbini, M.; Indio, V.; Ravegnini, G.; Nannini, M.; de Luca, M.; Tarantino, G.; Angelini, S.; Gronchi, A.; Vincenzi, B.; et al. Genome-wide analysis identifies MEN1 and MAX mutations and a neuroendocrine-like molecular heterogeneity in Quadruple WT GIST. Mol. Cancer Res. 2017, 15, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Belinsky, M.G.; Rink, L.; Cai, K.Q.; Capuzzi, S.J.; Hoang, Y.; Chien, J.; Godwin, A.K.; von Mehren, M. Somatic loss of function mutations in neurofibromin 1 and MYC associated factor X genes identified by exome-wide sequencing in a wild-type GIST case. BMC Cancer 2015, 15, 887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, N.; Ornitz, D.M. Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J. Biochem. 2011, 149, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbini, M.; Indio, V.; Tarantino, G.; Ravegnini, G.; Angelini, S.; Nannini, M.; Saponara, M.; Santini, D.; Ceccarelli, C.; Fiorentino, M.; et al. Gain of FGF4 is a frequent event in KIT/PDGFRA/SDH/RAS-P WT GIST. Genes Chromosomes Cancer 2019, 58, 636–642. [Google Scholar] [CrossRef] [Green Version]
- Flavahan, W.A.; Drier, Y.; Johnstone, S.E.; Hemming, M.L.; Tarjan, D.R.; Hegazi, E.; Shareef, S.J.; Javed, N.M.; Raut, C.P.; Eschle, B.K.; et al. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature 2019, 575, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Javidi-Sharifi, N.; Traer, E.; Martinez, J.; Gupta, A.; Taguchi, T.; Dunlap, J.; Heinrich, M.C.; Corless, C.L.; Rubin, B.P.; Druker, B.J.; et al. Crosstalk between KIT and FGFR3 promotes gastrointestinal stromal tumor cell growth and drug resistance. Cancer Res. 2015, 75, 880–891. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Hung, H.; Li, X.; Ruddy, D.A.; Wang, Y.; Ong, R.; Chow, P.; Qiu, S.; Tam, A.; Rakiec, D.P.; et al. FGFR-mediated reactivation of MAPK signaling attenuates antitumor effects of imatinib in gastrointestinal stromal tumors. Cancer Discov. 2015, 5, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Boichuk, S.; Dunaev, P.; Galembikova, A.; Mustafin, I.; Valeeva, E. Inhibition of fibroblast growth factor receptor-signaling sensitizes imatinib-resistant gastrointestinal stromal tumors to low doses of topoisomerase II inhibitors. Anti-Cancer Drugs 2018, 29, 549–559. [Google Scholar] [CrossRef]
- Boichuk, S.; Dunaev, P.; Galembikova, A.; Bikinieva, F.; Nurgatina, I.; Mustafin, I.; Aukhadieva, A.; Kurtasanov, R.; Andriutsa, N.; Shagimardanova, E.; et al. Inhibition of FGFR2-signaling attenuates a homology-mediated dna repair in gist and sensitizes them to DNA-topoisomerase II inhibitors. Int. J. Mol. Sci. 2020, 21, 352. [Google Scholar]
- Boichuk, S.; Galembikova, A.; Dunaev, P.; Valeeva, E.; Shagimardanova, E.; Gusev, O.; Khaiboullina, S. A novel receptor tyrosine kinase switch promotes gastrointestinal stromal tumor drug resistance. Molecules 2017, 22, 2152. [Google Scholar] [CrossRef] [Green Version]
- Boichuk, S.; Galembikova, A.; Dunaev, P.; Micheeva, E.; Valeeva, E.; Novikova, M.; Khromova, N.; Kopnin, P. Targeting of FGF-Signaling Re-Sensitizes Gastrointestinal Stromal Tumors (GIST) to Imatinib in Vitro and in Vivo. Molecules 2018, 23, 2643. [Google Scholar] [CrossRef] [Green Version]
- Itoh, N.; Ornitz, D.M. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004, 20, 563–569. [Google Scholar] [CrossRef]
- Trueb, B. Biology of FGFRL1, the fifth fibroblast growth factor receptor. Cell. Mol. Life Sci. 2011, 68, 951–964. [Google Scholar] [CrossRef] [Green Version]
- Kalinina, J.; Dutta, K.; Ilghari, D.; Beenken, A.; Goetz, R.; Eliseenkova, A.V.; Cowburn, D.; Mohammadi, M. The alternatively spliced acid box region plays a key role in FGF receptor autoinhibition. Structure 2012, 20, 77–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.E.; Lu, J.; Chen, H.; Werner, S.; Williams, L.T. The human fibroblast growth factor receptor genes: A common structural arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain. Mol. Cell. Biol. 1991, 11, 4627–4634. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Bottaro, D.P.; Fleming, T.P.; Smith, C.L.; Burgess, W.H.; Chan, A.M.L.; Aaronson, S.A. Determination of ligand-binding specificity by alternative splicing: Two distinct growth factor receptors encoded by a single gene. Proc. Natl. Acad. Sci. USA 1992, 89, 246–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Kan, M.; Xu, J.; Yan, G.; McKeehan, W.L. Ligand-specific structural domains in the fibroblast growth factor receptor. J. Biol. Chem. 1995, 270, 10222–10230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Holzmann, K.; Grunt, T.; Heinzle, C.; Sampl, S.; Steinhoff, H.; Reichmann, N.; Kleiter, M.; Hauck, M.; Marian, B. Alternative Splicing of Fibroblast Growth Factor Receptor IgIII Loops in Cancer. J. Nucleic Acids 2012, 2012, 950508. [Google Scholar] [CrossRef] [Green Version]
- Ornitz, D.M.; Itoh, N. The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ornitz, D.M. FGFs, heparan sulfate and FGFRs: Complex interactions essential for development. BioEssays 2000, 22, 108–112. [Google Scholar]
- Potthoff, M.J.; Kliewer, S.A.; Mangelsdorf, D.J. Endocrine fibroblast growth factors 15/19 and 21: From feast to famine. Genes Dev. 2012, 26, 312–324. [Google Scholar] [CrossRef] [Green Version]
- Brewer, J.R.; Mazot, P.; Soriano, P. Genetic insights into the mechanisms of Fgf signaling. Genes Dev. 2016, 30, 751–771. [Google Scholar] [CrossRef] [Green Version]
- Hallinan, N.; Finn, S.; Cuffe, S.; Rafee, S.; O’Byrne, K.; Gately, K. Targeting the fibroblast growth factor receptor family in cancer. Cancer Treat. Rev. 2016, 46, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Tanner, Y.; Grose, R.P. Dysregulated FGF signalling in neoplastic disorders. Semin. Cell Dev. Biol. 2016, 53, 126–135. [Google Scholar] [CrossRef]
- Yoon, K.; Nery, S.; Rutlin, M.L.; Radtke, F.; Fishell, G.; Gaiano, N. Fibroblast growth factor receptor signaling promotes radial glial identity and interacts with Notch1 signaling in telencephalic progenitors. J. Neurosci. 2004, 24, 9497–9506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lew, E.D.; Furdui, C.M.; Anderson, K.S.; Schlessinger, J. The precise sequence of FGF receptor autophosphorylation is kinetically driven and is disrupted by oncogenic mutations. Sci. Signal. 2009, 2, ra6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronca, R.; Ghedini, G.C.; Maccarinelli, F.; Sacco, A.; Locatelli, S.L.; Foglio, E.; Taranto, S.; Grillo, E.; Matarazzo, S.; Castelli, R.; et al. FGF trapping inhibits multiple myeloma growth through c-Myc degradation-induced mitochondrial oxidative stress. Cancer Res. 2020. [Google Scholar] [CrossRef] [Green Version]
- Raja, A.; Park, I.; Haq, F.; Ahn, S.-M. FGF19-FGFR4 Signaling in Hepatocellular Carcinoma. Cells 2019, 8, 536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhou, Y.; Huang, T.; Wu, F.; Pan, Y.; Dong, Y.; Wang, Y.; Chan, A.K.Y.; Liu, L.; Kwan, J.S.H.; et al. FGF18, a prominent player in FGF signaling, promotes gastric tumorigenesis through autocrine manner and is negatively regulated by miR-590-5p. Oncogene 2019, 38, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Dakhova, O.; Creighton, C.J.; Ittmann, M. Endocrine fibroblast growth factor FGF19 promotes prostate cancer progression. Cancer Res. 2013, 73, 2551–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matarazzo, S.; Melocchi, L.; Rezzola, S.; Grillo, E.; Maccarinelli, F.; Giacomini, A.; Turati, M.; Taranto, S.; Zammataro, L.; Cerasuolo, M.; et al. Long pentraxin-3 follows and modulates bladder cancer progression. Cancers (Basel) 2019, 11, 1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesluyes, T.; Pérot, G.; Largeau, M.R.; Brulard, C.; Lagarde, P.; Dapremont, V.; Lucchesi, C.; Neuville, A.; Terrier, P.; Vince-Ranchère, D.; et al. RNA sequencing validation of the Complexity INdex in SARComas prognostic signature. Eur. J. Cancer 2016, 57, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Grupo Espanol de Investigacion en Sarcomas Single Agent Regorafenib in First-line for Metastatic/Unresectable KIT/PDGFR Wild Type GIST (REGISTRI). Available online: https://clinicaltrials.gov/ct2/show/NCT02638766 (accessed on 9 April 2020).
- Ben-Ami, E.; Barysauskas, C.M.; von Mehren, M.; Heinrich, M.C.; Corless, C.L.; Butrynski, J.E.; Morgan, J.A.; Wagner, A.J.; Choy, E.; Yap, J.T.; et al. Long-term follow-up results of the multicenter phase II trial of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of standard tyrosine kinase inhibitor therapy. Ann. Oncol. 2016, 27, 1794–1799. [Google Scholar] [CrossRef]
- Sarker, D.; Molife, R.; Evans, T.R.J.; Hardie, M.; Marriott, C.; Butzberger-Zimmerli, P.; Morrison, R.; Fox, J.A.; Heise, C.; Louie, S.; et al. A phase I pharmacokinetic and pharmacodynamic study of TKI258, an oral, multitargeted receptor tyrosine kinase inhibitor in patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 2075–2081. [Google Scholar] [CrossRef] [Green Version]
- Joensuu, H.; Blay, J.-Y.; Comandone, A.; Martin-Broto, J.; Fumagalli, E.; Grignani, G.; del Muro, X.G.; Adenis, A.; Valverde, C.; Pousa, A.L.; et al. Dovitinib in patients with gastrointestinal stromal tumour refractory and/or intolerant to imatinib. Br. J. Cancer 2017, 117, 1278–1285. [Google Scholar] [CrossRef] [Green Version]
- Soria, J.C.; Massard, C.; Magné, N.; Bader, T.; Mansfield, C.D.; Blay, J.Y.; Bui, B.N.; Moussy, A.; Hermine, O.; Armand, J.P. Phase 1 dose-escalation study of oral tyrosine kinase inhibitor masitinib in advanced and/or metastatic solid cancers. Eur. J. Cancer 2009, 45, 2333–2341. [Google Scholar] [CrossRef]
- le Cesne, A.; Blay, J.Y.; Bui, B.N.; Bouché, O.; Adenis, A.; Domont, J.; Cioffi, A.; Ray-Coquard, I.; Lassau, N.; Bonvalot, S.; et al. Phase II study of oral masitinib mesilate in imatinib-naïve patients with locally advanced or metastatic gastro-intestinal stromal tumour (GIST). Eur. J. Cancer 2010, 46, 1344–1351. [Google Scholar] [CrossRef]
- Adenis, A.; Blay, J.-Y.; Bui-Nguyen, B.; Bouché, O.; Bertucci, F.; Isambert, N.; Bompas, E.; Chaigneau, L.; Domont, J.; Ray-Coquard, I.; et al. Masitinib in advanced gastrointestinal stromal tumor (GIST) after failure of imatinib: A randomized controlled open-label trial. Ann. Oncol. 2014, 25, 1762–1769. [Google Scholar] [CrossRef]
- Garner, A.P.; Gozgit, J.M.; Anjum, R.; Vodala, S.; Schrock, A.; Zhou, T.; Serrano, C.; Eilers, G.; Zhu, M.; Ketzer, J.; et al. Ponatinib inhibits polyclonal drug-resistant KIT oncoproteins and shows therapeutic potential in heavily pretreated gastrointestinal stromal tumor (GIST) patients. Clin. Cancer Res. 2014, 20, 5745–5755. [Google Scholar] [CrossRef] [Green Version]
- Ganjoo, K.N.; Villalobos, V.M.; Kamaya, A.; Fisher, G.A.; Butrynski, J.E.; Morgan, J.A.; Wagner, A.J.; D’Adamo, D.; McMillan, A.; Demetri, G.D.; et al. A multicenter phase II study of pazopanib in patients with advanced gastrointestinal stromal tumors (GIST) following failure of at least imatinib and sunitinib. Ann. Oncol. 2014, 25, 236–240. [Google Scholar] [CrossRef]
- Mir, O.; Cropet, C.; Toulmonde, M.; Cesne, A.L.; Molimard, M.; Bompas, E.; Cassier, P.; Ray-Coquard, I.; Rios, M.; Adenis, A.; et al. Pazopanib plus best supportive care versus best supportive care alone in advanced gastrointestinal stromal tumours resistant to imatinib and sunitinib (PAZOGIST): A randomised, multicentre, open-label phase 2 trial. Lancet. Oncol. 2016, 17, 632–641. [Google Scholar] [CrossRef]
- Centre Leon Berard Multicentre Placebo-controlled Double-blinded Phase II Study of Lenvatinib Efficacy in Patients With Locally Advanced or Metastatic GIST (Gastrointestinal Stromal Tumor) After Imatinib/Sunitinib Failure (LENVAGIST). Available online: https://clinicaltrials.gov/ct2/show/NCT04193553 (accessed on 9 April 2020).
- Kelly, C.M.; Shoushtari, A.N.; Qin, L.-X.; D’Angelo, S.P.; Dickson, M.A.; Gounder, M.M.; Keohan, M.L.; Mcfadyen, C.; Sjoberg, A.; Singer, S.; et al. A phase Ib study of BGJ398, a pan-FGFR kinase inhibitor in combination with imatinib in patients with advanced gastrointestinal stromal tumor. Investig. New Drugs 2019, 37, 282–290. [Google Scholar] [CrossRef]
- Matos, I.; Goyal, L.; Cleary, J.; Voss, M.; Oh, D.; Bernstam, F.M.; Ng, C.; Iyer, G.; Ishii, N.; Hu, Y.; et al. Debio 1347 in patients with gastrointestinal cancers harboring an FGFR gene fusion: Preliminary results. Ann. Oncol. 2019, 30 (Suppl. 4), iv122–iv123. [Google Scholar] [CrossRef]
- Gozgit, J.M.; Wong, M.J.; Moran, L.; Wardwell, S.; Mohemmad, Q.K.; Narasimhan, N.I.; Shakespeare, W.C.; Wang, F.; Clackson, T.; Rivera, V.M. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol. Cancer Ther. 2012, 11, 690–699. [Google Scholar] [CrossRef] [Green Version]

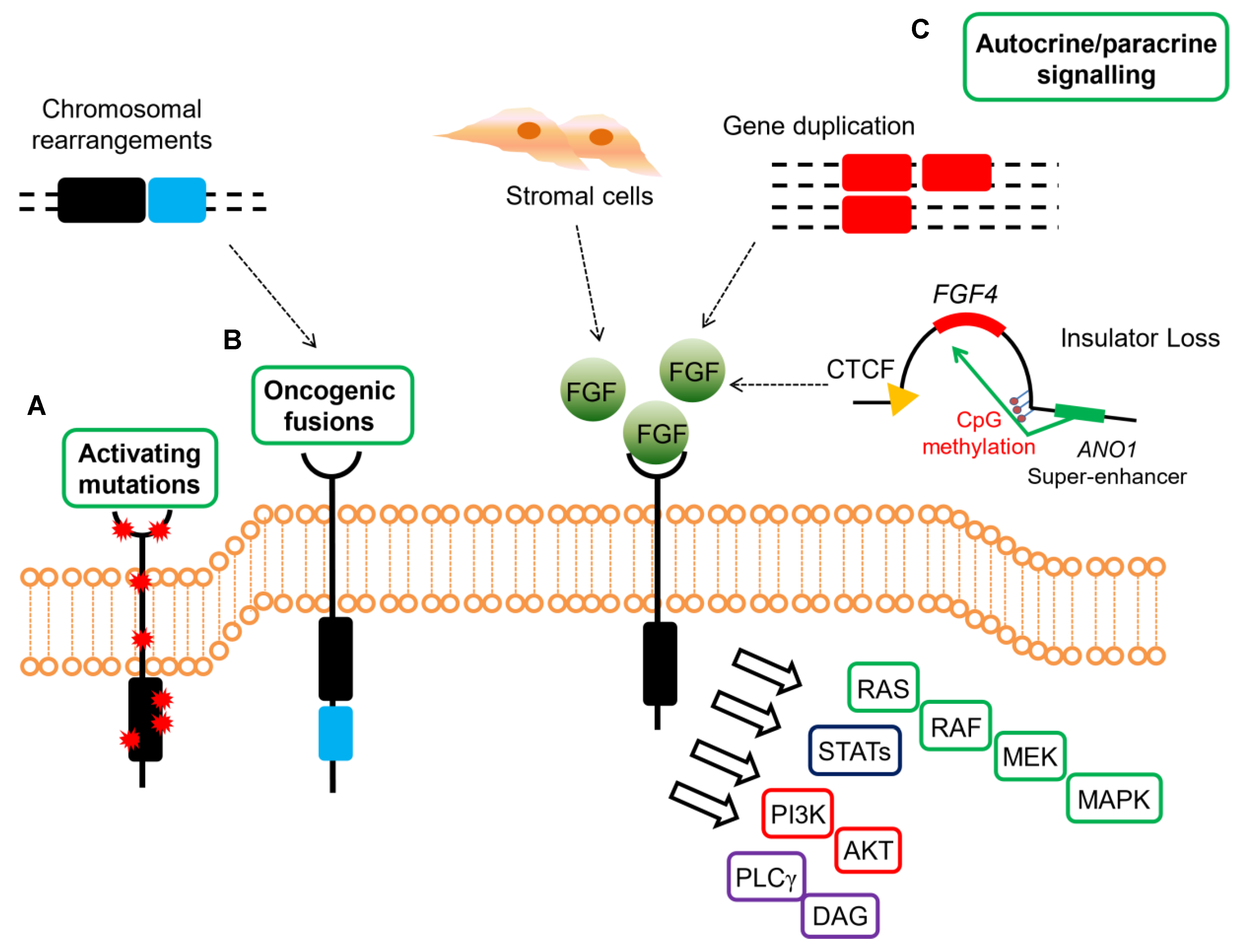


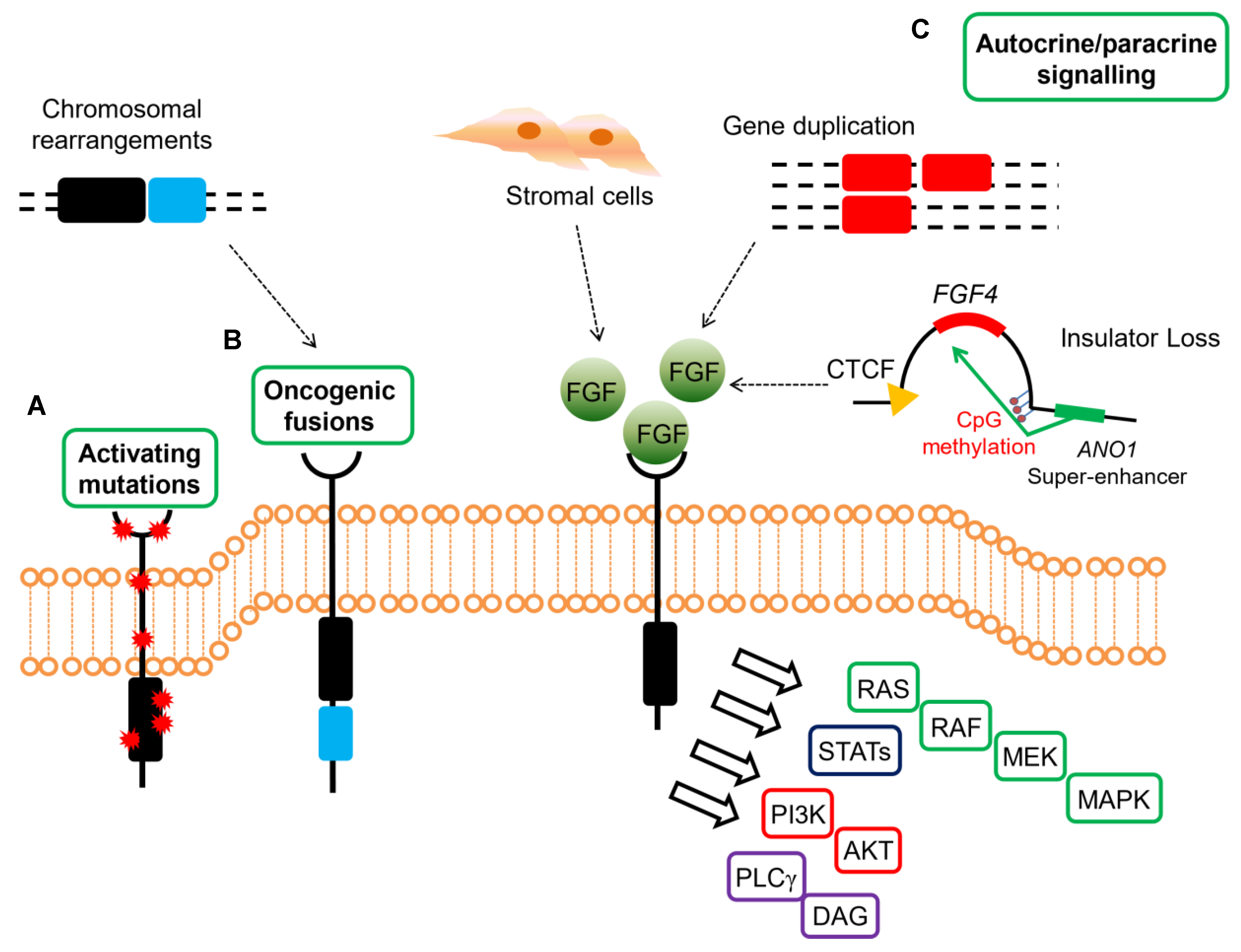
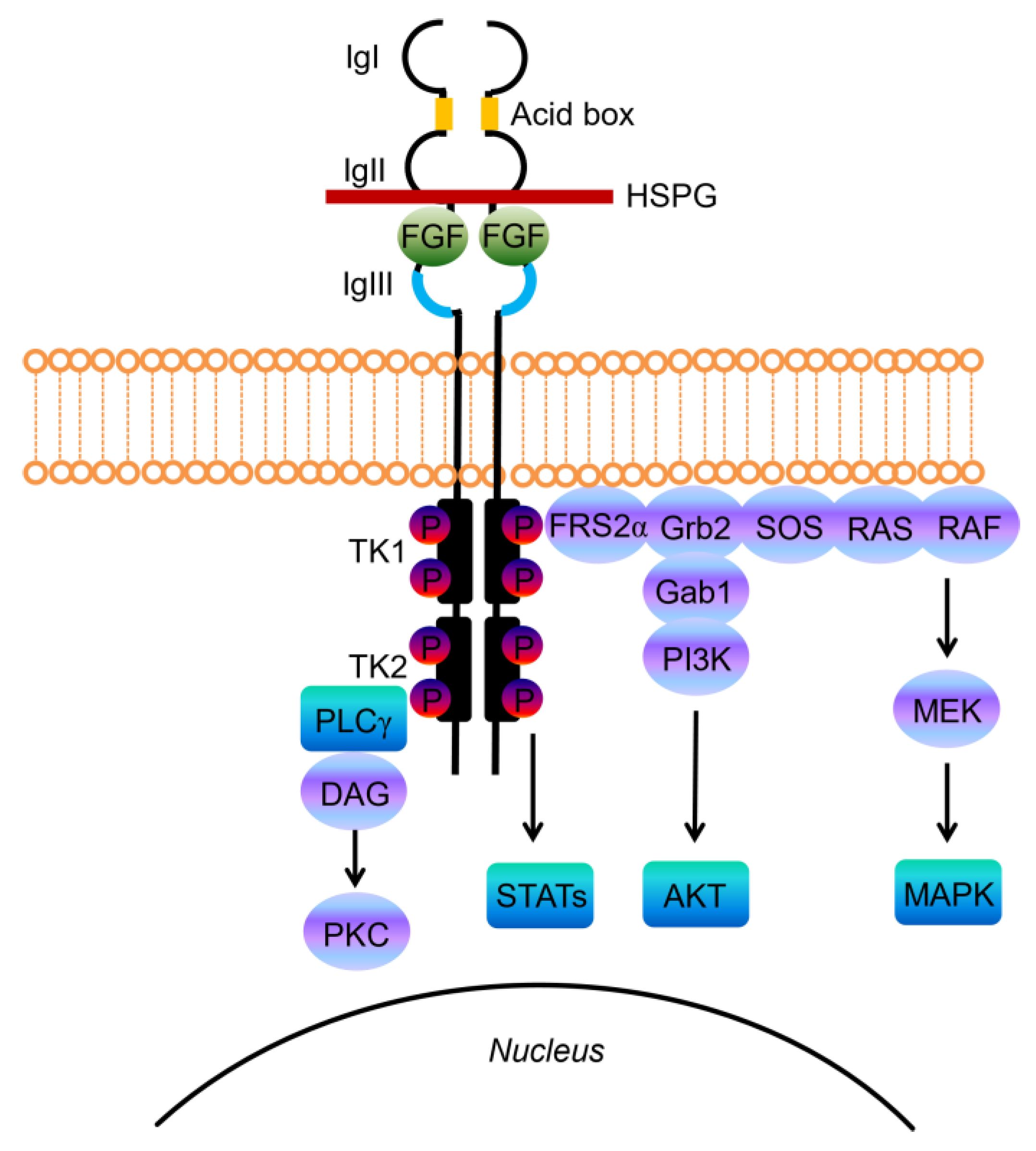{kind=link}
{kind=link}
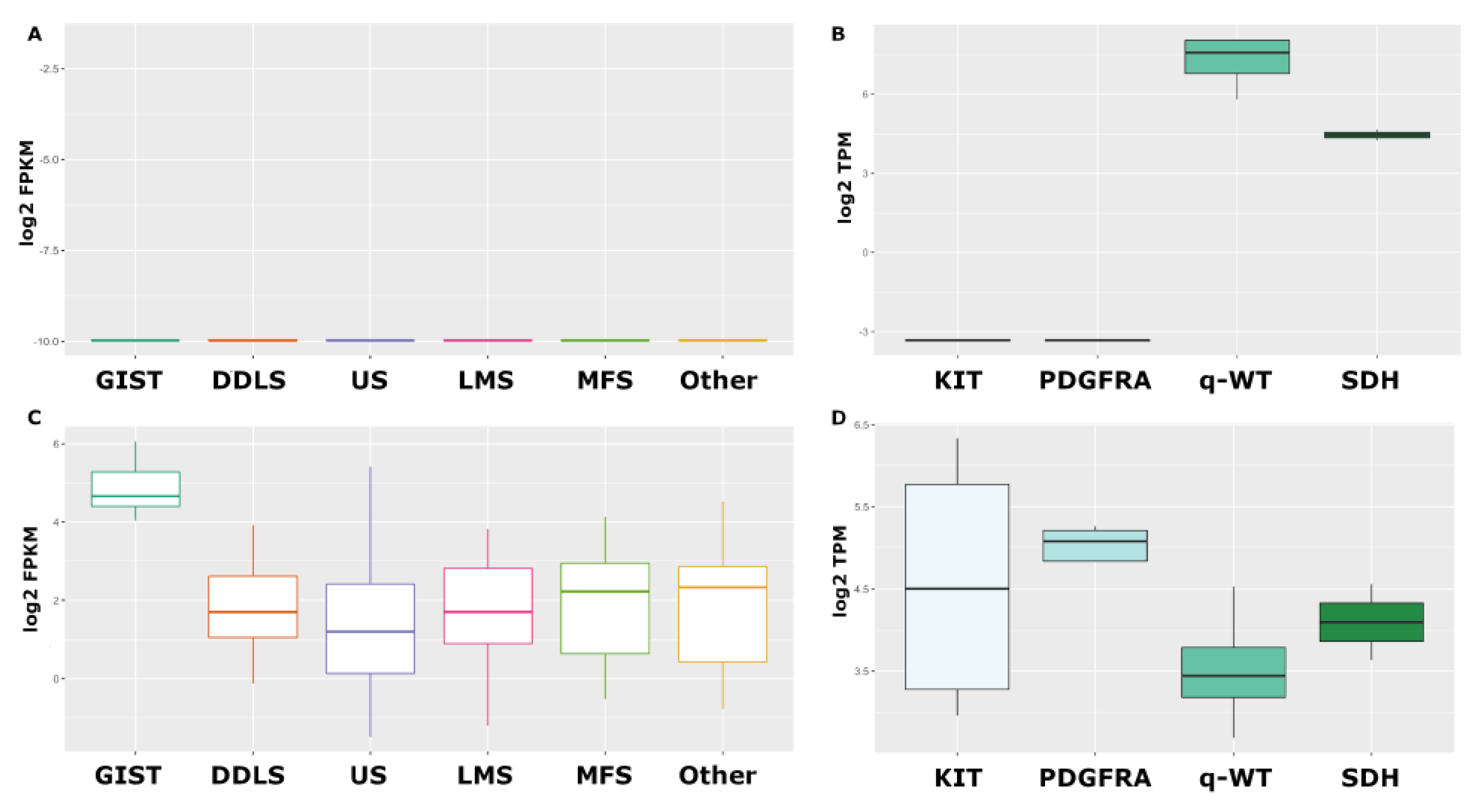{kind=link}
| n Cases | FGF/FGFR Alteration | Methodology | GIST Classification | |
|---|---|---|---|---|
| Shi et al., 2016 [21] | 1 | FGFR1 K656E | Target gene panel | quadruple WT GIST |
| 2 | FGFR1-TACC1 | Target gene panel | quadruple WT GIST | |
| 1 | FGFR1-HOOK3 | Target gene panel | quadruple WT GIST | |
| Pantaleo et al., 2017 [22] | 1 | FGFR1 N546K | Whole exome seq | quadruple WT GIST |
| Urbini et al., 2019 [25] | 6 | FGF4 duplication and overexpression | SNP-array, RNA sequencing | quadruple WT GIST |
| Flavahan et al., 2019 [26] | 19 * | FGF insulator methylation and FGF3/FGF4 overexpression | CTCF ChIP and bisulfite seq, HiC and 4C, RNA sequencing | SDH-deficient GIST |
| Drug | Target | References in GIST |
|---|---|---|
| Multi-kinase inhibitors | ||
| Regorafenib | FGFR1, VEGFR1,2.3 TIE2, KIT, RET, RAF-1, BRAF, and BRAF V600E, PDGFRβ | [3,56,57] |
| Dovitinib | FGFR1/3, VEGFR1/2/3, PDGFRβ, KIT, RET | [58,59] |
| Masitinib | FGFR3, PDGFRα/β, Lck, FAK | [60,61,62] |
| Ponatinib | FGFR1/2/3/4, Abl, PDGFRα/β, RET, KIT, CSF1R, FLT3, VEGFR1/2/3 | [63] |
| Pazopanib | FGFR1, VEGFR1/2/3, PDGFRβ, KIT | [64,65] |
| Lenvatinib | FGFR1/2/3, VEGFR1/2/3, RET, KIT | [66] |
| Nindetanib | FGFR1, VEGFR2/3, PDGFRα | - |
| Lucitanib | FGFR1, VEGFR1/2, PDGFRα/β,Src | - |
| FGFR inhibitors | ||
| Erdafitinib | FGFR1/2/3/4 | - |
| Futibatinib | FGFR1/2/3/4 | - |
| LY2874455 | FGFR1/2/3/4 | - |
| Rogaratinib | FGFR1/2/3/4 | - |
| PRN1371 | FGFR1/2/3/4 | - |
| Infigratinib | FGFR1/2/3 | [67] |
| Pemigatinib | FGFR1/2/3 | - |
| Debio 1347 | FGFR1/2/3 | [68] |
| AZD4547 | FGFR1/2/3 | - |
| Roblitinib | FGFR4 | - |
| H3B-6527 | FGFR4 | - |
| Fisogatinib | FGFR4 | - |
| Ligand trap and antibodies | ||
| FP-1039 | FGF2 | - |
| FPA114 | FGFR2-IIb | - |
| MFGR1877S | FGFR3 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Astolfi, A.; Pantaleo, M.A.; Indio, V.; Urbini, M.; Nannini, M. The Emerging Role of the FGF/FGFR Pathway in Gastrointestinal Stromal Tumor. Int. J. Mol. Sci. 2020, 21, 3313. https://doi.org/10.3390/ijms21093313
Astolfi A, Pantaleo MA, Indio V, Urbini M, Nannini M. The Emerging Role of the FGF/FGFR Pathway in Gastrointestinal Stromal Tumor. International Journal of Molecular Sciences. 2020; 21(9):3313. https://doi.org/10.3390/ijms21093313
Chicago/Turabian StyleAstolfi, Annalisa, Maria Abbondanza Pantaleo, Valentina Indio, Milena Urbini, and Margherita Nannini. 2020. "The Emerging Role of the FGF/FGFR Pathway in Gastrointestinal Stromal Tumor" International Journal of Molecular Sciences 21, no. 9: 3313. https://doi.org/10.3390/ijms21093313
APA StyleAstolfi, A., Pantaleo, M. A., Indio, V., Urbini, M., & Nannini, M. (2020). The Emerging Role of the FGF/FGFR Pathway in Gastrointestinal Stromal Tumor. International Journal of Molecular Sciences, 21(9), 3313. https://doi.org/10.3390/ijms21093313