
Sensitivity of Intra- and Intermolecular Interactions of Benzo[h]quinoline from Car–Parrinello Molecular Dynamics and Electronic Structure Inspection

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
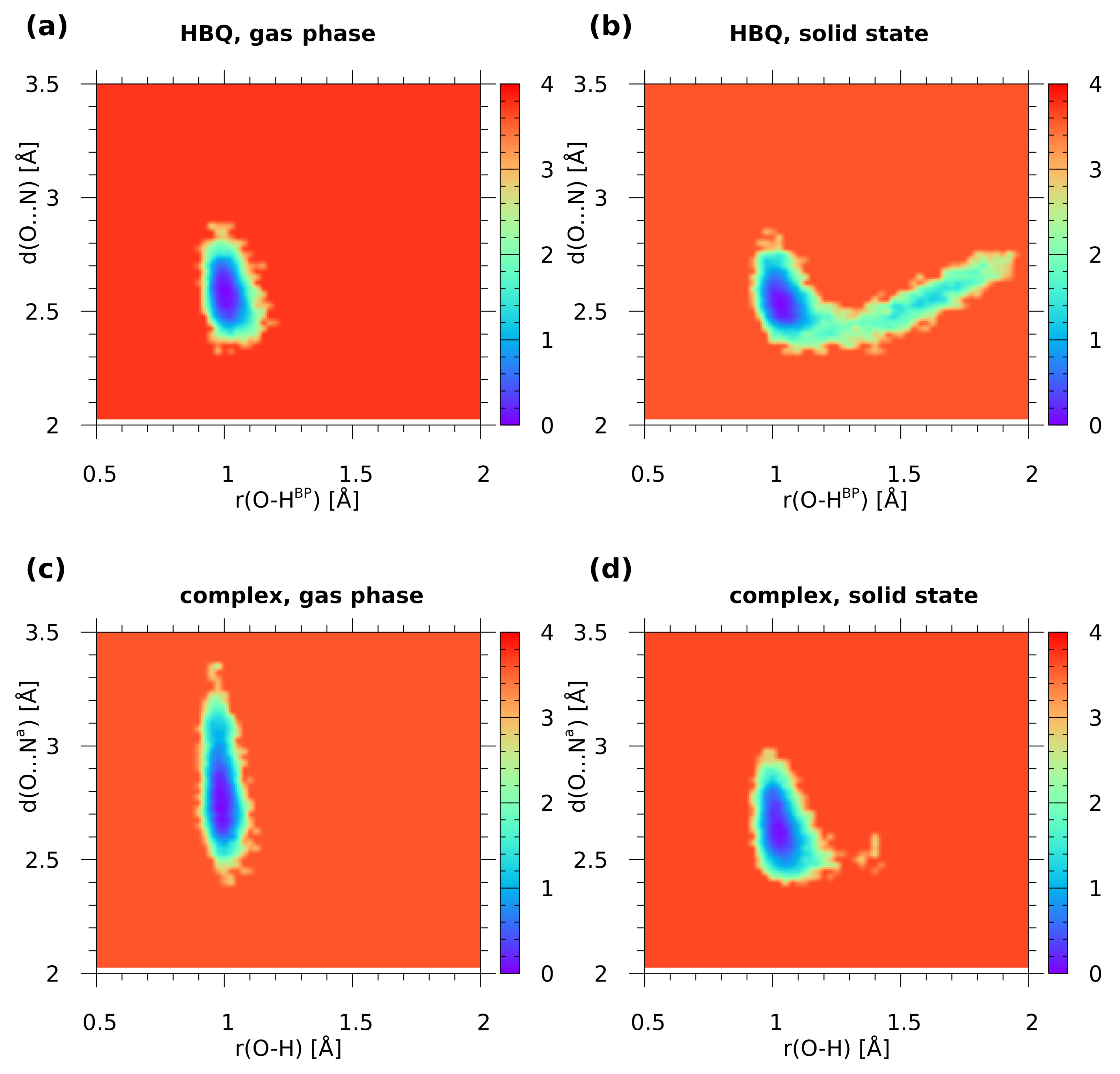
2.1. Car–Parrinello Molecular Dynamics in the Gas and Crystalline Phases—Structural and Vibrational Aspects
2.2. A Posteriori Inclusion of Quantum Effects to the O-H Stretching and Potential of Mean Force (Pmf)
2.3. Constrained Density Functional Theory (CDFT) Electronic Structure Analysis of Benzo[h]Quinoline-2-methylresorcinol Dimer and Trimers
2.4. Electron Localization Function (ELF) Topological Analysis of the Hydrogen Bonding Based on CPMD Trajectory
3. Computational Methods and Procedures
3.1. Car–Parrinello Molecular Dynamics in the Gas and Crystalline Phases
3.2. A Posteriori Inclusion of Quantum Effects of Nuclear Motion of the O-H Stretching
3.3. One-Dimensional (1D) and Two-Dimensional (2D) Potential of Mean Force (Pmf)
3.4. Constrained Density Functional Theory (CDFT) Method
3.5. Electron Localization Function (ELF)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CDFT | Constrained Density Functional Theory |
| CPMD | Car–Parrinello Molecular Dynamics |
| DFT | Density Functional Theory |
| ELF | Electron Localization Function |
| HB | Hydrogen bond |
| HBQ | 10-hydroxybenzo[h]quinoline |
| Pmf | Potential of mean force |
References
- Woodley, S.M.; Catlow, R. Crystal structure prediction from first principles. Nat. Mater. 2008, 7, 937–946. [Google Scholar] [CrossRef]
- Ryan, K.; Lengyel, J.; Shatruk, M. Crystal Structure Prediction via Deep Learning. J. Am. Chem. Soc. 2018, 140, 10158–10168. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. New Type of Halogen Bond: Multivalent Halogen Interacting with - and -Electrons. Molecules 2017, 22, 2150. [Google Scholar] [CrossRef] [Green Version]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Noncovalent Bonds through Sigma and Pi-Hole Located on the Same Molecule. Guiding Principles and Comparisons. Molecules 2021, 26, 1740. [Google Scholar] [CrossRef]
- Grabowski, S.J. Tetrel Bonds with -Electrons Acting as Lewis Bases—Theoretical Results and Experimental Evidences. Molecules 2018, 23, 1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabłoński, M.; Palusiak, M. Nature of a Hydride–Halogen Bond. A SAPT-, QTAIM-, and NBO-Based Study. J. Phys. Chem. A 2012, 116, 2322–2332. [Google Scholar] [CrossRef]
- Jabłoński, M. Ten years of charge-inverted hydrogen bonds. Struct. Chem. 2020, 31, 61–80. [Google Scholar] [CrossRef]
- Kubicki, M.; Borowiak, T.; Antkowiak, W.Z. 10-Hydroxybenzo[H]quinoline. Acta Cryst. C 1995, 51, 1173–1175. [Google Scholar] [CrossRef]
- Mir, N.A.; Dubey, R.; Tothadi, S.; Desiraju, G.R. Combinatorial crystal synthesis of ternary solids based on 2-methylresorcinol. CrystEngComm 2015, 17, 7866–7869. [Google Scholar] [CrossRef]
- Inazu, T. The Synthesis of Several Gold Chelates. Bull. Chem. Soc. Jpn. 1966, 39, 1065–1066. [Google Scholar] [CrossRef]
- Roberts, E.L.; Chou, P.T.; Alexander, T.A.; Agbaria, R.A.; Warner, I.M. Effects of Organized Media on the Excited-State Intramolecular Proton Transfer of 10-Hydroxybenzo[/z]quinoline. J. Phys. Chem. 1995, 99, 5431–5437. [Google Scholar] [CrossRef]
- Chou, P.T.; Wei, C.Y. Photophysics of 10-Hydroxybenzo[h]quinoline in Aqueous Solution. J. Phys. Chem. 1996, 100, 17059–17066. [Google Scholar] [CrossRef]
- Chou, P.T.; Chen, Y.C.; Yu, W.S.; Chou, Y.H.; Wei, C.Y.; Cheng, Y.M. Excited-State Intramolecular Proton Transfer in 10-Hydroxybenzo[h]quinoline. J. Phys. Chem. A 2001, 105, 1731–1740. [Google Scholar] [CrossRef]
- Takeuchi, S.; Tahara, T. Coherent Nuclear Wavepacket Motions in Ultrafast Excited-State Intramolecular Proton Transfer: Sub-30-fs Resolved Pump-Probe Absorption Spectroscopy of 10-Hydroxybenzo[h]quinoline in Solution. J. Phys. Chem. A 2005, 109, 10199–10207. [Google Scholar] [CrossRef]
- Schriever, C.; Barbatti, M.; Stock, K.; Aquino, A.J.; Tunega, D.; Lochbrunner, S.; Riedle, E.; de Vivie-Riedle, R.; Lischka, H. The interplay of skeletal deformations and ultrafast excited-state intramolecular proton transfer: Experimental and theoretical investigation of 10-hydroxybenzo[h]quinoline. Chem. Phys. 2008, 347, 446–461. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, M. A TDDFT study on the singlet and triplet excited-state hydrogen bonding and proton transfer of 10-hydroxybenzo[h]quinoline (HBQ) and 7, 9-diiodo-10-hydroxybenzo[h]quinoline (DIHBQ). Chem. Phys. Lett. 2011, 512, 35–39. [Google Scholar] [CrossRef]
- Higashi, M.; Saito, S. Direct Simulation of Excited-State Intramolecular Proton Transfer and Vibrational Coherence of 10-Hydroxybenzo[h]quinoline in Solution. J. Phys. Chem. Lett. 2011, 2, 2366–2371. [Google Scholar] [CrossRef]
- Paul, B.K.; Guchhait, N. TD–DFT investigation of the potential energy surface for Excited-State Intramolecular Proton Transfer (ESIPT) reaction of 10-hydroxybenzo[h]quinoline: Topological (AIM) and population (NBO) analysis of the intramolecular hydrogen bonding interaction. J. Lumin. 2011, 131, 1918–1926. [Google Scholar] [CrossRef]
- Chou, P.T.; Wu, G.R.; Liu, Y.I.; Yu, W.S.; Chiou, C.S. Proton-Transfer Tautomerism in 10-Hydroxybenzo[h]quinolines: Heavy Atom Effects and Non-Hydrogen-Bonded Photorotamer Formation in 77 K Glassy Matrixes. J. Phys. Chem. A 2002, 106, 5967–5973. [Google Scholar] [CrossRef]
- Piechowska, J.; Gryko, D.T. Preparation of a Family of 10-Hydroxybenzo[h]quinoline Analogues via a Modified Sanford Reaction and Their Excited State Intramolecular Proton Transfer Properties. J. Org. Chem. 2011, 76, 10220–10228. [Google Scholar] [CrossRef]
- Piechowska, J.; Huttunen, K.; Wróbel, Z.; Lemmetyinen, H.; Tkachenko, N.V.; Gryko, D.T. Excited State Intramolecular Proton Transfer in Electron-Rich and Electron-Poor Derivatives of 10-Hydroxybenzo[h]quinoline. J. Phys. Chem. A 2012, 116, 9614–9620. [Google Scholar] [CrossRef]
- Deperasińska, I.; Gryko, D.T.; Karpiuk, E.; Kozankiewicz, B.; Makarewicz, A.; Piechowska, J. Low-Temperature Spectra of the Analogues of 10-Hydroxybenzo[h]quinoline as an Indication of Barrierless ESIPT. J. Phys. Chem. A 2012, 116, 12049–12055. [Google Scholar] [CrossRef] [PubMed]
- Bardez, E.; Chatelain, A.; Larrey, B.; Valeur, B. Photoinduced Coupled Proton and Electron Transfers. 1. 6-Hydroxyquinoline. J. Phys. Chem. 1994, 98, 2357–2366. [Google Scholar] [CrossRef]
- Kaczmarek, L.; Borowicz, P.; Grabowska, A. Strongly modified [2,20-bipyridyl]-3,30-diol (BP(OH)2): A system undergoing excited state intramolecularproton transfer as a photostabilizer of polymersand as a solar energy collector. J. Photochem. Photobiol. A 2001, 138, 159–166. [Google Scholar] [CrossRef]
- Mohammed, O.; Pines, D.; Nibbering, E.; Pines, E. Base-Induced Solvent Switches in Acid–Base Reactions. Angew. Chem. Int. Ed. 2007, 46, 1458–1461. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kwon, O.H.; Kim, S.; Park, S.; Choi, M.G.; Cha, M.; Park, S.Y.; Jang, D.J. Imidazole-Based Excited-State Intramolecular Proton-Transfer Materials: Synthesis and Amplified Spontaneous Emission from a Large Single Crystal. J. Am. Chem. Soc. 2005, 127, 10070–10074. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kwon, J.E.; Kim, S.H.; Seo, J.; Chung, K.; Park, S.Y.; Jang, D.J.; Medina, B.M.; Gierschner, J.; Park, S.Y. A White-Light-Emitting Molecule: Frustrated Energy Transfer between Constituent Emitting Centers. J. Am. Chem. Soc. 2009, 131, 14043–14049. [Google Scholar] [CrossRef] [PubMed]
- Dubey, R.; Desiraju, G.R. Combinatorial Crystal Synthesis: Structural Landscape of Phloroglucinol:1, 2-bis(4-pyridyl)ethylene and Phloroglucinol:Phenazine. Angew. Chem. Int. Ed. 2014, 53, 13178–13182. [Google Scholar] [CrossRef]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Jezierska, A.; Kizior, B.; Szyja, B.M.; Panek, J.J. On the nature of inter- and intramolecular interactions involving benzo[h]quinoline and 10-hydroxybenzo[h]quinoline: Electronic ground state vs excited state study. J. Mol. Struct. 2021, 1234, 130126. [Google Scholar] [CrossRef]
- Denisov, G.S.; Mavri, J.; Sobczyk, L. Potential Energy Shape for the Proton Motion in Hydrogen Bonds Reflected in Infraredand NMR Spectra. In Hydrogen Bonding—New Insights, (Challenges and Advances in Computational Chemistry and Physics, 3), 1st ed.; Grabowski, S.J., Ed.; Springer: Dordrecht, The Netherlands, 2006; pp. 377–416. [Google Scholar]
- Filarowski, A.; Koll, A. Integrated intensity of νs(OH) absorption bands in bent hydrogen bonds in ortho-dialkylaminomethyl phenols. Vib. Spectrosc. 1996, 12, 15–24. [Google Scholar] [CrossRef]
- Filarowski, A.; Koll, A. Specificity of the intramolecular hydrogen bond. The differences in spectroscopic characteristics of the intermolecular and intramolecular H-bonds. Vib. Spectrosc. 1998, 17, 123–131. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [Green Version]
- Marx, D.; Tuckerman, M.E.; Parrinello, M. Solvated excess protons in water: Quantum effects on the hydration structure. J. Phys. Condens. Matter 2000, 12, A153–A159. [Google Scholar] [CrossRef]
- Ceriotti, M.; Cuny, J.; Parrinello, M.; Manolopoulos, D.E. Nuclear quantum effects and hydrogen bond fluctuations in water. Proc. Natl. Acad. Sci. USA 2013, 110, 15591–15596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaduk, B.; Kowalczyk, T.; Voorhis, T.V. Constrained Density Functional Theory. Chem. Rev. 2011, 112, 321–370. [Google Scholar] [CrossRef] [Green Version]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Clarendon: Oxford, UK, 1994. [Google Scholar]
- Gilli, G.; Bellucci, F.; Ferretti, V.; Bertolasi, V. Evidence for resonance-assisted hydrogen bonding from crystal-structure correlations on the enol form of the .beta.-diketone fragment. J. Am. Chem. Soc. 1989, 111, 1023–1028. [Google Scholar] [CrossRef]
- Balasubramanian, M.; Reynolds, A.; Blair, T.J.; Khalil, M. Probing ultrafast vibrational dynamics of intramolecular hydrogen bonds with broadband infrared pump-probe spectroscopy. Chem. Phys. 2019, 519, 38–44. [Google Scholar] [CrossRef]
- Gaigeot, M.P.; Sprik, M. Ab Initio Molecular Dynamics Computation of the Infrared Spectrum of Aqueous Uracil. J. Phys. Chem. B 2003, 107, 10344–10358. [Google Scholar] [CrossRef]
- Jezierska, A.; Panek, J.J. Theoretical study of intramolecular hydrogen bond in selected symmetric “proton sponges” on the basis of DFT and CPMD methods. J. Mol. Model. 2020, 26, 37. [Google Scholar] [CrossRef]
- Molčanov, K.; Stare, J.; Vener, M.V.; Kojić-Prodić, B.; Mali, G.; Grdadolnik, J.; Mohaček-Grošev, V. Nitranilic acid hexahydrate, a novel benchmark system of the Zundel cation in an intrinsically asymmetric environment: Spectroscopic features and hydrogen bond dynamics characterised by experimental and theoretical methods. Phys. Chem. Chem. Phys. 2014, 16, 998–1007. [Google Scholar] [CrossRef]
- Stare, J.; Gradišek, A.; Seliger, J. Nuclear quadrupole resonance supported by periodic quantum calculations: A sensitive tool for precise structural characterization of short hydrogen bonds. Phys. Chem. Chem. Phys. 2020, 22, 27681–27689. [Google Scholar] [CrossRef]
- Alikhani, M.E.; Fuster, F.; Silvi, B. What Can Tell the Topological Analysis of ELF on Hydrogen Bonding? Struct. Chem. 2005, 16, 203–210. [Google Scholar] [CrossRef]
- Fuster, F.; Silvi, B. Does the topological approach characterize the hydrogen bond? Theor. Chem. Acc. 2000, 104, 13–21. [Google Scholar] [CrossRef]
- Schlegel, H.B. Estimating the hessian for gradient-type geometry optimizations. Theor. Chim. Acta 1984, 66, 333–340. [Google Scholar] [CrossRef]
- Kittel, C. Introduction To Solid State Physics, 8th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Hockney, R.W. Potential Calculation and Some Applications. Methods Comput. Phys. 1970, 9, 135–211. [Google Scholar]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Jezierska, A.; Panek, J.J.; Koll, A.; Mavri, J. Car-Parrinello simulation of an O–H stretching envelope and potential of mean force of an intramolecular hydrogen bonded system: Application to a Mannich base in solid state and in vacuum. J. Chem. Phys. 2007, 126, 205101. [Google Scholar] [CrossRef] [PubMed]
- Jezierska, A.; Panek, J.J.; Koll, A. Spectroscopic Properties of a Strongly Anharmonic Mannich Base N-oxide. ChemPhysChem 2008, 9, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Stare, J.; Mavri, J. Numerical solving of the vibrational time-independent Schrödinger equation in one and two dimensions using the variational method. Comput. Phys. Commun. 2002, 143, 222–240. [Google Scholar] [CrossRef]
- Kirkwood, J.G. Statistical Mechanics of Fluid Mixtures. J. Chem. Phys. 1935, 3, 300–313. [Google Scholar] [CrossRef]
- Jezierska, A.; Panek, J.J. First-Principle Molecular Dynamics Study of Selected Schiff and Mannich Bases: Application of Two-Dimensional Potential of Mean Force to Systems with Strong Intramolecular Hydrogen Bonds. J. Chem. Theory Comput. 2008, 4, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Jezierska, A.; Panek, J.J. Investigations of an O-HS hydrogen bond via Car-Parrinello and path integral molecular dynamics. J. Comput. Chem. 2009, 30, 1241–1250. [Google Scholar] [CrossRef]
- CPMD 3.17.1. Copyright IBM Corp. (1990–2004) Copyright MPI für Festkoerperforschung Stuttgart (1997–2001). Available online: http://www.cpmd.org (accessed on 24 November 2020).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Mercury—Crystal Structure Visualisation. Available online: http://www.ccdc.cam.ac.uk/Solutions/CSDSystem/Pages/Mercury.aspx (accessed on 27 November 2020).
- Williams, T.; Kelley, C. Gnuplot 4.4: An Interactive Plotting Program. 2010. Available online: http://gnuplot.sourceforge.net/ (accessed on 27 November 2020).
- Holmberg, N.; Laasonen, K. Efficient Constrained Density Functional Theory Implementation for Simulation of Condensed Phase Electron Transfer Reactions. J. Chem. Theory Comput. 2017, 13, 587–601. [Google Scholar] [CrossRef] [Green Version]
- Holmberg, N.; Laasonen, K. Diabatic model for electrochemical hydrogen evolution based on constrained DFT configuration interaction. J. Chem. Phys. 2018, 149, 104702. [Google Scholar] [CrossRef] [Green Version]
- Kühne, T.D.; Iannuzzi, M.; Ben, M.D.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; et al. CP2K: An electronic structure and molecular dynamics software package—Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef] [PubMed]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. A multicenter numerical integration scheme for polyatomic molecules. J. Chem. Phys. 1988, 88, 2547–2553. [Google Scholar] [CrossRef]
- Andreussi, O.; Dabo, I.; Marzari, N. Revised self-consistent continuum solvation in electronic-structure calculations. J. Chem. Phys. 2012, 136, 064102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momma, K.; Izumi, F. VESTA 3for three-dimensional visualization of crystal, volumetric and morphology data. J. Apply. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian˜16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Panek, J.J.; Jezierska, A. N-oxide Derivatives: Car–Parrinello Molecular Dynamics and Electron Localization Function Study on Intramolecular Hydrogen Bonds. J. Phys. Chem. A 2018, 122, 6605–6614. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Kohout, M. DGrid, version 5.1; Max-Planck-Institut fuer Chemische Physik Fester Stoffe: Dresden, Germany, 2019. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | [eV] |
|---|---|
| dimer | 6.12 |
| trimer 1 | 5.03 |
| trimer 2 | 5.13 |
| Parameter | HBQ, Gas Phase | HBQ, Solid | Complex, Gas Phase |
|---|---|---|---|
| n(O-HBP...N) | 20 | 17 | 22 |
| V(O-HBP) | 1.764–1.838 | 1.759–1.842 | 1.647–1.835 |
| V(N) | 2.684–2.867 | 2.755–2.854 | 2.686–2.820 |
| n(O...HBP...N) | 2 | 2 | 0 |
| V(O) | 1.263–1.355 | 1.235–1.249 | – |
| V(HBP) | 0.486–0.554 | 0.529–0.547 | – |
| V(N) | 2.664–2.800 | 2.621–2.859 | – |
| n(O...HBP-N) | 0 | 3 | 0 |
| V(O) | – | 2.557–2.646 | – |
| V(HBP-N) | – | 2.453–2.908 | – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panek, J.J.; Zasada, J.; Szyja, B.M.; Kizior, B.; Jezierska, A. Sensitivity of Intra- and Intermolecular Interactions of Benzo[h]quinoline from Car–Parrinello Molecular Dynamics and Electronic Structure Inspection. Int. J. Mol. Sci. 2021, 22, 5220. https://doi.org/10.3390/ijms22105220
Panek JJ, Zasada J, Szyja BM, Kizior B, Jezierska A. Sensitivity of Intra- and Intermolecular Interactions of Benzo[h]quinoline from Car–Parrinello Molecular Dynamics and Electronic Structure Inspection. International Journal of Molecular Sciences. 2021; 22(10):5220. https://doi.org/10.3390/ijms22105220
Chicago/Turabian StylePanek, Jarosław J., Joanna Zasada, Bartłomiej M. Szyja, Beata Kizior, and Aneta Jezierska. 2021. "Sensitivity of Intra- and Intermolecular Interactions of Benzo[h]quinoline from Car–Parrinello Molecular Dynamics and Electronic Structure Inspection" International Journal of Molecular Sciences 22, no. 10: 5220. https://doi.org/10.3390/ijms22105220