
Involvement of the Protein Ras Homolog Enriched in the Striatum, Rhes, in Dopaminergic Neurons’ Degeneration: Link to Parkinson’s Disease

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Discovery of Rhes
1.1. Protein Structure
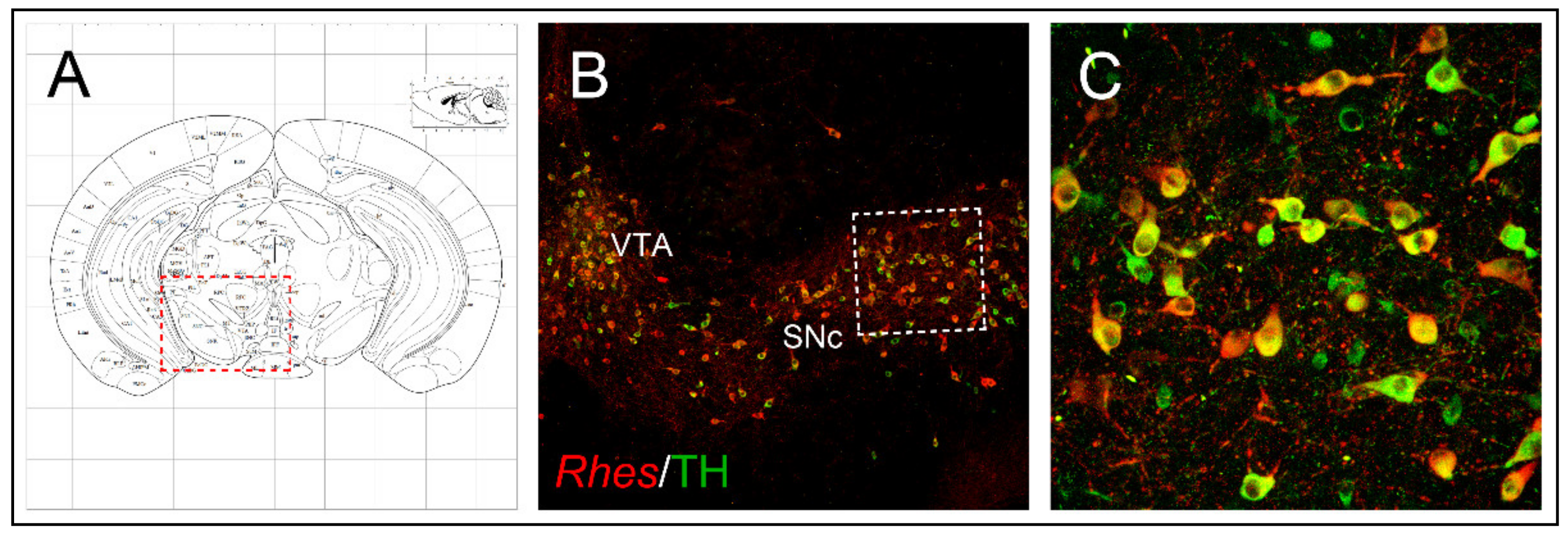
1.2. Anatomical Brain Localization
2. Ontogeny of Rhes and Its Striatal Regulation
2.1. Rhes Is Modulated by Thyroid Hormones
2.2. Rhes Expression Is Regulated by Dopamine Innervation
3. Rhes Intracellular Signaling
3.1. In Vitro Modulation of Rhes-Dependent cAMP/PKA Signaling
3.2. Rhes Affects Striatal cAMP/PKA Signaling in Mice
3.3. Rhes Affects the PI3K/Akt Signaling Pathway
4. Rhes Involvement in Huntington Disease and L-DOPA-Induced Dyskinesia
4.1. Rhes Acts as SUMO E3 Ligase for the Mutant Huntingtin
4.2. Role of Rhes in Modulating HD-Dependent Phenotypes in Animal Models
4.3. Rhes Affects L-DOPA-Induced Dyskinesia (LID) in PD Mouse Model
5. Involvement of Rhes in Parkinson’s Disease: Focus on Rhes Regulation of Nigrostriatal Neurons’ Survival
5.1. Rhes Counteracts Nigrostriatal Degeneration during Ageing in a Gender-Dependent Manner
5.2. Rhes Reduces the MDMA-Induced Dopaminergic Degeneration and Neuroinflammation Affecting the Nigrostriatal System
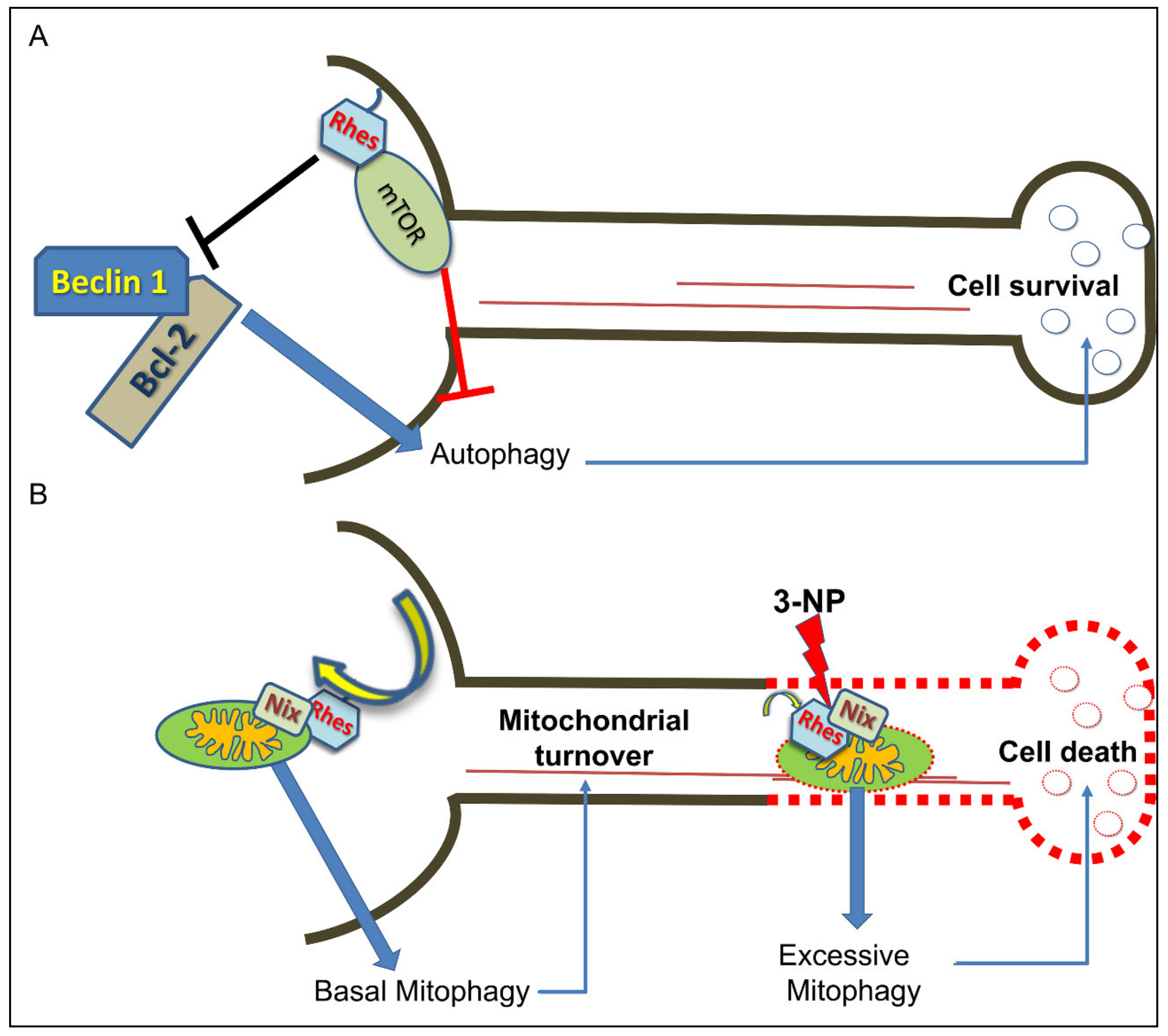
6. Rhes Influences Autophagy and Mitophagy Processes
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Usui, H.; Falk, J.D.; Dopazo, A.; De Lecea, L.; Erlander, M.G.; Sutcliffe, J.G. Isolation of clones of rat striatum-specific mRNAs by directional tag PCR subtraction. J. Neurosci. 1994, 14, 4915–4926. [Google Scholar] [CrossRef] [PubMed]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature 1991, 349, 117–127. [Google Scholar] [CrossRef]
- Thapliyal, A.; Verma, R.; Kumar, N. Small G proteins Dexras1 and RHES and their role in pathophysiological processes. Int. J. Cell Biol. 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Vargiu, P.; De Abajo, R.; Garcia-Ranea, J.A.; Valencia, A.; Santisteban, P.; Crespo, P.; Bernal, J. The small GTP-binding protein, Rhes, regulates signal transduction from G protein-coupled receptors. Oncogene 2004, 23, 559–568. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.; Goddard, A.; Ladds, G.; Davey, J. The cationic region of Rhes mediates its interactions with specific Gbeta subunits. Cell Physiol. Biochem. 2009, 23, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ehrenberg, A.J.; Leng, K.; Letourneau, K.N.; Hernandez, I.; Lew, C.; Seeley, W.W.; Spina, S.; Miller, B.; Heinsen, H.; Kampmann, M.; et al. Patterns of neuronal Rhes as a novel hallmark of tauopathies. Acta Neuropathol. 2021, 141, 651–666. [Google Scholar] [CrossRef]
- Falk, J.D.; Vargiu, P.; Foye, P.E.; Usui, H.; Perez, J.; Danielson, P.E.; Lerner, D.L.; Bernal, J.; Sutcliffe, J.G. Rhes: A striatal-specific Ras homolog related to Dexras1. J. Neurosci. Res. 1999, 57, 782–788. [Google Scholar] [CrossRef]
- Hernandez, I.; Luna, G.; Rauch, J.N.; Reis, S.A.; Giroux, M.; Karch, C.M.; Boctor, D.; Sibih, Y.E.; Storm, N.J.; Diaz, A.; et al. A farnesyltransferase inhibitor activates lysosomes and reduces tau pathology in mice with tauopathy. Sci. Transl. Med. 2019, 11, eaat3005. [Google Scholar] [CrossRef] [PubMed]
- Errico, F.; Santini, E.; Migliarini, S.; Borgkvist, A.; Centonze, D.; Nasti, V.; Carta, M.; De Chiara, V.; Prosperetti, C.; Spano, D.; et al. The GTP-binding protein Rhes modulates dopamine signalling in striatal medium spiny neurons. Mol. Cell. Neurosci. 2008, 37, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Ghiglieri, V.; Napolitano, F.; Pelosi, B.; Schepisi, C.; Migliarini, S.; Di Maio, A.; Pendolino, V.; Mancini, M.; Sciamanna, G.; Vitucci, D.; et al. Rhes influences striatal cAMP/PKA-dependent signaling and synaptic plasticity in a gender-sensitive fashion. Sci. Rep. 2015, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sciamanna, G.; Napolitano, F.; Pelosi, B.; Bonsi, P.; Vitucci, D.; Nuzzo, T.; Punzo, D.; Ghiglieri, V.; Ponterio, G.; Pasqualetti, M.; et al. Rhes regulates dopamine D2 receptor transmission in striatal cholinergic interneurons. Neurobiol. Dis. 2015, 78, 146–161. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.; LaHoste, G. Rhes, the Ras homolog enriched in striatum, is reduced under conditions of dopamine supersensitivity. Neuroscience 2006, 137, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.M.; LaHoste, G.J.; Ruskin, D.N. Ontogeny and dopaminergic regulation in brain of Ras homolog enriched in striatum (Rhes). Brain Res. 2008, 1245, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Szele, F.G.; Artymyshyn, R.; Molinoff, P.B.; Chesselet, M.-F. Heterogeneous distribution of dopamine D2 receptor mRNA in the rat striatum: A quantitative analysis with in situ hybridization histochemistry. Anat. Rec. 1991, 231, 548–558. [Google Scholar] [CrossRef]
- Pinna, A.; Napolitano, F.; Pelosi, B.; Di Maio, A.; Wardas, J.; Casu, M.A.; Costa, G.; Migliarini, S.; Calabresi, P.; Pasqualetti, M.; et al. The small GTP-binding protein Rhes influences nigrostriatal-dependent motor behavior during aging. Mov. Disord. 2016, 31, 583–589. [Google Scholar] [CrossRef] [Green Version]
- Vitucci, D.; Di Giorgio, A.; Napolitano, F.; Pelosi, B.; Blasi, G.; Errico, F.; Attrotto, M.T.; Gelao, B.; Fazio, L.; Taurisano, P.; et al. Rasd2 modulates prefronto-striatal phenotypes in humans and “Schizophrenia-like behaviors” in mice. Neuropsychopharmacology 2016, 41, 916–927. [Google Scholar] [CrossRef]
- Manzano, J.; Morte, B.; Scanlan, T.S.; Bernal, J. Differential effects of triiodothyronine and the thyroid hormone receptor beta-specific agonist GC-1 on thyroid hormone target genes in the b ain. Endocrinology 2003, 144, 5480–5487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallortigara, J.; Alfos, S.; Micheau, J.; Higueret, P.; Enderlin, V. T3 administration in adult hypothyroid mice modulates expression of proteins involved in striatal synaptic plasticity and improves motor behavior. Neurobiol. Dis. 2008, 31, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Vargiu, P.; Morte, B.; Manzano, J.; Perez, J.; De Abajo, R.; Sutcliffe, J.G.; Bernal, J. Thyroid hormone regulation of rhes, a novel Ras homolog gene expressed in the striatum. Brain Res. Mol. Brain Res. 2001, 94, 1–8. [Google Scholar] [CrossRef]
- Napolitano, F.; Warren, E.B.; Migliarini, S.; Punzo, D.; Errico, F.; Li, Q.; Thiolat, M.-L.; Vescovi, A.L.; Calabresi, P.; Bezard, E.; et al. Decreased Rhes mRNA levels in the brain of patients with Parkinson’s disease and MPTP-treated macaques. PLoS ONE 2017, 12, e0181677. [Google Scholar] [CrossRef] [Green Version]
- Harrison, L.M.; He, Y. Rhes and AGS1/Dexras1 affect signaling by dopamine D1 receptors through adenylyl cyclase. J. Neurosci. Res. 2011, 89, 874–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapliyal, A.; Bannister, R.A.; Hanks, C.; Adams, B.A. The monomeric G proteins AGS1 and Rhes selectively influence Galphai-dependent signaling to modulate N-type (CaV2.2) calcium channels. Am. J. Physiol. Cell Physiol. 2008, 295, C1417–C1426. [Google Scholar] [CrossRef]
- Shahani, N.; Swarnkar, S.; Giovinazzo, V.; Morgenweck, J.; Bohn, L.M.; Scharager-Tapia, C.; Pascal, B.; Martinez-Acedo, P.; Khare, K.; Subramaniam, S. RasGRP1 promotes amphetamine-induced motor behavior through a Rhes interaction network (“Rhesactome”) in the striatum. Sci. Signal. 2016, 9, ra111. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, F.; De Rosa, A.; Russo, R.; Di Maio, A.; Garofalo, M.; Federici, M.; Migliarini, S.; LeDonne, A.; Rizzo, F.R.; Avallone, L.; et al. The striatal-enriched protein Rhes is a critical modulator of cocaine-induced molecular and behavioral responses. Sci. Rep. 2019, 9, 15294. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, F.; D’Angelo, L.; de Girolamo, P.; Avallone, L.; de Lange, P.; Usiello, A. The thyroid hormone-target gene Rhes a novel crossroad for neurological and psychiatric disorders: New insights from animal models. Neuroscience 2018, 384, 419–428. [Google Scholar] [CrossRef]
- Bang, S.; Steenstra, C.; Kim, S.F. Striatum specific protein, Rhes regulates AKT pathway. Neurosci. Lett. 2012, 521, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Harrison, L.; Muller, S.; Spano, D. Effects of the Ras homolog Rhes on Akt/protein kinase B and glycogen synthase kinase 3 phosphorylation in striatum. Neuroscience 2013, 236, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, J.-M.; Gainetdinov, R.; Caron, M.G. The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol. Sci. 2007, 28, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.A.; Henley, J.M. Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 2010, 428, 133–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, D.B.; Zanella, C.A.; Henley, J.M.; Cimarosti, H. Sumoylation: Implications for Neurodegenerative Diseases. Adv. Exp. Med. Biol. 2017, 963, 261–281. [Google Scholar]
- Subramaniam, S.; Napolitano, F.; Mealer, R.G.; Kim, S.; Errico, F.; Barrow, R.; Shahani, N.; Tyagi, R.; Snyder, S.H.; Usiello, A. Rhes, a striatal-enriched small G protein, mediates mTOR signaling and L-DOPA-induced dyskinesia. Nat. Neurosci. 2011, 15, 191–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, S.; Sixt, K.M.; Barrow, R.; Snyder, S.H. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 2009, 324, 1327–1330. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Palacino, J. A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. FASEB J. 2013, 27, 1820–1829. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, S.; Mealer, R.G.; Sixt, K.M.; Barrow, R.K.; Usiello, A.; Snyder, S.H. Rhes, a physiologic regulator of sumoylation, enhances cross-sumoylation between the basic sumoylation enzymes E1 and Ubc9. J. Biol. Chem. 2010, 285, 20428–20432. [Google Scholar] [CrossRef] [Green Version]
- Mealer, R.G.; Subramaniam, S.; Snyder, S.H. Rhes deletion is neuroprotective in the 3-nitropropionic acid model of Huntington’s disease. J. Neurosci. 2013, 33, 4206–4210. [Google Scholar] [CrossRef] [PubMed]
- Baiamonte, B.A.; Lee, F.A.; Brewer, S.T.; Spano, D.; LaHoste, G.J. Attenuation of Rhes activity significantly delays the appearance of behavioral symptoms in a mouse model of Huntington’s disease. PLoS ONE 2013, 8, e53606. [Google Scholar] [CrossRef] [Green Version]
- Swarnkar, S.; Chen, Y.; Pryor, W.M.; Shahani, N.; Page, D.T.; Subramaniam, S. Ectopic expression of the striatal-enriched GTPase Rhes elicits cerebellar degeneration and an ataxia phenotype in Huntington’s disease. Neurobiol. Dis. 2015, 82, 66–77. [Google Scholar] [CrossRef]
- Liu, Q.; Cheng, S.; Yang, H.; Zhu, L.; Pan, Y.; Jing, L.; Tang, B.; Li, S.; Li, X.-J. Loss of Hap1 selectively promotes striatal degeneration in Huntington disease mice. Proc. Natl. Acad. Sci. USA 2020, 117, 20265–20273. [Google Scholar] [CrossRef]
- Sbodio, J.I.; Paul, B.D.; Machamer, C.E.; Snyder, S.H. Golgi protein ACBD3 mediates neurotoxicity associated with Huntington’s disease. Cell Rep. 2013, 4, 890–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Subramaniam, S. Rhes travels from cell to cell and transports Huntington disease protein via TNT-like protrusion. J. Cell. Biol. 2019, 218, 1972–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decressac, M.; Bjorklund, A. mTOR inhibition alleviates L-DOPA-induced dyskinesia in parkinsonian rats. J. Parkinsons Dis. 2013, 3, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Heiman, M.; Greengard, P.; Valjent, E.; Fisone, G. Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2009, 2, ra36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obeso, J.A.; Stamelou, M.; Goetz, C.G.; Poewe, W.; Lang, A.E.; Weintraub, D.; Burn, D.; Halliday, G.M.; Bezard, E.; Przedborski, S.; et al. Past, present, and future of Parkinson’s disease: A special essay on the 200th anniversary of the Shaking Palsy. Mov. Disord. 2017, 32, 1264–1310. [Google Scholar] [CrossRef]
- Calabresi, P.; Di Filippo, M.; Ghiglieri, V.; Tambasco, N.; Picconi, B. Levodopa-induced dyskinesias in patients with Parkinson’s disease: Filling the bench-to-bedside gap. Lancet Neurol. 2010, 9, 1106–1117. [Google Scholar] [CrossRef]
- Cenci, M.A. Dopamine dysregulation of movement control in L-DOPA-induced dyskinesia. Trends Neurosci. 2007, 30, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Stern, M.B.; Sethi, K. The scientific and clinical basis for the treatment of Parkinson disease. Neurology 2009, 72, S1–S136. [Google Scholar] [CrossRef] [Green Version]
- Picconi, B.; Centonze, D.; Håkansson, K.; Bernardi, G.; Greengard, P.; Fisone, G.; Cenci, M.A.; Calabresi, P. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat. Neurosci. 2003, 6, 501–506. [Google Scholar] [CrossRef]
- Jenner, P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat. Rev. Neurosci. 2008, 9, 665–677. [Google Scholar] [CrossRef]
- Subramaniam, S.; Snyder, S.H. Huntington’s disease is a disorder of the corpus striatum: Focus on Rhes (Ras homologue enriched in the striatum). Neuropharmacology 2011, 60, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Brugnoli, A.; Napolitano, F.; Usiello, A.; Morari, M. Genetic deletion of Rhes or pharmacological blockade of mTORC1 prevent striato-nigral neurons activation in levodopa-induced dyskinesia. Neurobiol. Dis. 2016, 85, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Eshraghi, M.; Ramírez-Jarquín, U.N.; Shahani, N.; Nuzzo, T.; De Rosa, A.; Swarnkar, S.; Galli, N.; Rivera, O.; Tsaprailis, G.; Scharager-Tapia, C.; et al. RasGRP1 is a causal factor in the development of l-DOPA-induced dyskinesia in Parkinson’s disease. Sci. Adv. 2020, 6, eaaz7001. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; Hastings, T.G. Biomedicine. Parkinson’s–Divergent causes, convergent mechanisms. Science 2004, 304, 1120–1122. [Google Scholar] [CrossRef]
- Schapira, A.H.; Jenner, P. Etiology and pathogenesis of Parkinson’s disease. Mov. Disord. 2011, 26, 1049–1055. [Google Scholar] [CrossRef]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [Green Version]
- Dhandapani, K.M.; Brann, D.W. Protective effects of estrogen and selective estrogen receptor modulators in the brain. Biol. Reprod. 2002, 67, 1379–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillies, G.E.; Pienaar, I.S.; Vohra, S.; Qamhawi, Z. Sex differences in Parkinson’s disease. Front. Neuroendocrinol. 2014, 35, 370–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartels, T.; De Schepper, S.; Hong, S. Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 2020, 370, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Henry, V.; Paillé, V.; Lelan, F.; Brachet, P.; Damier, P. Kinetics of microglial activation and degeneration of dopamine-containing neurons in a rat model of Parkinson disease induced by 6-hydroxydopamine. J. Neuropathol. Exp. Neurol. 2009, 68, 1092–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.-K.; Ischiropoulos, H.; Przedborski, S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. 2002, 22, 1763–1771. [Google Scholar] [CrossRef]
- Costa, G.; Pinna, A.; Porceddu, P.F.; Casu, M.A.; Di Maio, A.; Napolitano, F.; Usiello, A.; Morelli, M. Rhes counteracts dopamine neuron degeneration and neuroinflammation depending on gender and age. Front. Aging Neurosci. 2018, 10, 163. [Google Scholar] [CrossRef] [Green Version]
- Barrett, S.P.; Darredeau, C.; Pihl, R.O. Patterns of simultaneous polysubstance use in drug using university students. Hum. Psychopharmacol. 2006, 21, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Simola, N.; Costa, G.; De Luca, M.A.; Piras, G.; Marongiu, J.; Fattore, L. Neuronal and peripheral damages induced by synthetic psychoactive substances: An update of recent findings from human and animal studies. Neural Regen. Res. 2020, 15, 802–816. [Google Scholar] [CrossRef]
- Strote, J.; Lee, J.E.; Wechsler, H. Increasing MDMA use among college students: Results of a national survey. J. Adolesc. Health 2002, 30, 64–72. [Google Scholar] [CrossRef]
- Baumann, M.H.; Wang, X.; Rothman, R.B. 3,4-Methylenedioxymethamphetamine (MDMA) neurotoxicity in rats: A reappraisal of past and present findings. Psychopharmacology 2007, 189, 407–424. [Google Scholar] [CrossRef] [Green Version]
- Easton, N.; Marsden, C.A. Ecstasy: Are animal data consistent between species and can they translate to humans? J. Psychopharmacol. 2006, 20, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Gudelsky, G.A.; Yamamoto, B.K. Actions of 3,4-methylenedioxymethamphetamine (MDMA) on cerebral dopaminergic, serotonergic and cholinergic neurons. Pharmacol. Biochem. Behav. 2008, 90, 198–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrott, A.C. Human psychopharmacology of Ecstasy (MDMA): A review of 15 years of empirical research. Hum. Psychopharmacol. 2001, 16, 557–577. [Google Scholar] [CrossRef]
- Costa, G.; Morelli, M.; Simola, N. Progression and persistence of neurotoxicity induced by MDMA in dopaminergic regions of the mouse brain and association with noradrenergic, GABAergic, and serotonergic damage. Neurotox. Res. 2017, 32, 563–574. [Google Scholar] [CrossRef]
- Frau, L.; Simola, N.; Porceddu, P.F.; Morelli, M. Effect of crowding, temperature and age on glia activation and dopaminergic neurotoxicity induced by MDMA in the mouse brain. Neurotoxicology 2016, 56, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Granado, N.; O′shea, E.; Bove, J.; Vila, M.; Colado, M.I.; Moratalla, R. Persistent MDMA-induced dopaminergic neurotoxicity in the striatum and substantia nigra of mice. J. Neurochem. 2008, 107, 1102–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moratalla, R.; Khairnar, A.; Simola, N.; Granado, N.; García-Montes, J.R.; Porceddu, P.F.; Tizabi, Y.; Costa, G.; Morelli, M. Amphetamine-related drugs neurotoxicity in humans and in experimental animals: Main mechanisms. Prog. Neurobiol. 2017, 155, 149–170. [Google Scholar] [CrossRef] [Green Version]
- Costa, G.; Porceddu, P.F.; Serra, M.; Casu, M.A.; Schiano, V.; Napolitano, F.; Pinna, A.; Usiello, A.; Morelli, M. Lack of Rhes increases MDMA-induced neuroinflammation and dopamine neuron degeneration: Role of gender and age. Int. J. Mol. Sci. 2019, 20, 1556. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, M.C.; Grosso, R.A.; Fader, C.M. Hallmarks of aging: An autophagic perspective. Front. Endocrinol. 2018, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Klionsky, D.J. Autophagy and human disease. Cell Cycle 2007, 6, 1837–1849. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Bouhamida, E.; Danese, A.; Previati, M.; Pinton, P.; Patergnani, S. Relevance of autophagy and mitophagy dynamics and markers in neurodegenerative diseases. Biomedicines 2021, 9, 149. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [Green Version]
- Dehay, B.; Martinez-Vicente, M.; Caldwell, G.A.; Caldwell, K.A.; Yue, Z.; Cookson, M.; Klein, C.; Vila, M.; Bezard, E. Lysosomal impairment in Parkinson’s disease. Mov. Disord. 2013, 28, 725–732. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Lin, K.-J.; Chen, S.-D.; Liou, C.-W.; Chuang, Y.-C.; Lin, H.-Y.; Lin, T.-K. The overcrowded crossroads: Mitochondria, alpha-synuclein, and the endo-lysosomal system interaction in Parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 5312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Scorziello, A.; Borzacchiello, D.; Sisalli, M.J.; Di Martino, R.; Morelli, M.; Feliciello, A. Mitochondrial homeostasis and signaling in Parkinson’s disease. Front. Aging Neurosci. 2020, 12, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, S. Rhes tunnels: A radical new way of communication in the brain’s striatum? Bioessays 2020, 42, e1900231. [Google Scholar] [CrossRef] [PubMed]
- Mealer, R.G.; Murray, A.J.; Shahani, N.; Subramaniam, S.; Snyder, S.H. Rhes, a striatal-selective protein implicated in Huntington disease, binds beclin-1 and activates autophagy. J. Biol. Chem. 2014, 289, 3547–3554. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serra, M.; Pinna, A.; Costa, G.; Usiello, A.; Pasqualetti, M.; Avallone, L.; Morelli, M.; Napolitano, F. Involvement of the Protein Ras Homolog Enriched in the Striatum, Rhes, in Dopaminergic Neurons’ Degeneration: Link to Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 5326. https://doi.org/10.3390/ijms22105326
Serra M, Pinna A, Costa G, Usiello A, Pasqualetti M, Avallone L, Morelli M, Napolitano F. Involvement of the Protein Ras Homolog Enriched in the Striatum, Rhes, in Dopaminergic Neurons’ Degeneration: Link to Parkinson’s Disease. International Journal of Molecular Sciences. 2021; 22(10):5326. https://doi.org/10.3390/ijms22105326
Chicago/Turabian StyleSerra, Marcello, Annalisa Pinna, Giulia Costa, Alessandro Usiello, Massimo Pasqualetti, Luigi Avallone, Micaela Morelli, and Francesco Napolitano. 2021. "Involvement of the Protein Ras Homolog Enriched in the Striatum, Rhes, in Dopaminergic Neurons’ Degeneration: Link to Parkinson’s Disease" International Journal of Molecular Sciences 22, no. 10: 5326. https://doi.org/10.3390/ijms22105326