
PARP Power: A Structural Perspective on PARP1, PARP2, and PARP3 in DNA Damage Repair and Nucleosome Remodelling

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. DNA Damage Recognition by PARP Enzymes
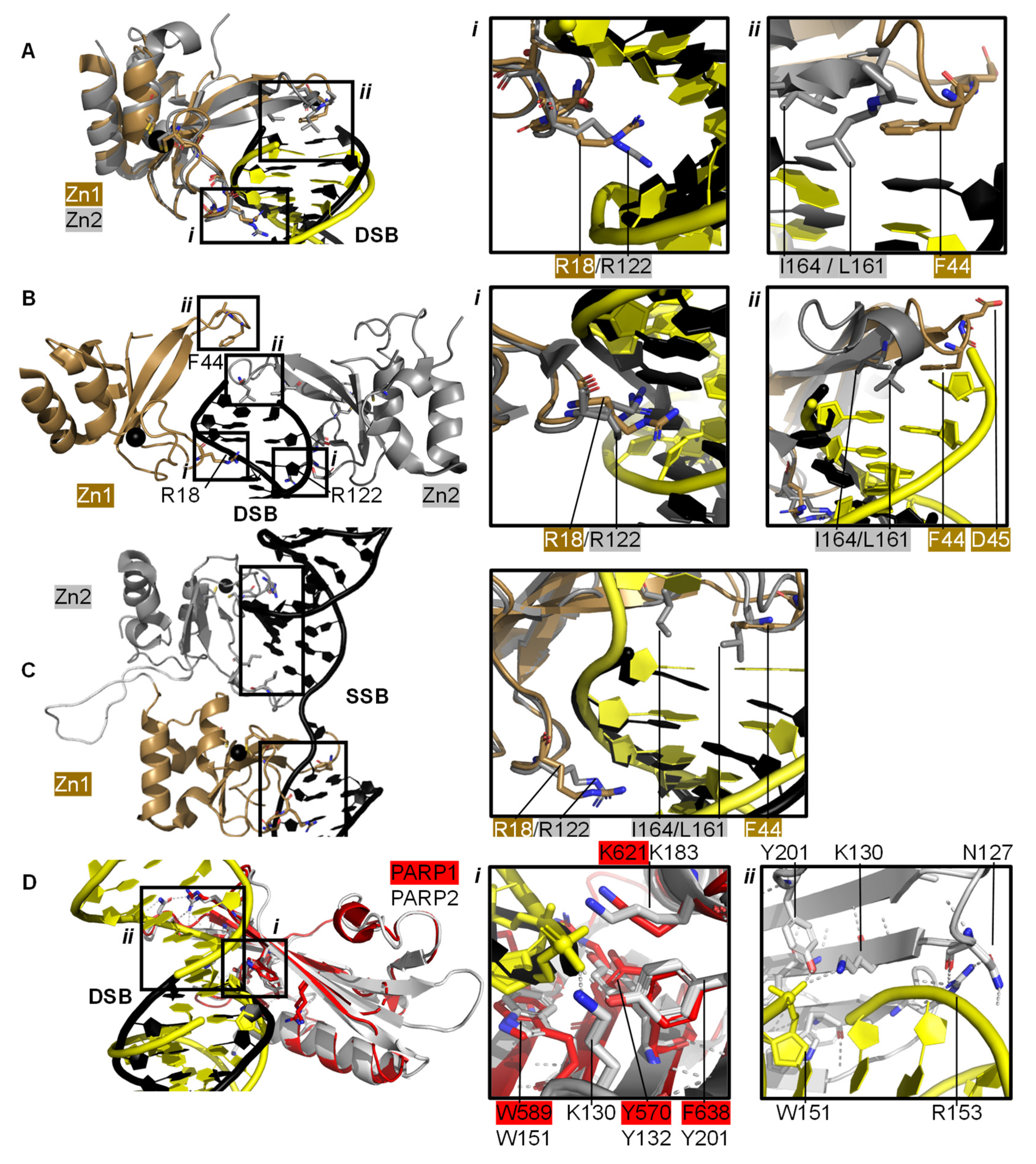
2.1. PARP1 Zn Fingers Bind a DNA Break
2.2. Recognition and Binding of Other DNA Breaks via PARP Domains
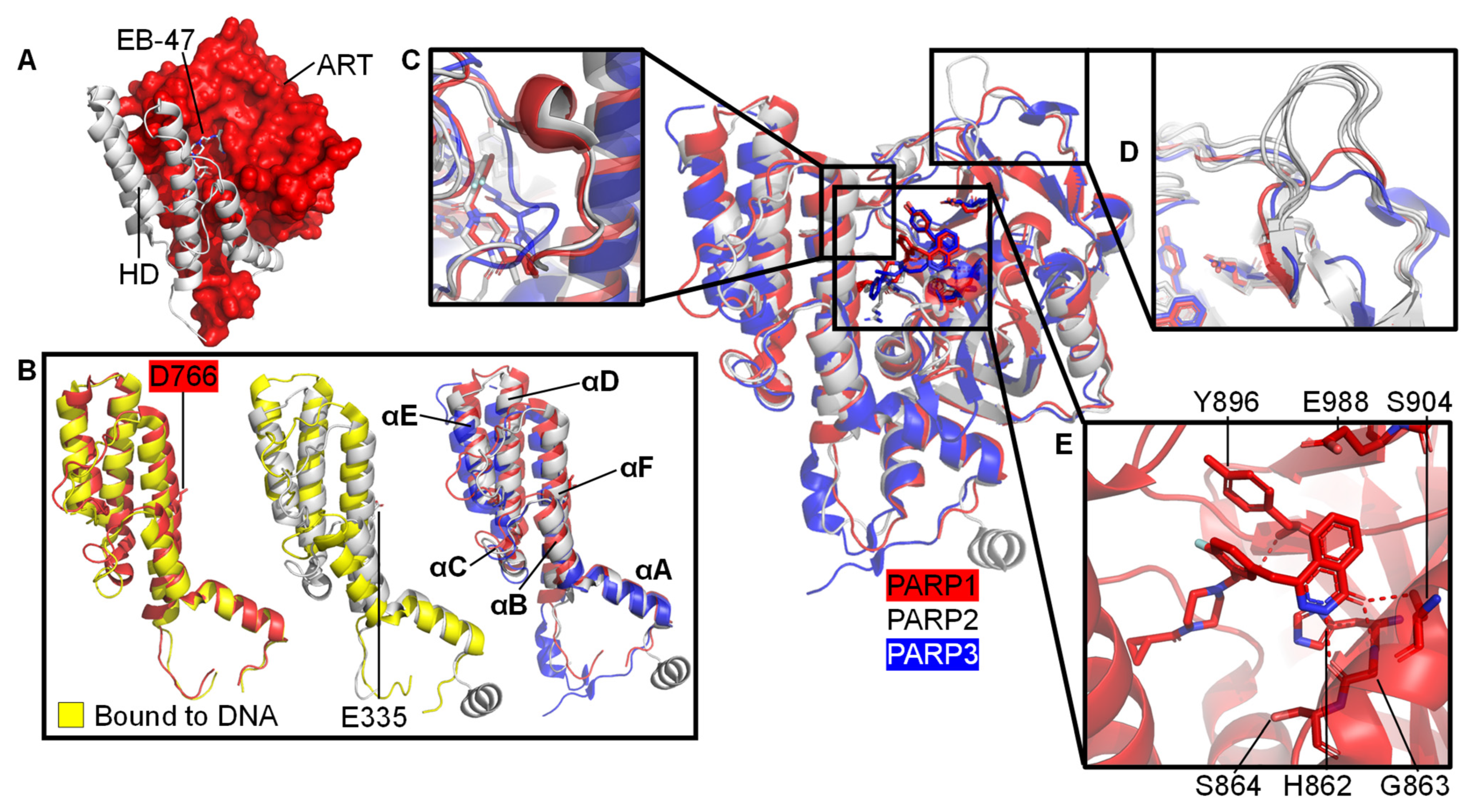
2.3. The PARP Paradox: Finding a Needle in a Haystack
3. PARP Activation and ADP-ribosylation
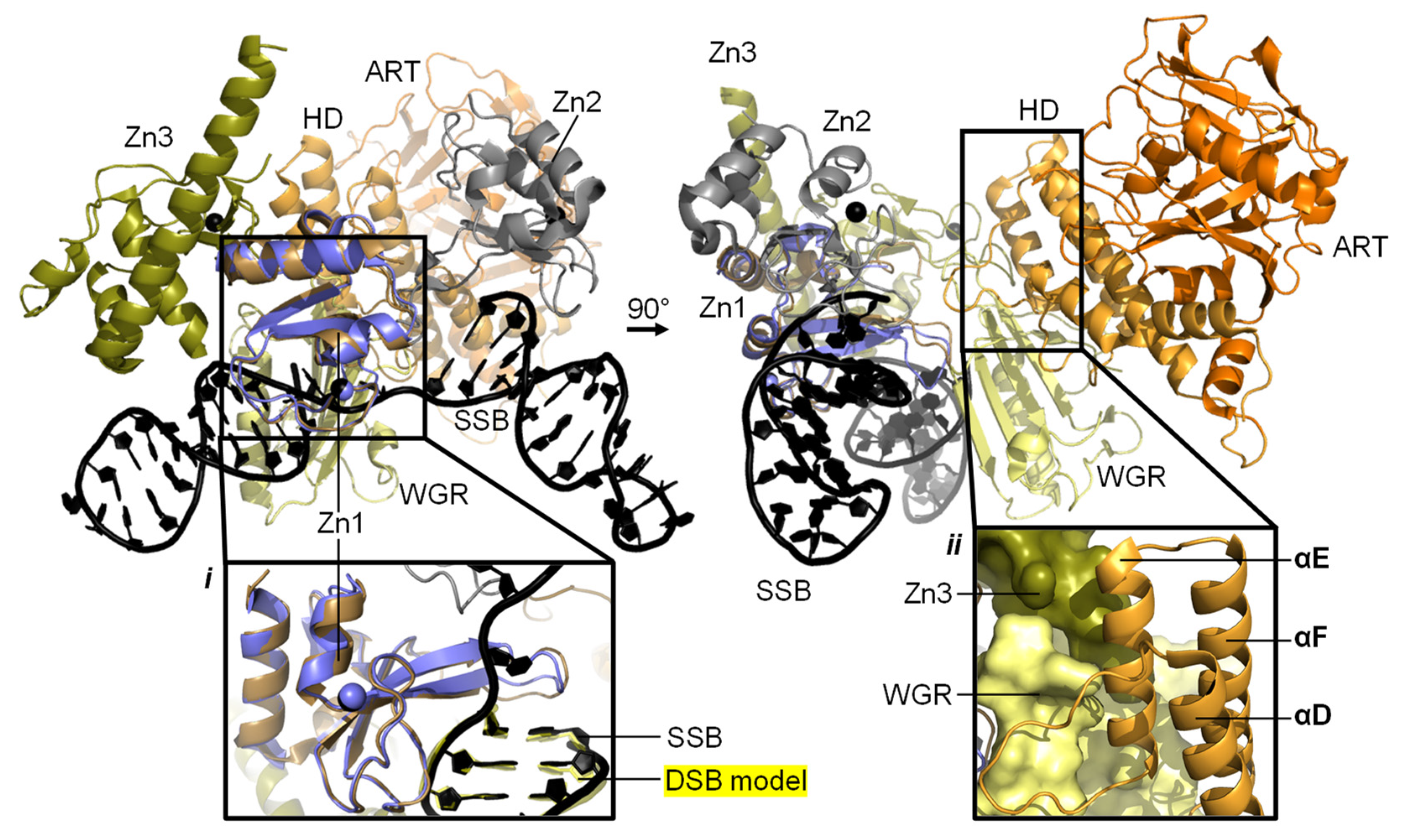
3.1. Understanding Activation of a Full-Length PARP1 on a SSB
3.2. Targets of ADP-Ribosylation
3.3. Variation in the Structure and Function of PAR Chains
4. PARP, HPF1, and Nucleosome Remodelling
4.1. The Role of HPF1
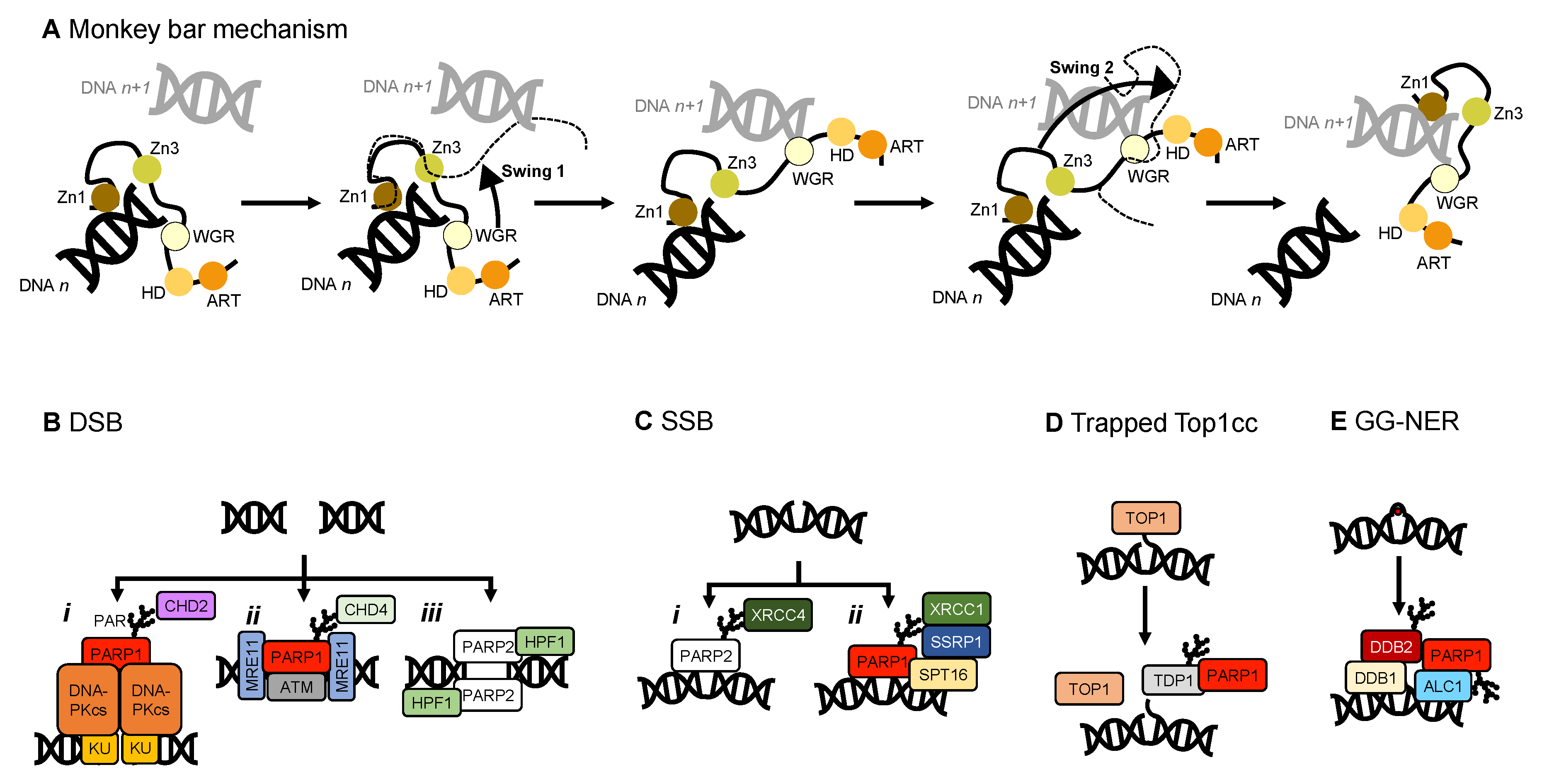
4.2. Histone Remodelling
4.3. Other Interaction Partners of PARP1
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| αA-F | α-helices A to F from HD |
| ADP | ADP-ribose |
| ADP | Adenosine diphosphate |
| ADPr | ADP-ribosylation |
| AFM | Atomic force microscopy |
| ALC1 | Amplified in liver cancer 1 |
| AP site | Apurinic/apyrimidinic site |
| ARH | ADP-ribose hydrolase |
| ART | ADP-ribosyl transferase |
| ATM | Ataxia telangiectasia mutated |
| BER | Base excision repair |
| BRCA1/2 | Breast cancer |
| BRCT | BRCA1 C-terminal domain |
| CAT | Catalytic domain |
| CHD2 | Chromodomain-helicase-DNA-binding protein 2 |
| CHD4 | Chromodomain-helicase-DNA-binding protein 4 |
| Cryo-EM | Cryo-electron microscopy |
| DDB1/2 | DNA damage binding proteins 1 and 2 |
| DDR | DNA damage response |
| D-loop | Donor loop of the active site |
| DNA | Deoxyribonucleic acid |
| DNAPKcs | DNA protein kinase catalytic subunit |
| DSB | Double-strand break |
| ETD | Electron transfer dissociation |
| FACT | Facilitate chromatin transcription |
| gap | Missing nucleotide, form of DNA break |
| GG-NER | Global genome nucleotide excision repair |
| H1 | Linker histone |
| H2A, H2B, H3, H4 | Core histones |
| HD | Helical domain |
| HDX-MS | Hydrogen/deuterium exchange-mass spectrometry |
| HeLa | Cervical cancer cell line from Henrietta Lacks |
| HPF1 | Histone PARylation factor 1 |
| HR | Homologous recombination |
| HYE | Conserved histidone, tyrosine, glutamic acid motif |
| IC50 | Half maximal inhibitory concentration |
| Kd | Dissociation constant |
| MAR | Mono (ADP-ribose) |
| MARylation | Mono-ADP-ribosylation |
| MRE11 | Meotic recombination 11 |
| MS | Mass spectrometry |
| NAD+ | Nicotinamide adenine dinucleotide |
| NHEJ | Non-homologous end joining |
| NMNAT-1 | Nicotinamide/nicotinic acid mononucleotide adenylyltransferase 1 |
| NMR | Nuclear magnetic resonance |
| NOESY | Nuclear overhauser effect spectroscopy |
| NuRD | Nucleosome remodeling and deacetylase |
| p53 | Tumour suppressor named after its apparent MW on SDS-PAGE gel, 53 kDa |
| PAR | Poly (ADP-ribose) |
| PARG | Poly(ADP-ribose) glycohydrolase |
| PARP1 | Poly (ADP-ribose) polymerase 1 |
| PARP2 | Poly (ADP-ribose) polymerase 2 |
| PARP3 | Poly (ADP-ribose) polymerase 3 |
| PARPi | PARP inhibitor |
| PARylation | Poly-ADP-ribosylation |
| PDB | Protein data bank |
| PTM | Post-translational modification |
| RMSD | Root mean square deviation |
| RNA | Ribonucleic acid |
| SAP | SAF/acinus/PIAS |
| SNF2 | Sucrose non-fermenting protein 2 |
| SSB | Single-strand break |
| TDP1 | Tyrosyl-DNA phosphodiesterase 1 |
| Top1cc | TOP1 cleavage complexes |
| V(D)J | Variable, diversity and joining |
| WGR | Conserved tryptophan, glycine, arginine-motif domain |
| XRCC1 | X-ray repair cross complementing group 1 |
| Zn1 | Zinc finger 1 |
| Zn2 | Zinc finger 2 |
| Zn3 | Zinc finger 3 |
References
- Gibson, B.A.; Kraus, W.L. New Insights into the Molecular and Cellular Functions of Poly(ADP-Ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Kraus, W.L. PARPs and ADP-Ribosylation: 50 Years … and Counting. Mol. Cell 2015, 58, 902–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.S.; Chang, P. Insights into the Biogenesis, Function, and Regulation of ADP-Ribosylation. Nat. Chem. Biol. 2018, 14, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Barkauskaite, E.; Jankevicius, G.; Ahel, I. Structures and Mechanisms of Enzymes Employed in the Synthesis and Degradation of PARP-Dependent Protein ADP-Ribosylation. Mol. Cell 2015, 58, 935–946. [Google Scholar] [CrossRef] [Green Version]
- Gupte, R.; Liu, Z.; Kraus, W.L. PARPs and ADP-Ribosylation: Recent Advances Linking Molecular Functions to Biological Outcomes. Genes Dev. 2017, 31, 101–126. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, A.R.; Nussenzweig, A. The Multifaceted Roles of PARP1 in DNA Repair and Chromatin Remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Hoch, N.C.; Polo, L.M. ADP-Ribosylation: From Molecular Mechanisms to Human Disease. Genet. Mol. Biol. 2020, 43, e20190075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, R.C.; Gill, D.M. ADP-Ribosylation in Mammalian Cell Ghosts. Dependence of Poly(ADP-Ribose) Synthesis on Strand Breakage in DNA. J. Biol. Chem. 1980, 255, 10493–10501. [Google Scholar] [CrossRef]
- Durkacz, B.W.; Omidiji, O.; Gray, D.A.; Shall, S. (ADP-Ribose)n Participates in DNA Excision Repair. Nature 1980, 283, 593–596. [Google Scholar] [CrossRef]
- Dockery, L.; Gunderson, C.; Moore, K. Rucaparib: The Past, Present, and Future of a Newly Approved PARP Inhibitor for Ovarian Cancer. OncoTargets Ther. 2017, 10, 3029–3037. [Google Scholar] [CrossRef] [Green Version]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-Ribose Polymerase Inhibition: Frequent Durable Responses in BRCA Carrier Ovarian Cancer Correlating With Platinum-Free Interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.-T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Tuli, R.; Shiao, S.L.; Nissen, N.; Tighiouart, M.; Kim, S.; Osipov, A.; Bryant, M.; Ristow, L.; Placencio-Hickok, V.; Hoffman, D.; et al. A Phase 1 Study of Veliparib, a PARP-1/2 Inhibitor, with Gemcitabine and Radiotherapy in Locally Advanced Pancreatic Cancer. EBioMedicine 2019, 40, 375–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Hassa, P.O. The Diverse Biological Roles of Mammalian PARPS, a Small but Powerful Family of Poly-ADP-Ribose Polymerases. Front. Biosci. 2008, 13, 3046. [Google Scholar] [CrossRef] [Green Version]
- Cho, C.C.; Chien, C.Y.; Chiu, Y.C.; Lin, M.H.; Hsu, C.H. Structural and Biochemical Evidence Supporting Poly ADP-Ribosylation in the Bacterium Deinococcus Radiodurans. Nat. Commun. 2019, 10, 14. [Google Scholar] [CrossRef]
- Mennella, M.R.F. The Dichotomy of the Poly(ADP-Ribose) Polymerase-Like Thermozyme from Sulfolobus Solfataricus. Challenges 2018, 9, 5. [Google Scholar] [CrossRef] [Green Version]
- Nishizuka, Y.; Ueda, K.; Nakazawa, K.; Hayaishi, O. Studies on the Polymer of Adenosine Diphosphate Ribose. J. Biol. Chem. 1967, 242, 3164–3171. [Google Scholar] [CrossRef]
- Fujimura, S.; Hasegawa, S.; Shimizu, Y.; Sugimura, T. Polymerization of the Adenosine 5′-Diphosphate-Ribose Moiety of Nicotinamide-Adenine Dinucleotide by Nuclear Enzyme. I. Enzymatic Reactions. Biochim. Biophys Acta 1967, 145, 247–259. [Google Scholar] [CrossRef]
- Ueda, K.; Reeder, R.H.; Honjo, T.; Nishizuka, Y.; Hayaishi, O. Poly Adenosine Diphosphate Ribose Synthesis Associated with Chromatin. Biochem. Biophys. Res. Commun. 1968, 31, 379–385. [Google Scholar] [CrossRef]
- Chambon, P.; Weill, J.D.; Doly, J.; Strosser, M.T.; Mandel, P. On the Formation of a Novel Adenylic Compound by Enzymatic Extracts of Liver Nuclei. Biochem. Biophys. Res. Commun. 1966, 25, 638–643. [Google Scholar] [CrossRef]
- Yamada, M.; Miwa, M.; Sugimura, T. Studies on Poly (Adenosine Diphosphate-Ribose) X. Properties of a Partially Purified Poly (Adenosine Diphosphate-Ribose) Polymerase’. Arch. Biochem. Biophys. 1971, 146, 579–586. [Google Scholar] [CrossRef]
- Juarez-Salinas, H.; Sims, J.L.; Jacobson, M.K. Poly(ADP-Ribose) Levels in Carcinogen-Treated Cells. Nature 1979, 282, 740–741. [Google Scholar] [CrossRef]
- Otake, H.; Miwa, M.; Fujimura, S.; Sugimura, T. Binding of ADP-ribose polymer with histone. J. Biochem. 1969, 65. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Auer, B.; Stingl, L.; Berghammer, H.; Haidacher, D.; Schweiger, M.; Wagner, E.F. Mice Lacking ADPRT and Poly(ADP-Ribosyl)Ation Develop Normally but Are Susceptible to Skin Disease. Genes Dev. 1995, 9, 509–520. [Google Scholar] [CrossRef] [Green Version]
- De Murcia, J.M.; Niedergang, C.; Trucco, C.; Ricoul, M.; Dutrillaux, B.; Mark, M.; Oliver, F.J.; Masson, M.; Dierich, A.; LeMeur, M.; et al. Requirement of Poly(ADP-Ribose) Polymerase in Recovery from DNA Damage in Mice and in Cells. Proc. Natl. Acad. Sci. USA 1997, 94, 7303–7307. [Google Scholar] [CrossRef] [Green Version]
- Masutani, M.; Suzuki, H.; Kamada, N.; Watanabe, M.; Ueda, O.; Nozaki, T.; Jishage, K.-I.; Watanabe, T.; Sugimoto, T.; Nakagama, H.; et al. Poly(ADP-Ribose) Polymerase Gene Disruption Conferred Mice Resistant to Streptozotocin-Induced Diabetes. Proc. Natl. Acad. Sci. USA 1999, 96, 2301–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-Ribosyl)Ation Reactions in the Regulation of Nuclear Functions. Biochem. J. 1999, 342, 249–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, V.; Dantzer, F.; Ame, J.-C.; de Murcia, G. Poly(ADP-Ribose): Novel Functions for an Old Molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Hakmé, A.; Wong, H.; Dantzer, F.; Schreiber, V. The Expanding Field of Poly(ADP-ribosyl)Ation Reactions. EMBO Rep. 2008, 9, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Langelier, M.-F.; Servent, K.M.; Rogers, E.E.; Pascal, J.M. A Third Zinc-Binding Domain of Human Poly(ADP-Ribose) Polymerase-1 Coordinates DNA-Dependent Enzyme Activation. J. Biol. Chem. 2008, 283, 4105–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langelier, M.-F.; Ruhl, D.D.; Planck, J.L.; Kraus, W.L.; Pascal, J.M. The Zn3 Domain of Human Poly(ADP-Ribose) Polymerase-1 (PARP-1) Functions in Both DNA-Dependent Poly(ADP-Ribose) Synthesis Activity and Chromatin Compaction*. J. Biol. Chem. 2010, 285, 18877–18887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langelier, M.-F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural Basis for DNA Damage-Dependent Poly(ADP-Ribosyl)Ation by Human PARP-1. Science 2012, 336, 728–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amé, J.-C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Höger, T.; Murcia, J.M.; de Murcia, G. PARP-2, a Novel Mammalian DNA Damage-Dependent Poly(ADP-Ribose) Polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Léger, K.; Bär, D.; Savić, N.; Santoro, R.; Hottiger, M.O. ARTD2 Activity Is Stimulated by RNA. Nucleic Acids Res. 2014, 42, 5072–5082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, P.N.; Goel, A.; Johnson, M.A. Poly(ADP-Ribose) Polymerases in Host-Pathogen Interactions, Inflammation, and Immunity. Microbiol. Mol. Biol. Rev. 2018, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravind, L.; Koonin, E.V. SAP—A Putative DNA-Binding Motif Involved in Chromosomal Organization. Trends Biochem. Sci. 2000, 25, 112–114. [Google Scholar] [CrossRef]
- Song, J.; Keppler, B.D.; Wise, R.R.; Bent, A.F. PARP2 Is the Predominant Poly(ADP-Ribose) Polymerase in Arabidopsis DNA Damage and Immune Responses. PLoS Genet. 2015, 11, e1005200. [Google Scholar] [CrossRef]
- Rissel, D.; Peiter, E. Poly(ADP-Ribose) Polymerases in Plants and Their Human Counterparts: Parallels and Peculiarities. Int. J. Mol. Sci. 2019, 20, 1638. [Google Scholar] [CrossRef] [Green Version]
- Okubo, S.; Hara, F.; Tsuchida, Y.; Shimotakahara, S.; Suzuki, S.; Hatanaka, H.; Yokoyama, S.; Tanaka, H.; Yasuda, H.; Shindo, H. NMR Structure of the N-Terminal Domain of SUMO Ligase PIAS1 and Its Interaction with Tumor Suppressor P53 and A/T-Rich DNA Oligomers. J. Biol. Chem. 2004, 279, 31455–31461. [Google Scholar] [CrossRef] [Green Version]
- Riccio, A.A.; Cingolani, G.; Pascal, J.M. PARP-2 Domain Requirements for DNA Damage-Dependent Activation and Localization to Sites of DNA Damage. Nucleic Acids Res. 2016, 44, 1691–1702. [Google Scholar] [CrossRef] [Green Version]
- The PyMOL Molecular Graphics System; Schrödinger, LLC: New York, NY, USA, 2021.
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Ruf, A.; de Murcia, J.M.; de Murcia, G.; Schulz, G.E. Structure of the Catalytic Fragment of Poly(ADP-Ribose) Polymerase from Chicken. Proc. Natl. Acad. Sci. USA 1996, 93, 7481–7485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruf, A.; de Murcia, G.; Schulz, G.E. Inhibitor and NAD + Binding to Poly(ADP-Ribose) Polymerase As Derived from Crystal Structures and Homology Modeling †, ‡. Biochemistry 1998, 37, 3893–3900. [Google Scholar] [CrossRef]
- Ruf, A.; Rolli, V.; de Murcia, G.; Schulz, G.E. The Mechanism of the Elongation and Branching Reaction of Poly(ADP-Ribose) Polymerase as Derived from Crystal Structures and Mutagenesis. J. Mol. Biol. 1998, 278, 57–65. [Google Scholar] [CrossRef]
- Oliver, A.W.; Amé, J.-C.; Roe, S.M.; Good, V.; de Murcia, G.; Pearl, L.H. Crystal Structure of the Catalytic Fragment of Murine Poly(ADP-Ribose) Polymerase-2. Nucleic Acids Res. 2004, 32, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, T.; Nakanishi, I.; Warizaya, M.; Iwashita, A.; Kido, Y.; Hattori, K.; Fujii, T. Inhibitor-Induced Structural Change of the Active Site of Human Poly(ADP-Ribose) Polymerase. FEBS Lett. 2004, 556, 43–46. [Google Scholar] [CrossRef] [Green Version]
- Karlberg, T.; Hammarström, M.; Schütz, P.; Svensson, L.; Schüler, H. Crystal Structure of the Catalytic Domain of Human PARP2 in Complex with PARP Inhibitor ABT-888. Biochemistry 2010, 49, 1056–1058. [Google Scholar] [CrossRef]
- Lehtiö, L.; Jemth, A.-S.; Collins, R.; Loseva, O.; Johansson, A.; Markova, N.; Hammarström, M.; Flores, A.; Holmberg-Schiavone, L.; Weigelt, J.; et al. Structural Basis for Inhibitor Specificity in Human Poly(ADP-Ribose) Polymerase-3 †. J. Med. Chem. 2009, 52, 3108–3111. [Google Scholar] [CrossRef]
- Bilokapic, S.; Suskiewicz, M.J.; Ahel, I.; Halic, M. Bridging of DNA Breaks Activates PARP2–HPF1 to Modify Chromatin. Nature 2020, 585, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Kraus, W.L.; Hottiger, M.O. PARP-1 and Gene Regulation: Progress and Puzzles. Mol. Asp. Med. 2013, 34, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.W.; Kim, D.-S.; Kraus, W.L. New Facets in the Regulation of Gene Expression by ADP-Ribosylation and Poly(ADP-Ribose) Polymerases. Chem. Rev. 2015, 115, 2453–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascal, J.M.; Ellenberger, T. The Rise and Fall of Poly(ADP-Ribose): An Enzymatic Perspective. DNA Repair 2015, 32, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langelier, M.-F.; Zandarashvili, L.; Aguiar, P.M.; Black, B.E.; Pascal, J.M. NAD+ Analog Reveals PARP-1 Substrate-Blocking Mechanism and Allosteric Communication from Catalytic Center to DNA-Binding Domains. Nat. Commun. 2018, 9, 844. [Google Scholar] [CrossRef]
- Pascal, J.M. The Comings and Goings of PARP-1 in Response to DNA Damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, W.; Wang, Y. PARP-1 and Its Associated Nucleases in DNA Damage Response. DNA Repair 2019, 81, 102651. [Google Scholar] [CrossRef]
- Eisemann, T.; Pascal, J.M. Poly(ADP-Ribose) Polymerase Enzymes and the Maintenance of Genome Integrity. Cell. Mol. Life Sci. 2020, 77, 19–33. [Google Scholar] [CrossRef]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Combined Strategies with Poly (ADP-Ribose) Polymerase (PARP) Inhibitors for the Treatment of Ovarian Cancer: A Literature Review. Diagnostics 2019, 9, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, S.A.; Tinker, A.V. PARP Inhibitors and the Evolving Landscape of Ovarian Cancer Management: A Review. BioDrugs 2019, 33, 255–273. [Google Scholar] [CrossRef]
- Murai, J.; Pommier, Y. PARP Trapping Beyond Homologous Recombination and Platinum Sensitivity in Cancers. Annu. Rev. Cancer Biol. 2019, 3, 131–150. [Google Scholar] [CrossRef]
- Xie, H.; Wang, W.; Xia, B.; Jin, W.; Lou, G. Therapeutic Applications of PARP Inhibitors in Ovarian Cancer. Biomed. Pharmacother. 2020, 127, 110204. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a Trap to Kill Cancer Cells: PARP Inhibitors and Their Mechanisms of Action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.I.; Majuelos-Melguizo, J.; Martí Martín-Consuegra, J.M.; Ruiz de Almodóvar, M.; López-Rivas, A.; Javier Oliver, F. Deciphering the Insights of Poly(ADP-Ribosylation) in Tumor Progression: Insights of poly(ADP-ribosylation) in tumor progression. Med. Res. Rev. 2015, 35, 678–697. [Google Scholar] [CrossRef] [PubMed]
- Swindall, A.; Stanley, J.; Yang, E. PARP-1: Friend or Foe of DNA Damage and Repair in Tumorigenesis? Cancers 2013, 5, 943–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donàs, F.; Chiodi, I.; Belgiovine, C.; Raineri, T.; Ricotti, R.; Mondello, C.; Scovassi, A.I. Poly(ADP-Ribosylation) and Neoplastic Transformation: Effect of PARP Inhibitors. Curr. Pharm. Biotechnol. 2013, 14, 524–536. [Google Scholar] [CrossRef]
- Bai, P.; Cantó, C. The Role of PARP-1 and PARP-2 Enzymes in Metabolic Regulation and Disease. Cell Metab. 2012, 16, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Kunze, F.A.; Hottiger, M.O. Regulating Immunity via ADP-Ribosylation: Therapeutic Implications and Beyond. Trends Immunol. 2019, 40, 159–173. [Google Scholar] [CrossRef]
- Vida, A.; Abdul-Rahman, O.; Mikó, E.; Brunyánszki, A.; Bai, P. Poly(ADP-Ribose) Polymerases in Aging—Friend or Foe? Curr. Protein Pept. Sci. 2016, 17, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Vida, A.; Márton, J.; Mikó, E.; Bai, P. Metabolic Roles of Poly(ADP-Ribose) Polymerases. Semin. Cell Dev. Biol. 2017, 63, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Caldecott, K.W. Single-Strand Break Repair and Genetic Disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- White, R.R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Mol. Cell 2016, 63, 729–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, E.; Ideker, T. Transcriptional Responses to DNA Damage. DNA Repair 2019, 79, 40–49. [Google Scholar] [CrossRef]
- Langelier, M.-F.; Riccio, A.A.; Pascal, J.M. PARP-2 and PARP-3 Are Selectively Activated by 5′ Phosphorylated DNA Breaks Through an Allosteric Regulatory Mechanism Shared with PARP-1. Nucleic Acids Res. 2014, 42, 7762–7775. [Google Scholar] [CrossRef] [Green Version]
- Hiom, K. Coping with DNA Double Strand Breaks. DNA Repair 2010, 9, 1256–1263. [Google Scholar] [CrossRef]
- Caldecott, K.W. XRCC1 Protein; Form and Function. DNA Repair 2019, 81, 102664. [Google Scholar] [CrossRef]
- Das, B.B.; Huang, S.N.; Murai, J.; Rehman, I.; Amé, J.-C.; Sengupta, S.; Das, S.K.; Majumdar, P.; Zhang, H.; Biard, D.; et al. PARP1–TDP1 Coupling for the Repair of Topoisomerase I–Induced DNA Damage. Nucleic Acids Res. 2014, 42, 4435–4449. [Google Scholar] [CrossRef]
- Spagnolo, L.; Barbeau, J.; Curtin, N.J.; Morris, E.P.; Pearl, L.H. Visualization of a DNA-PK/PARP1 Complex. Nucleic Acids Res. 2012, 40, 4168–4177. [Google Scholar] [CrossRef] [Green Version]
- Roth, D.B. V(D)J Recombination: Mechanism, Errors, and Fidelity. In Mobile DNA III; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Haince, J.-F.; Kozlov, S.; Dawson, V.L.; Dawson, T.M.; Hendzel, M.J.; Lavin, M.F.; Poirier, G.G. Ataxia Telangiectasia Mutated (ATM) Signaling Network Is Modulated by a Novel Poly(ADP-Ribose)-Dependent Pathway in the Early Response to DNA-Damaging Agents. J. Biol. Chem. 2007, 282, 16441–16453. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Quesada, R.; Muñoz-Gámez, J.; Martín-Oliva, D.; Peralta, A.; Valenzuela, M.T.; Matínez-Romero, R.; Quiles-Pérez, R.; Murcia, J.; de Murcia, G.; de Almodóvar, M.; et al. Interaction between ATM and PARP-1 in Response to DNA Damage and Sensitization of ATM Deficient Cells Through PARP Inhibition. BMC Mol. Biol. 2007, 8, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haince, J.-F.; McDonald, D.; Rodrigue, A.; Déry, U.; Masson, J.-Y.; Hendzel, M.J.; Poirier, G.G. PARP1-Dependent Kinetics of Recruitment of MRE11 and NBS1 Proteins to Multiple DNA Damage Sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabarz, A.; Barascu, A.; Guirouilh-Barbat, J.; Lopez, B.S. Initiation of DNA Double Strand Break Repair: Signaling and Single-Stranded Resection Dictate the Choice Between Homologous Recombination, Non-Homologous End-Joining and Alternative End-Joining. Am. J. Cancer Res. 2012, 2, 249–268. [Google Scholar] [PubMed]
- Langelier, M.-F.; Planck, J.L.; Roy, S.; Pascal, J.M. Crystal Structures of Poly(ADP-Ribose) Polymerase-1 (PARP-1) Zinc Fingers Bound to DNA: Structural and Functional Insights into DNA-Dependent PARP-1 Activity. J. Biol. Chem. 2011, 286, 10690–10701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.A.E.; Timinszky, G.; Arribas-Bosacoma, R.; Kozlowski, M.; Hassa, P.O.; Hassler, M.; Ladurner, A.G.; Pearl, L.H.; Oliver, A.W. The Zinc-Finger Domains of PARP1 Cooperate to Recognize DNA Strand Breaks. Nat. Struct. Mol. Biol. 2012, 19, 685–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eustermann, S.; Videler, H.; Yang, J.-C.; Cole, P.T.; Gruszka, D.; Veprintsev, D.; Neuhaus, D. The DNA-Binding Domain of Human PARP-1 Interacts with DNA Single-Strand Breaks as a Monomer through Its Second Zinc Finger. J. Mol. Biol. 2011, 407, 149–170. [Google Scholar] [CrossRef] [Green Version]
- Eustermann, S.; Wu, W.-F.; Langelier, M.-F.; Yang, J.-C.; Easton, L.E.; Riccio, A.A.; Pascal, J.M.; Neuhaus, D. Structural Basis of Detection and Signaling of DNA Single-Strand Breaks by Human PARP-1. Mol. Cell 2015, 60, 742–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obaji, E.; Haikarainen, T.; Lehtiö, L. Structural Basis for DNA Break Recognition by ARTD2/PARP2. Nucleic Acids Res. 2018, 46, 12154–12165. [Google Scholar] [CrossRef] [Green Version]
- Trucco, C.; Flatter, E.; Fribourg, S.; de Murcia, G.; Ménissier-de Murcia, J. Mutations in the Amino-Terminal Domain of the Human Poly(ADP-Ribose) Polymerase That Affect Its Catalytic Activity but Not Its DNA Binding Capacity. FEBS Lett. 1996, 399, 313–316. [Google Scholar] [CrossRef] [Green Version]
- Rulten, S.L.; Fisher, A.E.O.; Robert, I.; Zuma, M.C.; Rouleau, M.; Ju, L.; Poirier, G.; Reina-San-Martin, B.; Caldecott, K.W. PARP-3 and APLF Function Together to Accelerate Nonhomologous End-Joining. Mol. Cell 2011, 41, 33–45. [Google Scholar] [CrossRef]
- Zarkovic, G.; Belousova, E.A.; Talhaoui, I.; Saint-Pierre, C.; Kutuzov, M.M.; Matkarimov, B.T.; Biard, D.; Gasparutto, D.; Lavrik, O.I.; Ishchenko, A.A. Characterization of DNA ADP-Ribosyltransferase Activities of PARP2 and PARP3: New Insights into DNA ADP-Ribosylation. Nucleic Acids Res. 2018, 46, 2417–2431. [Google Scholar] [CrossRef]
- Liu, L.; Kong, M.; Gassman, N.R.; Freudenthal, B.D.; Prasad, R.; Zhen, S.; Watkins, S.C.; Wilson, S.H.; Van Houten, B. PARP1 Changes from Three-Dimensional DNA Damage Searching to One-Dimensional Diffusion after Auto-PARylation or in the Presence of APE1. Nucleic Acids Res. 2017, 45, 12834–12847. [Google Scholar] [CrossRef] [PubMed]
- Milo, R.; Jorgensen, P.; Moran, U.; Weber, G.; Springer, M. BioNumbers—the Database of Key Numbers in Molecular and Cell Biology. Nucleic Acids Res. 2010, 38, D750–D753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.D.; Gerace, L. The Size-Wise Nucleus: Nuclear Volume Control in Eukaryotes. J. Cell. Biol. 2007, 179, 583–584. [Google Scholar] [CrossRef]
- Sukhanova, M.V.; Abrakhi, S.; Joshi, V.; Pastre, D.; Kutuzov, M.M.; Anarbaev, R.O.; Curmi, P.A.; Hamon, L.; Lavrik, O.I. Single Molecule Detection of PARP1 and PARP2 Interaction with DNA Strand Breaks and Their Poly(ADP-Ribosyl)Ation Using High-Resolution AFM Imaging. Nucleic Acids Res. 2016, 44, e60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukhanova, M.V.; Hamon, L.; Kutuzov, M.M.; Joshi, V.; Abrakhi, S.; Dobra, I.; Curmi, P.A.; Pastre, D.; Lavrik, O.I. A Single-Molecule Atomic Force Microscopy Study of PARP1 and PARP2 Recognition of Base Excision Repair DNA Intermediates. J. Mol. Biol. 2019, 431, 2655–2673. [Google Scholar] [CrossRef]
- Rudolph, J.; Mahadevan, J.; Dyer, P.; Luger, K. Poly(ADP-Ribose) Polymerase 1 Searches DNA via a ‘Monkey Bar’ Mechanism. eLife 2018, 7, e37818. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, J.; Mahadevan, J.; Luger, K. Probing the Conformational Changes Associated with DNA Binding to PARP1. Biochemistry 2020, 59, 2003–2011. [Google Scholar] [CrossRef]
- Matta, E.; Kiribayeva, A.; Khassenov, B.; Matkarimov, B.T.; Ishchenko, A.A. Insight into DNA Substrate Specificity of PARP1-Catalysed DNA Poly(ADP-Ribosyl)Ation. Sci. Rep. 2020, 10, 3699. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Matic, I.; Uchima, L.; Rood, J.; Zaja, R.; Hay, R.T.; Ahel, I.; Chang, P. Family-Wide Analysis of Poly(ADP-Ribose) Polymerase Activity. Nat. Commun. 2014, 5, 4426. [Google Scholar] [CrossRef] [Green Version]
- Fouquerel, E.; Goellner, E.M.; Yu, Z.; Gagné, J.-P.; de Moura, M.B.; Feinstein, T.; Wheeler, D.; Redpath, P.; Li, J.; Romero, G.; et al. ARTD1/PARP1 Negatively Regulates Glycolysis by Inhibiting Hexokinase 1 Independent of NAD+ Depletion. Cell Rep. 2014, 8, 1819–1831. [Google Scholar] [CrossRef] [Green Version]
- Rack, J.G.M.; Palazzo, L.; Ahel, I. (ADP-Ribosyl)Hydrolases: Structure, Function, and Biology. Genes Dev. 2020, 23. [Google Scholar] [CrossRef]
- Li, C.; Debing, Y.; Jankevicius, G.; Neyts, J.; Ahel, I.; Coutard, B.; Canard, B. Viral Macro Domains Reverse Protein ADP-Ribosylation. J. Virol. 2016, 90, 8478–8486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bütepage, M.; Preisinger, C.; von Kriegsheim, A.; Scheufen, A.; Lausberg, E.; Li, J.; Kappes, F.; Feederle, R.; Ernst, S.; Eckei, L.; et al. Nucleolar-Nucleoplasmic Shuttling of TARG1 and Its Control by DNA Damage-Induced Poly-ADP-Ribosylation and by Nucleolar Transcription. Sci. Rep. 2018, 8, 17. [Google Scholar] [CrossRef]
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP)–Function in Genome Maintenance and Relevance of Inhibitors for Anti-Cancer Therapy. Front. Mol. Biosci. 2020, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Zandarashvili, L.; Langelier, M.-F.; Velagapudi, U.K.; Hancock, M.A.; Steffen, J.D.; Billur, R.; Hannan, Z.M.; Wicks, A.J.; Krastev, D.B.; Pettitt, S.J.; et al. Structural Basis for Allosteric PARP-1 Retention on DNA Breaks. Science 2020, 368, eaax6367. [Google Scholar] [CrossRef]
- Ogden, T.E.H.; Yang, J.-C.; Schimpl, M.; Easton, L.E.; Underwood, E.; Rawlins, P.B.; McCauley, M.M.; Langelier, M.-F.; Pascal, J.M.; Embrey, K.J.; et al. Dynamics of the HD Regulatory Subdomain of PARP-1; Substrate Access and Allostery in PARP Activation and Inhibition. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef]
- Wahlberg, E.; Karlberg, T.; Kouznetsova, E.; Markova, N.; Macchiarulo, A.; Thorsell, A.-G.; Pol, E.; Frostell, Å.; Ekblad, T.; Öncü, D.; et al. Family-Wide Chemical Profiling and Structural Analysis of PARP and Tankyrase Inhibitors. Nat. Biotechnol. 2012, 30, 283–288. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, L.O.; Rulten, S.L.; Cranston, A.N.; Odedra, R.; Brown, H.; Jaspers, J.E.; Jones, L.; Knights, C.; Evers, B.; Ting, A.; et al. The PARP Inhibitor AZD2461 Provides Insights into the Role of PARP3 Inhibition for Both Synthetic Lethality and Tolerability with Chemotherapy in Preclinical Models. Cancer Res. 2016, 76, OF1–OF11. [Google Scholar]
- Chen, Q.; Kassab, M.A.; Dantzer, F.; Yu, X. PARP2 Mediates Branched Poly ADP-Ribosylation in Response to DNA Damage. Nat. Commun. 2018, 9, 3233. [Google Scholar] [CrossRef]
- Steffen, J.D.; Brody, J.R.; Armen, R.S.; Pascal, J.M. Structural Implications for Selective Targeting of PARPs. Front. Oncol. 2013, 3, 301. [Google Scholar] [CrossRef] [Green Version]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-Ribosyl)Ation by PARP1: Reaction Mechanism and Regulatory Proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [Green Version]
- Thorsell, A.-G.; Ekblad, T.; Karlberg, T.; Löw, M.; Pinto, A.F.; Trésaugues, L.; Moche, M.; Cohen, M.S.; Schüler, H. Structural Basis for Potency and Promiscuity in Poly(ADP-Ribose) Polymerase (PARP) and Tankyrase Inhibitors. J. Med. Chem. 2017, 60, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, A.E.G.; Karlberg, T.; Thorsell, A.-G.; Hesse, M.; Spjut, S.; Ekblad, T.; Andersson, C.D.; Pinto, A.F.; Weigelt, J.; Hottiger, M.O.; et al. PARP Inhibitor with Selectivity toward ADP-Ribosyltransferase ARTD3/PARP3. ACS Chem. Biol. 2013, 8, 1698–1703. [Google Scholar] [CrossRef]
- Papeo, G.; Posteri, H.; Borghi, D.; Busel, A.A.; Caprera, F.; Casale, E.; Ciomei, M.; Cirla, A.; Corti, E.; D’Anello, M.; et al. Discovery of 2-[1-(4,4-Difluorocyclohexyl)Piperidin-4-Yl]-6-Fluoro-3-Oxo-2,3-Dihydro-1H-Isoindole-4-Carboxamide (NMS-P118): A Potent, Orally Available, and Highly Selective PARP-1 Inhibitor for Cancer Therapy. J. Med. Chem. 2015, 58, 6875–6898. [Google Scholar] [CrossRef] [PubMed]
- Dawicki-McKenna, J.M.; Langelier, M.-F.; DeNizio, J.E.; Riccio, A.A.; Cao, C.D.; Karch, K.R.; McCauley, M.; Steffen, J.D.; Black, B.E.; Pascal, J.M. PARP-1 Activation Requires Local Unfolding of an Autoinhibitory Domain. Mol. Cell 2015, 60, 755–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ Homeostasis in Health and Disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J.; Ding, M.; Yu, Y. Site-Specific Characterization of the Asp- and Glu-ADP-Ribosylated Proteome. Nat. Methods 2013, 10, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Buch-Larsen, S.C.; Hendriks, I.A.; Lodge, J.M.; Rykær, M.; Furtwängler, B.; Shishkova, E.; Westphall, M.S.; Coon, J.J.; Nielsen, M.L. Mapping Physiological ADP-Ribosylation Using Activated Ion Electron Transfer Dissociation. Cell Rep. 2020, 32, 108176. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Larsen, S.C.; Nielsen, M.L. An Advanced Strategy for Comprehensive Profiling of ADP-Ribosylation Sites Using Mass Spectrometry-Based Proteomics*. Mol. Cell. Proteom. 2019, 18, 1010–1026. [Google Scholar] [CrossRef]
- Larsen, S.C.; Hendriks, I.A.; Lyon, D.; Jensen, L.J.; Nielsen, M.L. Systems-Wide Analysis of Serine ADP-Ribosylation Reveals Widespread Occurrence and Site-Specific Overlap with Phosphorylation. Cell Rep. 2018, 24, 2493–2505.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, M.; Hanashiro, K.; Kim, S.-H.; Hanai, S.; Boulares, A.H.; Miwa, M.; Fukasawa, K. Inhibition of Crm1–P53 Interaction and Nuclear Export of P53 by Poly(ADP-Ribosyl)Ation. Nat. Cell Biol. 2007, 9, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Zerfaoui, M.; Errami, Y.; Naura, A.S.; Suzuki, Y.; Kim, H.; Ju, J.; Liu, T.; Hans, C.P.; Kim, J.G.; Elmageed, Z.Y.A.; et al. Poly(ADP-Ribose) Polymerase-1 Is a Determining Factor in Crm1-Mediated Nuclear Export and Retention of P65 NF-KB upon TLR4 Stimulation. J. Immunol. 2010, 185, 1894–1902. [Google Scholar] [CrossRef]
- Heo, K.; Kim, H.; Choi, S.H.; Choi, J.; Kim, K.; Gu, J.; Lieber, M.R.; Yang, A.S.; An, W. FACT-Mediated Exchange of Histone Variant H2AX Regulated by Phosphorylation of H2AX and ADP-Ribosylation of Spt16. Mol. Cell 2008, 30, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Lonskaya, I.; Potaman, V.N.; Shlyakhtenko, L.S.; Oussatcheva, E.A.; Lyubchenko, Y.L.; Soldatenkov, V.A. Regulation of Poly(ADP-Ribose) Polymerase-1 by DNA Structure-Specific Binding. J. Biol. Chem. 2005, 280, 17076–17083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivelo, C.A.; Leung, A.K.L. Proteomics Approaches to Identify Mono(ADP-Ribosyl)Ated and Poly(ADP-Ribosyl)Ated Proteins. Proteomics 2015, 15, 203–217. [Google Scholar] [CrossRef] [Green Version]
- Gibbs-Seymour, I.; Fontana, P.; Rack, J.G.M.; Ahel, I. HPF1/C4orf27 Is a PARP-1-Interacting Protein That Regulates PARP-1 ADP-Ribosylation Activity. Mol. Cell 2016, 62, 432–442. [Google Scholar] [CrossRef]
- Bonfiglio, J.J.; Fontana, P.; Zhang, Q.; Colby, T.; Gibbs-Seymour, I.; Atanassov, I.; Bartlett, E.; Zaja, R.; Ahel, I.; Matic, I. Serine ADP-Ribosylation Depends on HPF1. Mol. Cell 2017, 65, 932–940.e6. [Google Scholar] [CrossRef] [Green Version]
- Steffen, J.D.; McCauley, M.M.; Pascal, J.M. Fluorescent Sensors of PARP-1 Structural Dynamics and Allosteric Regulation in Response to DNA Damage. Nucleic Acids Res. 2016, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger, A.; Bürkle, A.; Hauser, K.; Mangerich, A. Real-Time Monitoring of PARP1-Dependent PARylation by ATR-FTIR Spectroscopy. Nat. Commun. 2020, 11, 2174. [Google Scholar] [CrossRef] [PubMed]
- Pion, E.; Ullmann, G.M.; Ame, J.-C.; Gerard, D.; de Murcia, G.; Bombarda, E. DNA-Induced Dimerization of Poly(ADP-Ribose) Polymerase-1 Triggers Its Activation. Biochemistry 2005, 44, 14670–14681. [Google Scholar] [CrossRef]
- Mendoza-Alvarez, H.; Alvarez-Gonzalez, R. Poly(ADP-Ribose) Polymerase Is a Catalytic Dimer and the Automodification Reaction Is Intermolecular. J. Biol. Chem. 1993, 268, 22575–22580. [Google Scholar] [CrossRef]
- Tao, Z.; Gao, P.; Liu, H. Identification of the ADP-Ribosylation Sites in the PARP-1 Automodification Domain: Analysis and Implications. J. Am. Chem. Soc. 2009, 131, 14258–14260. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.D.; Gagné, J.-P.; Poirier, G.G.; Goodlett, D.R. Mapping PARP-1 Auto-ADP-Ribosylation Sites by Liquid Chromatography–Tandem Mass Spectrometry. J. Proteome Res. 2013, 12, 1868–1880. [Google Scholar] [CrossRef]
- Gagné, J.-P.; Ethier, C.; Defoy, D.; Bourassa, S.; Langelier, M.-F.; Riccio, A.A.; Pascal, J.M.; Moon, K.-M.; Foster, L.J.; Ning, Z.; et al. Quantitative Site-Specific ADP-Ribosylation Profiling of DNA-Dependent PARPs. DNA Repair 2015, 30, 68–79. [Google Scholar] [CrossRef]
- Talhaoui, I.; Lebedeva, N.A.; Zarkovic, G.; Saint-Pierre, C.; Kutuzov, M.M.; Sukhanova, M.V.; Matkarimov, B.T.; Gasparutto, D.; Saparbaev, M.K.; Lavrik, O.I.; et al. Poly(ADP-Ribose) Polymerases Covalently Modify Strand Break Termini in DNA Fragments in Vitro. Nucleic Acids Res. 2016, 44, 9279–9295. [Google Scholar] [CrossRef] [Green Version]
- Munnur, D.; Ahel, I. Reversible Mono-ADP-Ribosylation of DNA Breaks. FEBS J. 2017, 284, 4002–4016. [Google Scholar] [CrossRef] [Green Version]
- Rolli, V.; O’Farrell, M.; Ménissier-de Murcia, J.; de Murcia, G. Random Mutagenesis of the Poly(ADP-Ribose) Polymerase Catalytic Domain Reveals Amino Acids Involved in Polymer Branching. Biochemistry 1997, 36, 12147–12154. [Google Scholar] [CrossRef]
- Aberle, L.; Krüger, A.; Reber, J.M.; Lippmann, M.; Hufnagel, M.; Schmalz, M.; Trussina, I.R.E.A.; Schlesiger, S.; Zubel, T.; Schütz, K.; et al. PARP1 Catalytic Variants Reveal Branching and Chain Length-Specific Functions of Poly(ADP-Ribose) in Cellular Physiology and Stress Response. Nucleic Acids Res. 2020, 48, 10015–10033. [Google Scholar] [CrossRef] [PubMed]
- De Murcia, G.; Jongstra-Bilen, J.; Ittel, M.E.; Mandel, P.; Delain, E. Poly(ADP-Ribose) Polymerase Auto-Modification and Interaction with DNA: Electron Microscopic Visualization. EMBO J. 1983, 2, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Tanaka, M.; Shimada, T.; Miwa, M.; Sugimura, T. Size and Shape of Poly(ADP-Ribose): Examination by Gel Filtration, Gel Electrophoresis and Electron Microscopy. Biochem. Biophys. Res. Commun. 1983, 112, 102–107. [Google Scholar] [CrossRef]
- Panzeter, P.L.; Realini, C.A.; Althaus, F.R. Noncovalent Interactions of Poly(Adenosine Diphosphate Ribose) with Histones. Biochemistry 1992, 31, 1379–1385. [Google Scholar] [CrossRef]
- Fahrer, J.; Kranaster, R.; Altmeyer, M.; Marx, A.; Bürkle, A. Quantitative Analysis of the Binding Affinity of Poly(ADP-Ribose) to Specific Binding Proteins as a Function of Chain Length. Nucleic Acids Res. 2007, 35, e143. [Google Scholar] [CrossRef] [PubMed]
- Ikejima, M.; Marsischky, G.; Gill, D.M. Direction of Elongation of Poly(ADP-Ribose) Chains. Addition of Residues at the Polymerase-Proximal Terminus. J. Biol. Chem. 1987, 262, 17641–17650. [Google Scholar] [CrossRef]
- Taniguchi, T. Reaction Mechanism for Automodification of Poly(ADP-Ribos Synthetase. Biochem. Biophys. Res. Commun. 1987, 147, 1008–1012. [Google Scholar] [CrossRef]
- Poirier, G.G.; de Murcia, G.; Jongstra-Bilen, J.; Niedergang, C.; Mandel, P. Poly(ADP-Ribosyl)Ation of Polynucleosomes Causes Relaxation of Chromatin Structure. Proc. Natl. Acad. Sci. USA 1982, 79, 3423–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazzo, L.; Leidecker, O.; Prokhorova, E.; Dauben, H.; Matic, I.; Ahel, I. Serine Is the Major Residue for ADP-Ribosylation upon DNA Damage. eLife 2018, 7. [Google Scholar] [CrossRef]
- Suskiewicz, M.J.; Zobel, F.; Ogden, T.E.H.; Fontana, P.; Ariza, A.; Yang, J.-C.; Zhu, K.; Bracken, L.; Hawthorne, W.J.; Ahel, D.; et al. HPF1 Completes the PARP Active Site for DNA Damage-Induced ADP-Ribosylation. Nature 2020, 579, 598–602. [Google Scholar] [CrossRef]
- De Oliveira, T.M.; van Beek, L.; Shilliday, F.; Debreczeni, J.É.; Phillips, C. Cryo-EM: The Resolution Revolution and Drug Discovery. SLAS DISCOV. Adv. Sci. Drug Discov. 2021, 26, 17–31. [Google Scholar] [CrossRef]
- Van Drie, J.H.; Tong, L. Cryo-EM as a Powerful Tool for Drug Discovery. Bioorg. Med. Chem. Lett. 2020, 30, 127524. [Google Scholar] [CrossRef]
- Benjin, X.; Ling, L. Developments, Applications, and Prospects of Cryo-electron Microscopy. Protein Soc. 2019, 29, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Fontana, P.; Bonfiglio, J.J.; Palazzo, L.; Bartlett, E.; Matic, I.; Ahel, I. Serine ADP-Ribosylation Reversal by the Hydrolase ARH3. eLife 2017, 6. [Google Scholar] [CrossRef]
- Abplanalp, J.; Leutert, M.; Frugier, E.; Nowak, K.; Feurer, R.; Kato, J.; Kistemaker, H.V.A.; Filippov, D.V.; Moss, J.; Caflisch, A.; et al. Proteomic Analyses Identify ARH3 as a Serine Mono-ADP-Ribosylhydrolase. Nat. Commun. 2017, 8, 2055. [Google Scholar] [CrossRef] [Green Version]
- Leidecker, O.; Bonfiglio, J.J.; Colby, T.; Zhang, Q.; Atanassov, I.; Zaja, R.; Palazzo, L.; Stockum, A.; Ahel, I.; Matic, I. Serine Is a New Target Residue for Endogenous ADP-Ribosylation on Histones. Nat. Chem. Biol. 2016, 12, 998–1000. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Lin, S.; Garcia, B.A.; Zhao, Y. Quantitative Proteomic Analysis of Histone Modifications. Chem. Rev. 2015, 115, 2376–2418. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, E.; Bonfiglio, J.J.; Prokhorova, E.; Colby, T.; Zobel, F.; Ahel, I.; Matic, I. Interplay of Histone Marks with Serine ADP-Ribosylation. Cell Rep. 2018, 24, 3488–3502.e5. [Google Scholar] [CrossRef] [Green Version]
- Liszczak, G.; Diehl, K.L.; Dann, G.P.; Muir, T.W. Acetylation Blocks DNA Damage-Induced Chromatin ADP-Ribosylation. Nat. Chem. Biol. 2018, 14, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Daniels, C.M.; Ong, S.-E.; Leung, A.K.L. Phosphoproteomic Approach to Characterize Protein Mono- and Poly(ADP-Ribosyl)Ation Sites from Cells. J. Proteome Res. 2014, 13, 3510–3522. [Google Scholar] [CrossRef] [Green Version]
- Martello, R.; Leutert, M.; Jungmichel, S.; Bilan, V.; Larsen, S.C.; Young, C.; Hottiger, M.O.; Nielsen, M.L. Proteome-Wide Identification of the Endogenous ADP-Ribosylome of Mammalian Cells and Tissue. Nat. Commun. 2016, 7, 12917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, F.; Nanni, P.; Barkow-Oesterreicher, S.; Hottiger, M.O. Optimization of LTQ-Orbitrap Mass Spectrometer Parameters for the Identification of ADP-Ribosylation Sites. J. Proteome Res. 2015, 14, 4072–4079. [Google Scholar] [CrossRef]
- Karch, K.R.; Langelier, M.-F.; Pascal, J.M.; Garcia, B.A. The Nucleosomal Surface Is the Main Target of Histone ADP-Ribosylation in Response to DNA Damage. Mol. BioSyst. 2017, 13, 2660–2671. [Google Scholar] [CrossRef]
- Huang, D.; Camacho, C.V.; Setlem, R.; Ryu, K.W.; Parameswaran, B.; Gupta, R.K.; Kraus, W.L. Functional Interplay between Histone H2B ADP-Ribosylation and Phosphorylation Controls Adipogenesis. Mol. Cell 2020, 79, 934–949.e14. [Google Scholar] [CrossRef]
- Yang, G.; Chen, Y.; Wu, J.; Chen, S.-H.; Liu, X.; Singh, A.K.; Yu, X. Poly(ADP-Ribosyl)Ation Mediates Early Phase Histone Eviction at DNA Lesions. Nucleic Acids Res. 2020, 48, 3001–3013. [Google Scholar] [CrossRef]
- Chou, D.M.; Adamson, B.; Dephoure, N.E.; Tan, X.; Nottke, A.C.; Hurov, K.E.; Gygi, S.P.; Colaiácovo, M.P.; Elledge, S.J. A Chromatin Localization Screen Reveals Poly (ADP Ribose)-Regulated Recruitment of the Repressive Polycomb and NuRD Complexes to Sites of DNA Damage. Proc. Natl. Acad. Sci. USA 2010, 107, 18475–18480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.P.G.; Ryan, D.P.; Galanty, Y.; Low, J.K.K.; Vandevenne, M.; Jackson, S.P.; Mackay, J.P. The N-Terminal Region of Chromodomain Helicase DNA-Binding Protein 4 (CHD4) Is Essential for Activity and Contains a High Mobility Group (HMG) Box-like-Domain That Can Bind Poly(ADP-Ribose). J. Biol. Chem. 2016, 291, 924–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basta, J.; Rauchman, M. The Nucleosome Remodeling and Deacetylase (NuRD) Complex in Development and Disease. Transl. Res. 2015, 165, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Sellou, H.; Lebeaupin, T.; Chapuis, C.; Smith, R.; Hegele, A.; Singh, H.R.; Kozlowski, M.; Bultmann, S.; Ladurner, A.G.; Timinszky, G.; et al. The Poly(ADP-Ribose)-Dependent Chromatin Remodeler Alc1 Induces Local Chromatin Relaxation upon DNA Damage. Mol. Biol. Cell 2016, 27, 3791–3799. [Google Scholar] [CrossRef] [PubMed]
- Ahel, D.; Hořejší, Z.; Wiechens, N.; Polo, S.E.; Garcia-Wilson, E.; Ahel, I.; Flynn, H.; Skehel, M.; West, S.C.; Jackson, S.P.; et al. Poly(ADP-Ribose)–Dependent Regulation of DNA Repair by the Chromatin Remodeling Enzyme ALC1. Science 2009, 325, 1240–1243. [Google Scholar] [CrossRef] [Green Version]
- Juhász, S.; Smith, R.; Schauer, T.; Spekhardt, D.; Mamar, H.; Zentout, S.; Chapuis, C.; Huet, S.; Timinszky, G. The Chromatin Remodeler ALC1 Underlies Resistance to PARP Inhibitor Treatment. Sci. Adv. 2020, 6, eabb8626. [Google Scholar] [CrossRef]
- Hewitt, G.; Borel, V.; Segura-Bayona, S.; Takaki, T.; Ruis, P.; Bellelli, R.; Lehmann, L.C.; Sommerova, L.; Vancevska, A.; Tomas-Loba, A.; et al. Defective ALC1 Nucleosome Remodeling Confers PARPi Sensitization and Synthetic Lethality with HRD. Mol. Cell 2020. [Google Scholar] [CrossRef]
- Lehmann, L.C.; Bacic, L.; Hewitt, G.; Brackmann, K.; Sabantsev, A.; Gaullier, G.; Pytharopoulou, S.; Degliesposti, G.; Okkenhaug, H.; Tan, S.; et al. Mechanistic Insights into Regulation of the ALC1 Remodeler by the Nucleosome Acidic Patch. Cell Rep. 2020, 33. [Google Scholar] [CrossRef]
- Luijsterburg, M.S.; de Krijger, I.; Wiegant, W.W.; Shah, R.G.; Smeenk, G.; de Groot, A.J.L.; Pines, A.; Vertegaal, A.C.O.; Jacobs, J.J.L.; Shah, G.M.; et al. PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-Homologous End-Joining. Mol. Cell 2016, 61, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Hanzlikova, H.; Gittens, W.; Krejcikova, K.; Zeng, Z.; Caldecott, K.W. Overlapping Roles for PARP1 and PARP2 in the Recruitment of Endogenous XRCC1 and PNKP into Oxidized Chromatin. Nucleic Acids Res. 2017, 45, 2546–2557. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, C.; Wei, L.; Teng, Y.; Nakajima, S.; Chen, X.; Xu, J.; Legar, B.; Ma, H.; Spagnol, S.T.; et al. SSRP1 Cooperates with PARP and XRCC1 to Facilitate Single Strand DNA Break Repair by Chromatin Priming. Cancer Res. 2017, 77, 2674–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pines, A.; Vrouwe, M.G.; Marteijn, J.A.; Typas, D.; Luijsterburg, M.S.; Cansoy, M.; Hensbergen, P.; Deelder, A.; de Groot, A.; Matsumoto, S.; et al. PARP1 Promotes Nucleotide Excision Repair through DDB2 Stabilization and Recruitment of ALC1. J. Cell. Biol. 2012, 199, 235–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Beek, L.; McClay, É.; Patel, S.; Schimpl, M.; Spagnolo, L.; Maia de Oliveira, T. PARP Power: A Structural Perspective on PARP1, PARP2, and PARP3 in DNA Damage Repair and Nucleosome Remodelling. Int. J. Mol. Sci. 2021, 22, 5112. https://doi.org/10.3390/ijms22105112
van Beek L, McClay É, Patel S, Schimpl M, Spagnolo L, Maia de Oliveira T. PARP Power: A Structural Perspective on PARP1, PARP2, and PARP3 in DNA Damage Repair and Nucleosome Remodelling. International Journal of Molecular Sciences. 2021; 22(10):5112. https://doi.org/10.3390/ijms22105112
Chicago/Turabian Stylevan Beek, Lotte, Éilís McClay, Saleha Patel, Marianne Schimpl, Laura Spagnolo, and Taiana Maia de Oliveira. 2021. "PARP Power: A Structural Perspective on PARP1, PARP2, and PARP3 in DNA Damage Repair and Nucleosome Remodelling" International Journal of Molecular Sciences 22, no. 10: 5112. https://doi.org/10.3390/ijms22105112
APA Stylevan Beek, L., McClay, É., Patel, S., Schimpl, M., Spagnolo, L., & Maia de Oliveira, T. (2021). PARP Power: A Structural Perspective on PARP1, PARP2, and PARP3 in DNA Damage Repair and Nucleosome Remodelling. International Journal of Molecular Sciences, 22(10), 5112. https://doi.org/10.3390/ijms22105112