Abstract
Genome-wide association studies (GWAS) found locus 3p21.31 associated with severe COVID-19. CCR5 resides at the same locus and, given its known biological role in other infection diseases, we investigated if common noncoding and rare coding variants, affecting CCR5, can predispose to severe COVID-19. We combined single nucleotide polymorphisms (SNPs) that met the suggestive significance level (P ≤ 1 × 10−5) at the 3p21.31 locus in public GWAS datasets (6406 COVID-19 hospitalized patients and 902,088 controls) with gene expression data from 208 lung tissues, Hi-C, and Chip-seq data. Through whole exome sequencing (WES), we explored rare coding variants in 147 severe COVID-19 patients. We identified three SNPs (rs9845542, rs12639314, and rs35951367) associated with severe COVID-19 whose risk alleles correlated with low CCR5 expression in lung tissues. The rs35951367 resided in a CTFC binding site that interacts with CCR5 gene in lung tissues and was confirmed to be associated with severe COVID-19 in two independent datasets. We also identified a rare coding variant (rs34418657) associated with the risk of developing severe COVID-19. Our results suggest a biological role of CCR5 in the progression of COVID-19 as common and rare genetic variants can increase the risk of developing severe COVID-19 by affecting the functions of CCR5.
1. Introduction
On 11 March 2020, World Health Organization (WHO) declared COVID-19 a pandemic. From the first case registered in December 2019 in Wuhan, over 160 million cases were registered in the world with over 3 million deaths (Access date: 19 May 2021, https://COVID19.who.int). These high numbers are mainly due to the strong power of SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus-2) transmissibility through the airways via human-to-human contact or droplet transfer [1]. Common symptoms of COVID-19 are fever or chills, cough, fatigue, muscle or muscle pain, headache, and loss of taste or smell [1]. In severe cases, the infection leads to breathing difficulties with dyspnea [2], and the most serious complication is the acute respiratory distress syndrome (ARDS), which is characterized by bilateral infiltrates, severe hypoxemia, and pulmonary edema [2]. Respiratory failure represents the principal cause of hospitalization of affected patients, which leads in the most cases to intubation with consequent mechanical ventilation [2,3]. The mortality rate caused by COVID-19 increases exponentially with age, which is the first risk factor [4,5]. The other factors that influence the course of the disease are pre-existent pathologies such as hypertension, diabetes, cardiovascular disease, and respiratory diseases [4,5]. Genetic predisposition may partly explain the severity of the phenotype in some COVID-19 patients.
A recent genome-wide association study (GWAS) has identified a genetic association between severe respiratory failure in COVID-19 hospitalized patients and the chromosome 3p21.31 [6] harboring multiple genes (LC6A20, LZTFL1, CCR9, FYCO1, CXCR6, XCR1, CCR1, CCR3, CCR2 and CCR5) that could be functionally implicated in COVID-19 [6]. The genetic association of 3p21.31 locus with severe COVID-19 was recently confirmed by another independent GWAS, strengthening the role of this locus in disease susceptibility, and novel risk variants at chromosome 19p13.3, 12q24.13, and 21q22.1 were reported [7]. Another study has shown that inactivating rare mutations in genes belonging to the type I interferon pathway predispose to life-threatening COVID-19 pneumonia [8].
Once a disease-predisposing locus has been identified through a GWAS, in order to unravel the molecular basis of risk etiology, the main challenges to be faced are the identification of the causal genetic variants (i.e., those that actually contribute to the development of the disease) and the detection of the genes whose function is influenced by the same causal variants [9]. Therefore, post GWAS analyses of chromosome 3p21.31 are needed to unravel functional genetic variants and genes that may have a role in COVID-19 pathology.
The CCR5 gene, located at the same locus risk 3p21.31, encodes a chemokine receptor and is expressed by immune system cell such as T cells and macrophages [10,11,12]. The deletion of 32 bp (CCR5-Delta32, rs333) in CCR5, which presents with an allelic frequency of about 10% in the Caucasian population, reduces susceptibility in Human Immunodeficiency Virus (HIV) infection in homozygosity [13,14,15]. CCR5 is also a target of the maraviroc drug used to treat patients with HIV [16]. CCR5 seems to be of considerable importance in specific lung infections [17]; indeed, CCR5 null mice show hyperacute inflammatory response following influenza A infection and mycobacterium tuberculosis, with an increased number of T lymphocyte and dendritic cell in the lung itself [18].
Based on these observations, we reasoned that functional genetic variants at identified GWAS risk 3p21.31 locus may affect the CCR5 functions and predispose to severe disease. Most of the risk variants identified by GWASs localize to noncoding regions of DNA and exert their effects by influencing transcriptional output through multiple mechanisms. However, if CCR5 has a key role in COVID-19 pathology, it is presumable that deleterious coding variants might be risk factors for disease as well.
Here, using summary statistics from GWAS meta-analysis of severe COVID-19 combined with expression Quantitative Traits Loci (eQTLs), chromosome conformation capture (Hi-C) data, and whole exome sequencing (WES) data, we aimed at identifying causal genetic variants that can specifically affect CCR5 in order to understand the underlying mechanism of SARS-CoV-2 infection in patients with severe clinically manifestation of disease.
2. Results
2.1. CCR5 Is Highly Expressed in Lung and Bronchus
Starting from the hypothesis that CCR5 has an important role in lung infection, we have analyzed CCR5 expression in different tissues. CCR5 was shown to be highly expressed in lung in two independent gene expression datasets of normal tissues profiled by GTEx consortium using RNA-Seq (Figure 1A) and by Roth et al. [19] using microarray (Figure 1B) technology. We also observed CCR5 as highly expressed in bronchus (Figure 1B).
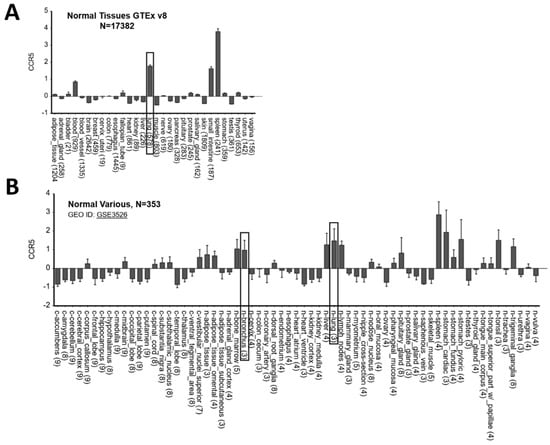
Figure 1.
CCR5 is highly expressed in lung and bronchus. (A) Gene expression data of 17,382 samples categorized in 30 different normal tissues obtained from GTEx database and (B) of 353 samples categorized in 69 different normal tissues obtained from Roth et al. study. The gray boxes highlight tissues from lung and bronchus. Data are plotted by using R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl, accessed on 10 May 2021).
2.2. CCR5 Expression Is Affected by SNPs at Chromosome Risk Locus 3p21.31
To verify if SNPs at the genome-wide associated locus 3p21.31 [6] may affect CCR5 functions, we used GWAS data of 6406 hospitalized COVID-19 patients and 902,088 controls of European origin from ‘The COVID-19 Host Genetics Initiative’ [20]. In the region on chromosome 3, which appears to be significantly associated with severe COVID-19 at the genome-wide level (https://www.covid19hg.org/results, accessed on 10 May 2021), we selected 52 SNPs with suggestive statistical significance (P ≤ 1 × 10−5) and that were eQTLs for CCR5 in lung (208 samples from GTEx database) (Supplementary Table S1). The three SNPs with the highest eQTLs p values in lung were rs9845542 (P = 4.9 × 10−6), rs12639314 (P = 6.5 × 10−6), and rs35951367 (P = 2.2 × 10−5) (Supplementary Table S1). These three SNPs are in complete linkage disequilibrium (LD) with each other in European and South Asian populations (Figure 2A, Figure S1) but not in Africans, mixed Americans, and East Asians (Figure S1).
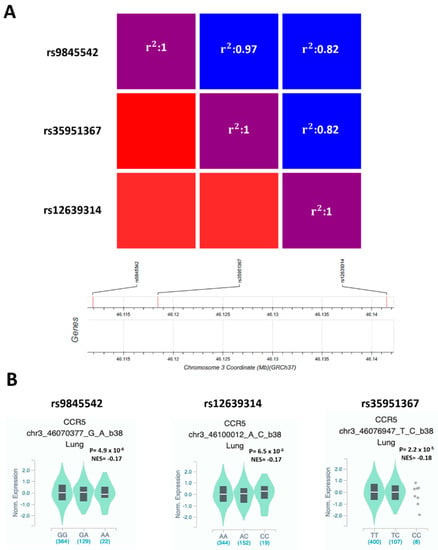
Figure 2.
rs9845542, rs12639314, and rs35951367 SNPs are in LD in the European population, and all the minor alleles are associated with low expression of CCR5 in lung. (A) Heatmap shows LD among the selected SNPs in the European population. (B) Violin plot showing the effect of eQTLs SNPs on CCR5 expression in lung. NES: normalized effect size.
In particular, the frequency of minor allele of rs35951367 varies considerably according to the analyzed population: the minor allele C is more frequent in the European populations (non-Finnish: 16.1%) than other populations of non-European origin such as African, where the frequency is 10.1% or East and South Asian, where the allele frequency drops from 4.9% to 0% (Figure S2). However, all the minor alleles were associated with increased risk of developing severe COVID-19 (rs9845542 P = 4.2 × 10−20, OR = 1.33, 95% CI: 1.25–1.41; rs12639314 P = 7.1 × 10−9, OR = 1.23, 95% CI: 1.14–1.31; rs35951367 P = 3.2 × 10−19, OR = 1.32, 95% CI: 1.24–1.40) (Supplementary Table S1) and correlated with lower expression levels of CCR5 in lung (Figure 2B).
To further investigate the potential role of the selected SNPs in regulating CCR5 expression, we used Hi-C data from 3DIV database [20] to explore the genomic interaction between the selected SNPs and our gene of interest. We compared Hi-C data of the three SNPs among all tissues present in the database (Figure 3A–C, Figure S3).
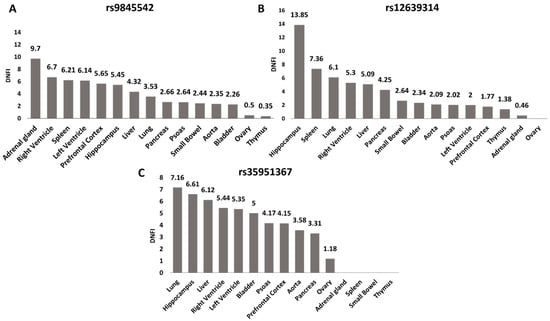
Figure 3.
rs35951367 is located in the genomic region with the highest level of interaction with CCR5 in lung. For all SNPs, the level of interaction with CCR5 as Distance Normalized Interaction Frequency (DNIF) in all tissues is shown. (A) DNIF of rs9845542; (B) DNIF of rs12639314 and (C) DNIF for rs35951367. The highest levels of interaction with CCR5 in lung were found for the (C) rs35951367 SNP. Data were retrieved from the 3DIV database (http://kobic.kr/3divv1/, accessed on 10 May 2021).
The rs35951367 SNP region showed the highest level of interaction with CCR5 than the regions of the other three SNPs with a Distance Normalized Interaction Frequency (DNFI) of 7.16 in lung (Figure 3C, Figure S3, Supplementary Table S2). Notably, in lung, the value of interaction frequency of rs35951367-associated region was the highest value observed (Figure 3, Supplementary Table S2). When we repeated the same analysis using normal lung fibroblast cell line (IMR90), we again observed the strongest interaction between rs35951367-associated region and CCR5 (Supplementary Table S3). Based on our results, we focused our attention on rs35951367, which was likely to be a regulatory SNP of CCR5.
Chip data from the ENCODE catalogue of two lung cell lines (IMR90 and NHLF) showed that rs35951367 falls in a CTCF binding region localized in the intron of XCR1 gene (Figure 4A,B).
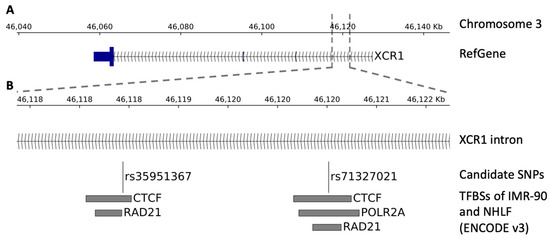
Figure 4.
Genomic view of the region surrounding the SNPs. The figure shows the transcription factor binding sites (TFBSs) within the region surrounding the SNPs. (A) Genomic view of the SNP localization. (B) Zoom-in view showing, from top to bottom, the SNP localization, the candidate SNPs, and the TFBSs of the ENCODE catalogue (v3) belonging to the IMR-90 and NHLF lung cell lines.
This SNP is an eQTL only for CCR5 in lung and not for XCR1 in lung or other tissues (Supplementary Table S4). By in-depth inspection analysis, closely located to rs35951367, we found the rs71327021 SNP that was previously excluded from our initial QTL analysis, as it did not pass the stringent levels of statistical correction. However, we verified that the rs71327021 SNP correlated with CCR5 expression with a nominal level of significance (Figure S4A). Interestingly, this SNP is in LD with our candidate rs35951367 SNP (r2 = 0.76) and in addition, it falls in binding sites of CTFC (Figure 4B). However, analysis of Hi-C data showed the strongest interaction between rs71327021-associated region and CCR5 was observed in spleen tissue and not in lung (Supplementary Table S5, Figure S4B).
2.3. Replication Studies
To confirm the association found in GWAS data for rs35951367, we conducted two replication studies. The first replication study was carried out using the genetic data from the 23andMe study [21]. In this cohort, including 1131 hospitalized COVID-19 patients, the minor allele C of rs35951367 was confirmed to be a risk factor for COVID-19 severe (OR = 1.41, P = 3.79 × 10−6); indeed, it was more frequent in cases (20%) than controls (15%). The second replication study was carried out using our Italian cohort of 202 hospitalized COVID-19 patients and 929 controls. Again, we confirmed the minor allele of rs35951367 as a risk factor for the more severe form of disease (OR = 1.307, P = 0.043) (Table 1).

Table 1.
Association of common regulatory variant rs35951367 with severe COVID-19 in the Italian cohort.
As rs35951367 showed a different allelic frequency in different populations (Figure S2), we used GWAS data of 2244 critically ill patients with COVID-19 and UK biobank controls from the GenoMICC study [7] to validate the association of rs35951367 with severe COVID-19 phenotype in non-European populations. The association was not confirmed in Africans (P = 0.43, OR = 1.24, 95% CI 0.59–1.89), East Asians (P = 0.75, OR = 1.14, 95% CI: 0.22–2.06), and South Asians (P = 0.18, OR = 1.18, 95% CI: 0.89–1.47).
2.4. Coding Variants in CCR5
In parallel with investigation of CCR5 locus in GWAS data, we conducted WES on 147 hospitalized COVID-19 patients of Southern Italy. In this WES dataset, we selected rare missense and INDEL variants (MAF < 0.01%) of CCR5. We found four variants in six patients (Table 2), and the rs34418657 variant was found in three patients (3/147, 2.04%).
Table 2.
Coding variants found in CCR5.
The minor allele of rs34418657 showed a frequency of 0.03% in European (non-Finnish) population and was predicted to be pathogenic by seven different tools (Supplementary Table S6). Using the DUET tool [22], rs34418657 was predicted to destabilize the protein (ΔΔG = −0.893 Kcal/mol), suggesting its potential role as loss of function.
Motivated by these data, an additional 74 COVID-19 hospitalized samples and 1084 controls with Italian origin were typed using TaqMan assay for the rare variant rs34418657. Combining data from WES and TaqMan assay, we found that heterozygotes are more frequent in 221 patients with severe COVID-19 than in controls (PGT = 0.027) (Table 3).
Table 3.
Association of rare coding variant rs34418657 with severe COVID-19 in the Italian cohort.
We also verified the frequency of 32 bp deletion (CCR5-Delta32, rs333), which was already associated with HIV resistance [15] using our 147 COVID-19 hospitalized patients and an Italian cohort of controls from public exomes data of 1095 Italian individuals (Network for Italian Genomes) [23]. No significant difference of frequency of CCR5-Delta32 was observed between controls (10.22%) and COVID-19 cases (10.20%), (P = 0.99; OR = 0.99, 95% CI: 0.57–1.73). To verify the association in GWAS dataset, we used the rs113341849 SNP in LD with rs333 (r2 = 0.97) and confirmed no association between CCR5-Delta32 (P = 0.11, OR = 0.92, 95% CI: 0.82–1.02).
3. Discussion
The clinical manifestations of COVID-19 are very heterogeneous; indeed, they can range from completely asymptomatic subjects up to patients who are hospitalized and undergo mechanical ventilation [1]. Numerous studies have focused on discovering the causes of this spectrum of phenotypes and on the evaluation of the presence of genetic differences in the predisposition to this pathology [24,25,26].
For example, two recent GWAS studies have demonstrated a genetic association between severe COVID-19 and the blood groups, where group O is a protective factor, and there are common variants at 3p21.31, 12q24.13, 19p13.2, 19p13.3 loci [6,7].
The 3p21.31 has been the first locus to be associated with the risk of developing aggressive forms of COVID-19 [6] and to be replicated in a different cohort of cases and controls [7]. The severe COVID-19 3p21.31 risk locus harbors several common variants and contains 17 known protein-coding genes including LC6A20, LZTFL1, CCR9, FYCO1, CXCR6, XCR1, CCR1, CCR3, CCR2, and CCR5. However, so far, causative variants and genes at this risk locus, influencing COVID-19 pathogenesis, remain to be investigated. Here, in order to study the potential biological role of the 3p21.31 risk locus, we focused our attention on the CCR5 gene based on the motivations described below.
CCR5 is a chemokine receptor and is known in the literature for its role in infection pathogenesis, especially in HIV type 1 (HIV-1), where a deletion of 32 bp, present with an allele frequency of 12% in the northern Europe population, confers resistance at the HIV-1 infection in homozygous [13,27,28,29]. A study conducted on public genomes from Neanderthals to modern humans identified a total of 262 polymorphisms in the CCR5 gene, with an SNP frequency per individual that ranged from 14 to 24 [30,31]. This high allelic heterogeneity is attributable to its role in the immune system as a result of co-evolution between humans and pathogens and is a peculiarity of only humans and not of other primates [31].
In leukocytes, CCR5 is a key receptor for chemotaxis from the bloodstream to the site of inflammation [32,33]; in particular, CCR5 is important for the rapid recruitment of memory CD8+ T cells to the lung airways during virus infection for limiting virus replication [34]. However, in addition to leukocytes, CCR5 is variably expressed in many tissues [32,35,36,37,38]. Our in silico analysis shows that it is highly expressed in the lung from healthy individuals, suggesting its key biological role in the pathogenesis of COVID-19. Additional studies to verify the expression of CCR5 in tissues from subjects with severe COVID-19 might further support its role in disease initiation and progression.
Starting from this evidence, we decided to investigate the CCR5 role in new SARS-CoV-2 infection using a dualistic approach: we first investigated the SNPs in noncoding regions, exploiting the freely available GWAS data, and then evaluated the coding variants obtained by WES of our cohort of severe cases.
We hypothesized that common variants at 3p21.31 may act as risk factors of severe COVID-19 by affecting the expression of CCR5. Combining public GWAS data with eQTLs data from the GTEx project, we have demonstrated that the minor allele of the rs35951367 SNP confers risk of severe COVID-19 and correlates with a lower expression of CCR5 in lung. In accord with our results, a preprint paper reports a transcription-wide association study that found CCR5 expression to be regulated by variants at 3p21.31 only in lung tissue [39].
Interestingly, Hi-C data obtained in lung tissue and cell lines showed that the rs35951367 SNP is located in a DNA region interacting with CCR5 and marked by CTCF binding in two lung cell lines (IMR90 and NHLF). CTCF can function as a transcriptional activator, a repressor, or an insulator protein, blocking the communication between enhancers and promoters [40]. It plays a relevant role in contributing to genome organization and gene expression [40,41]. Therefore, our data suggest that the rs35951367 SNP may functionally act by altering topological domains and thus the normal transcriptional program of the CCR5 gene. In vitro experimental studies are needed to confirm these findings and, in addition, to further genetic studies of variants already found to be associated with the different expression of CCR5 [42,43,44].
Very close to rs35951367, we found the rs71327021 SNP, which also falls into the CTCF-binding region in lung cell lines, but it is not an eQTL for CCR5 in the lungs. However, the rs71327021 SNP may be a good candidate to be tested in future genetic and functional studies.
Replication study is the golden standard for validating genome-wide association findings; thus, we used the genetic data from the 23andMe study and typed the rs35951367 SNP by TaqMan assay in cases and controls from Southern Italy; in both cohorts, the genetic association was confirmed. However, the same variant was not found to be associated with severity of COVID-19 illness in non-European subjects such as Africans and Asians, which was likely because of the lower frequency of the variant observed in these populations with respect to Europeans. So, we can speculate that this genetic risk factor may be one of the causes linked to the different rate of mortality observed across the different populations. Future studies will help to better define the effect of genetic variants at the CCR5 locus on the clinical subgroups of COVID-19 disease—for instance, performing association analyses on patients stratified by disease aggressiveness.
Together, our findings provide evidence that common functional SNPs can predispose to clinically severe COVID-19 by long-range interaction with CCR5 and that this gene can play a relevant role in the pathogenesis of SARS-CoV-2 infection.
In accordance with our findings, it was observed that the deficiency of CCR5 in West Nile virus infection, which shares with COVID-19 the multitude of phenotypes ranging from the subclinical to the most severe cases, represents a risk factor to develop severe symptoms [45]. Furthermore, in vivo studies indicate that mice CCR5−/−, infected with West Nile virus, have an elevated mortality rate and viral loads compared with wild-type mice [46].
In addition, the lung appears to be a particularly sensitive tissue to low CCR5 expression as demonstrated in studies of influenza A virus infection, where CCR5 deficiency is associated with poor outcome and with an increased mortality [18,47]. Notably, null mice of CCR5 show an accelerated macrophage accumulation in lungs with an increased expression of RANTES, MCP-1, and IP-10 [18]. The latter is one of three pro-inflammatory cytokines that correlate with rapid disease progression in hospitalized COVID-19 patients [48]. The above-mentioned scientific evidence is in agreement with our findings according to which a reduced expression of CCR5 in the lungs can promote COVID-19 progression.
Since CCR5 is known in the literature for its numerous polymorphisms, we decided to also investigate if there are rare pathogenic coding variants associated with the severe COVID-19 phenotype. In our cohort of hospitalized patients profiled by WES, we found an association of a rare variant in exon 3 (rs34418657) predicted to destabilize the protein product. This variant has been already described in association study with unexplained recurrent pregnancy loss. This association was explained, as CCR5 is necessary for maternal–fetal tolerance, and rs34418657 was predicted to destabilize the protein [49]. Our observations further suggest that genetic alterations that reduce the CCR5 function predispose to COVID-19 progression and highlight once again the key role of CCR5 in SARS-CoV-2 infection.
Conversely, the CCR5-Delta32 (rs333) common coding variants, known to protect from HIV infection [50], seem to be not associated with a minor risk of developing severe forms of COVID-19, as indicated by both our WES and GWAS results. These data provide evidence that rare and common coding variants of CCR5 can have different and independent risk effects on the onset and progression of infectious diseases.
In conclusion, we demonstrate that common noncoding regulatory variants at the previously identified 3p21.31 risk locus and rare pathogenic coding variants in CCR5 can contribute to severe clinical manifestations of COVID-19 by affecting CCR5 functions. Therefore, these findings suggest that CCR5 is a good candidate to be experimentally studied for unravelling the biological basis of COVID-19 progression and for developing novel therapeutic interventions.
4. Materials and Methods
4.1. GWAS Data
Summary statistics (P-value, and effect size) were obtained from the GWAS dataset “B2_ALL_eur_leave_23andme”, deposited in the fourth round of GWAS meta-analysis of COVID-19 Host Genetics Initiative website (www.COVID19hg.org, accessed on 10 May 2021) [51], available since 20 October 2020. The GWAS dataset includes 6406 hospitalized COVID-19 patients and 902,088 members of the general population with European genetic ancestry and does not include 23andMe cohort. The COVID-19 Host Genetics Initiative was a global initiative to elucidate the role of host genetic factors in the susceptibility and severity of the SARS-CoV-2 virus pandemic.
4.2. Replication Study of rs35951367 SNP
To validate the association between rs35951367 and a severe form of COVID-19 disease, we use two independent cohorts of cases and controls.
The first cohort data were obtained from ‘23andMe COVID-19 Study’ [21]. This study includes 1131 COVID-19 patients reported to have a positive test and hospitalization following SARS-CoV-2 infection. Participants answered web questions about symptoms of cold or flu-like illnesses, COVID-19 diagnosis and testing, hospitalization, and severity of illness. Participants provided informed consent and participated in the research online, under a protocol approved by the external AAHRPP-accredited IRB, Ethical & Independent Review Services (E&I Review). Participants were included in the analysis on the basis of consent status as checked at the time that data analyses were initiated. The full GWAS summary statistics for the 23andMe discovery dataset will be made available through 23andMe to qualified researchers under an agreement with 23andMe that protects the privacy of the 23andMe participants. Please visit https://research.23andme.com/collaborate/#dataset-access/ (accessed on 10 May 2021) for more information and to apply to access the data.
The second cohort included 929 healthy controls and 212 hospitalized COVID-19 collected from our research group that was genotyped by TaqMan® SNP Genotyping Assay for rs34418657 SNP (Applied Biosystems by Thermo Fisher Scientific, Waltham, MA, USA). Twenty DNA samples failed the genotyping. The healthy controls cohort was composed of 39 subjects that resulted to be positive for anti-SARS-CoV2-IgG antibodies but without any symptoms and 890 healthy controls recruited before the COVID-19 pandemic. Hardy–Weinberg equilibrium was evaluated using the goodness-of-fit chi-square test in control and cases subjects (P > 0.05). Clinical characteristics of cases and controls are reported in Supplementary Table S7.
Exomes of 1095 Italian individuals were retrieved from the web database Network for Italian Genomes (NIG) (http://nigdb.cineca.it/index.php, accessed on 10 May 2021) [23] for evaluating the association of rs333 in our cohort of patients.
4.3. In Silico Analysis
Allele and genotype frequencies were obtained from gnomAD (https://gnomad.broadinstitute.org/, accessed on 10 May 2021) [47]; eQTLs analysis was performed by using public data from Genotype-Tissue Expression (GTEx) Portal (https://gtexportal.org/home, accessed on 10 May 2021). ChIP data of transcription factor in two lung cell lines (IMR90 and NHLF) was obtained from ENCODE catalogue [48,49]. CCR5 gene expression was obtained from NCBI Gene Expression Omnibus (GEO) Database [50] and plotted using R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl, accessed on 10 May 2021). 3DIV was used for the analysis of Hi-C data (http://kobic.kr/3divv1/, accessed on 10 May 2021) [21]. DUET was used to evaluate effect of missense variants on CCR5 protein (http://biosig.unimelb.edu.au/duet/stability, accessed on 10 May 2021) [22].
4.4. Whole Exome Sequencing
We collected COVID-19 samples from several hospitals in Southern Italy: Azienda ospedaliera specialistica dei colli, Monaldi-Cotugno; Azienda Ospedaliera di Rilievo Nazionale Antonio Cardarelli, P.O. Ospedale Boscotrecase, Villa dei Fiori S.r.l.- Acerra. The genomic DNA of COVID-19 patients was extracted from peripheral blood using a Maxwell® RSC Blood DNA Kit (Promega, Madison, WI, USA), and DNA concentration and purity were evaluated using a NanoDrop™ 8000 Spectrophotometer. For library preparation, we measured the DNA concentration of 147 COVID-19 patients with a Qubit® DNA Assay Kit in Qubit® 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA), and 1.0 μg of DNA was used for library preparation. Genomic DNA was sonicated to generate 180–250 bp fragments and then end-polished, A-tailed, and ligated with the full-length adapter with further PCR amplification. PCR products were purified using AMPure XP system (Beckman Coulter, Brea, CA, USA) and quantified using the Agilent high sensitivity DNA assay on the Agilent Bioanalyzer 2100 system. The whole exome was captured with Agilent SureSelect Human All Exome V6 (Agilent Tecnologies, Santa Clara, CA, USA), and the sequencing was performed on an Illumina NovaSeq 6000 Systems.
4.5. Bioinformatic Analysis of Sequencing Data
The paired end (2 × 150 bp) sequencing returned an average of 42 million raw reads per sample. After removing sequencing artifacts, we retained, on average, the 98.35% of raw reads. The percentage of bases with quality scores above 20 and above 30 (Q20 and Q30) was 97.7% and 93.8%, respectively. The cleaned reads were aligned versus the reference genome (GRCh37/hg19) and mapping BAM files were obtained with BWA-mem (version 0.7.17) and SAMTools (version 1.8). On average, 99.89% of reads were mapped and 20.47% of duplicate reads were removed with Picard (version 2.18.9). We covered 99.63% of the target regions, and the average sequencing depth on target was 149.06x. Most (95.08%) of the target regions were covered with at least 20 reads, which were sufficient for reliable variant calls. SNVs and small insertions and deletions (INDELs) were detected with GATK HaplotypeCaller, and the functional annotation of variants was performed with ANNOVAR. We obtained 189,625 raw SNVs and 32,089 raw INDELs per sample. We excluded off-target variants (e.g., intergenic, intronic, etc.) and single nucleotide polymorphisms (SNPs) with allele frequencies greater than 1% in non-Finnish European populations of the 1000 Genomes Project, ExAC (v3) and GnomAD (v2.1.1) databases. To remove possible false positives, we also discarded variants falling in genomic duplicated regions. Then, the set of exonic variants was filtered to remove synonymous SNVs.
4.6. Association Study of the Coding Rare Variant rs34418657
In order to assess the association of the rare variant rs34418657, identified through WES, we genotyped by TaqMan® SNP Genotyping Assay (Applied Biosystems by Thermo Fisher Scientific, Waltham, MA, USA) 1084 healthy controls and an additional 74 COVID-19 hospitalized patients not analyzed by WES. Fifty controls and one COVID-19 patient failed the genotyping. Healthy controls cohort was composed of 379 people that were shown to be positive for anti-SARS-CoV2-IgG antibodies but without any symptoms and 705 healthy controls recruited before the COVID-19 pandemic. A higher number of healthy controls with respect to common variant rs35951367 was typed in order to gain more statistical power to detect significant genetic differences since the rs34418657 variant is very rare in the general population. Hardy–Weinberg equilibrium was evaluated using the goodness-of-fit chi-square test in control and case subjects (P > 0.05). Clinical characteristics of this large cohort of controls are also reported in Supplementary Table S7.
DNA samples from patients were obtained after they signed informed consent and according to the Declaration of Helsinki. Local university ethical committees approved the study.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/ijms22105372/s1. Figure S1: Top CCR5 eQTLs SNPs in lung are not in LD in African and AMR populations. Figure S2: Minor allele frequencies of the selected SNPs in different populations. Figure S3: rs35951367 shows the highest level of interaction with CCR5 in lung. Figure S4: Minor allele of rs71327021 was associated with low expression of CCR5 in lung, but this last one is not tissue, where interaction with CCR5 is stronger. Table S1: eQTL SNPs for CCR5 in lung. Table S2: Results of analysis of Hi-C data of rs35951367 region in lung. Table S3: Results of analysis of Hi-C data for rs35951367 region in lung cell line (IMR90). Table S4: Results of eQTLs analysis of rs35951367 in all tissues. Table S5: Results of the analysis of Hi-C data for the rs71327021 region in different normal tissues. Table S6: Rare coding variants of the CCR5 gene in 147 COVID-19 hospitalized cases. Table S7: Clinical characteristics of COVID-19 hospitalized patients and healthy controls.
Author Contributions
S.C. and M.C. designed and conducted the study and prepared the manuscript; V.A.L. and G.D. analyzed the data; R.R., I.A. and B.E.R. sampled genomic DNA from COVID-19 patients; S.C. genotyped COVID-19 patients and in-house controls; G.F. (Giulia Frisso), P.A., G.M.C., G.S., I.G., C.P., M.D.M., G.F. (Giuseppe Fiorentino), G.R., P.C., C.B. and B.P. cared for COVID-19 patients, A.I. and M.Z. provided a critical review of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the project “CEINGE TASK-FORCE COVID19”, code D64I200003800 by Regione Campania for the fight against Covid-19 (DGR n. 140 del 17 Marzo 2020).
Institutional Review Board Statement
Local university ethical committees approved the study.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The GWAS dataset ‘B2_ALL_eur_leave_23andme’ is accessible at www.COVID19hg.org (accessed on 10 May 2021). The full GWAS summary statistics for the 23andMe discovery dataset will be made available through 23andMe to qualified researchers under an agreement with 23andMe that protects the privacy of the 23andMe participants at https://research.23andme.com/collaborate/#dataset-access/ (accessed on 10 May 2021) for more information and to apply to access the data. Exomes of healthy Italian individuals are accessible via the database Network for Italian Genomes (NIG) (http://nigdb.cineca.it/index.php, accessed on 20 March 2021).
Acknowledgments
We would like to thank the research participants and employees of 23andMe for making this work possible. We are grateful to Roberta Campochiaro for helping with the data management.
Conflicts of Interest
The authors declare that there are no competing interests.
References
- Baloch, S.; Baloch, M.A.; Zheng, T.; Pei, X. The Coronavirus Disease 2019 (COVID-19) Pandemic. Tohoku J. Exp. Med. 2020, 250, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Berlin, D.A.; Gulick, R.M.; Martinez, F.J. Severe Covid-19. N. Engl. J. Med. 2020, 383, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Bhatraju, P.K.; Ghassemieh, B.J.; Nichols, M.; Kim, R.; Jerome, K.R.; Nalla, A.K.; Greninger, A.L.; Pipavath, S.; Wurfel, M.M.; Evans, L.; et al. Covid-19 in Critically Ill Patients in the Seattle Region—Case Series. N. Engl. J. Med. 2020, 382, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Peng, F.; Xu, B.; Zhao, J.; Liu, H.; Peng, J.; Li, Q.; Jiang, C.; Zhou, Y.; Liu, S.; et al. Risk factors of critical & mortal COVID-19 cases: A systematic literature review and meta-analysis. J. Infect. 2020, 81, e16–e25. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernandez, J.; Prati, D.; Baselli, G.; Asselta, R.; et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370. [Google Scholar] [CrossRef] [PubMed]
- Freedman, M.L.; Monteiro, A.N.; Gayther, S.A.; Coetzee, G.A.; Risch, A.; Plass, C.; Casey, G.; De Biasi, M.; Carlson, C.; Duggan, D.; et al. Principles for the post-GWAS functional characterization of cancer risk loci. Nat. Genet. 2011, 43, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Gornalusse, G.G.; Mummidi, S.; Gaitan, A.A.; Jimenez, F.; Ramsuran, V.; Picton, A.; Rogers, K.; Manoharan, M.S.; Avadhanam, N.; Murthy, K.K.; et al. Epigenetic mechanisms, T-cell activation, and CCR5 genetics interact to regulate T-cell expression of CCR5, the major HIV-1 coreceptor. Proc. Natl. Acad. Sci. USA 2015, 112, E4762–E4771. [Google Scholar] [CrossRef]
- Bleul, C.C.; Wu, L.; Hoxie, J.A.; Springer, T.A.; Mackay, C.R. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 1925–1930. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, Z.R.; Christmann, B.S.; Stafford, J.D.; Moran, J.M.; Buller, R.M.L.; Corbett, J.A. CCR5 is a required signaling receptor for macrophage expression of inflammatory genes in response to viral double-stranded RNA. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 316, R525–R534. [Google Scholar] [CrossRef] [PubMed]
- Solloch, U.V.; Lang, K.; Lange, V.; Bohme, I.; Schmidt, A.H.; Sauter, J. Frequencies of gene variant CCR5-Delta32 in 87 countries based on next-generation sequencing of 1.3 million individuals sampled from 3 national DKMS donor centers. Hum. Immunol. 2017, 78, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Paxton, W.A.; Wolinsky, S.M.; Neumann, A.U.; Zhang, L.; He, T.; Kang, S.; Ceradini, D.; Jin, Z.; Yazdanbakhsh, K.; et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat. Med. 1996, 2, 1240–1243. [Google Scholar] [CrossRef]
- Ni, J.; Wang, D.; Wang, S. The CCR5-Delta32 Genetic Polymorphism and HIV-1 Infection Susceptibility: A Meta-analysis. Open Med. (Wars.) 2018, 13, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Woollard, S.M.; Kanmogne, G.D. Maraviroc: A review of its use in HIV infection and beyond. Drug Des. Dev. Ther. 2015, 9, 5447–5468. [Google Scholar] [CrossRef]
- Algood, H.M.; Flynn, J.L. CCR5-deficient mice control Mycobacterium tuberculosis infection despite increased pulmonary lymphocytic infiltration. J. Immunol. 2004, 173, 3287–3296. [Google Scholar] [CrossRef]
- Dawson, T.C.; Beck, M.A.; Kuziel, W.A.; Henderson, F.; Maeda, N. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am. J. Pathol. 2000, 156, 1951–1959. [Google Scholar] [CrossRef]
- Roth, R.B.; Hevezi, P.; Lee, J.; Willhite, D.; Lechner, S.M.; Foster, A.C.; Zlotnik, A. Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics 2006, 7, 67–80. [Google Scholar] [CrossRef]
- Yang, D.; Jang, I.; Choi, J.; Kim, M.S.; Lee, A.J.; Kim, H.; Eom, J.; Kim, D.; Jung, I.; Lee, B. 3DIV: A 3D-genome Interaction Viewer and database. Nucleic Acids Res. 2018, 46, D52–D57. [Google Scholar] [CrossRef]
- Shelton, J.F.; Shastri, A.J.; Ye, C.; Weldon, C.H.; Filshtein-Sonmez, T.; Coker, D.; Symons, A.; Esparza-Gordillo, J.; The 23andMe COVID-19 Team; Aslibekyan, S.; et al. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat. Genet. 2021. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef] [PubMed]
- Daga, S.; Fallerini, C.; Baldassarri, M.; Fava, F.; Valentino, F.; Doddato, G.; Benetti, E.; Furini, S.; Giliberti, A.; Tita, R.; et al. Employing a systematic approach to biobanking and analyzing clinical and genetic data for advancing COVID-19 research. Eur. J. Hum. Genet. 2021. [Google Scholar] [CrossRef] [PubMed]
- LoPresti, M.; Beck, D.B.; Duggal, P.; Cummings, D.A.T.; Solomon, B.D. The Role of Host Genetic Factors in Coronavirus Susceptibility: Review of Animal and Systematic Review of Human Literature. Am. J. Hum. Genet. 2020, 107, 381–402. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; Andolfo, I.; Lasorsa, V.A.; Iolascon, A.; Capasso, M. Genetic Analysis of the Coronavirus SARS-CoV-2 Host Protease TMPRSS2 in Different Populations. Front. Genet. 2020, 11, 872. [Google Scholar] [CrossRef]
- Andolfo, I.; Russo, R.; Lasorsa, A.V.; Cantalupo, S.; Rosato, B.E.; Bonfiglio, F.; Frisso, G.; Pasquale, A.; Cassese, G.M.; Servillo, G.; et al. Common variants at 21q22.3 locus influence MX1 gene expression and susceptibility to severe COVID-19. medRxiv 2020, 24, 102322. [Google Scholar] [CrossRef]
- Lawhorn, C.; Yuferov, V.; Randesi, M.; Ho, A.; Morgello, S.; Kreek, M.J.; Levran, O. Genetic diversity and linkage disequilibrium in the chemokine receptor CCR2-CCR5 region among individuals and populations. Cytokine 2013, 64, 571–576. [Google Scholar] [CrossRef][Green Version]
- Trecarichi, E.M.; Tumbarello, M.; de Gaetano Donati, K.; Tamburrini, E.; Cauda, R.; Brahe, C.; Tiziano, F.D. Partial protective effect of CCR5-Delta 32 heterozygosity in a cohort of heterosexual Italian HIV-1 exposed uninfected individuals. AIDS Res. Ther. 2006, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Brelot, A.; Chakrabarti, L.A. CCR5 Revisited: How Mechanisms of HIV Entry Govern AIDS Pathogenesis. J. Mol. Biol. 2018, 430, 2557–2589. [Google Scholar] [CrossRef] [PubMed]
- Zeberg, H.; Paabo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 2020, 587, 610–612. [Google Scholar] [CrossRef] [PubMed]
- Hoover, K.C. Intragenus (Homo) variation in a chemokine receptor gene (CCR5). PLoS ONE 2018, 13, e0204989. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, E.J.; Boisvert, J.; Murphy, K.; Vierra, M.A.; Genovese, M.C.; Wardlaw, A.J.; Greenberg, H.B.; Hodge, M.R.; Wu, L.; Butcher, E.C.; et al. Expression of the chemokine receptors CCR4, CCR5, and CXCR3 by human tissue-infiltrating lymphocytes. Am. J. Pathol. 2002, 160, 347–355. [Google Scholar] [CrossRef]
- Glass, W.G.; Lim, J.K.; Cholera, R.; Pletnev, A.G.; Gao, J.L.; Murphy, P.M. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J. Exp. Med. 2005, 202, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Kohlmeier, J.E.; Miller, S.C.; Smith, J.; Lu, B.; Gerard, C.; Cookenham, T.; Roberts, A.D.; Woodland, D.L. The chemokine receptor CCR5 plays a key role in the early memory CD8+ T cell response to respiratory virus infections. Immunity 2008, 29, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Eitner, F.; Cui, Y.; Hudkins, K.L.; Anderson, D.M.; Schmidt, A.; Morton, W.R.; Alpers, C.E. Chemokine receptor (CCR5) expression in human kidneys and in the HIV infected macaque. Kidney Int. 1998, 54, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Dorf, M.E.; Berman, M.A.; Tanabe, S.; Heesen, M.; Luo, Y. Astrocytes express functional chemokine receptors. J. Neuroimmunol. 2000, 111, 109–121. [Google Scholar] [CrossRef]
- Singh, S.K.; Mishra, M.K.; Eltoum, I.A.; Bae, S.; Lillard, J.W., Jr.; Singh, R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci. Rep. 2018, 8, 1323. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.; Rezaie, P.; Newcombe, J.; Cuzner, M.L.; Male, D.; Woodroofe, M.N. Expression of the beta-chemokine receptors CCR2, CCR3 and CCR5 in multiple sclerosis central nervous system tissue. J. Neuroimmunol. 2000, 108, 192–200. [Google Scholar] [CrossRef]
- Pathak, G.A.; Singh, K.; Miller-Fleming, T.W.; Wendt, F.; Ehsan, N.; Hou, K.; Johnson, R.; Lu, Z.; Gopalan, S.; Dimbou, L.Y.; et al. Integrative analyses identify susceptibility genes underlying COVID-19 hospitalization. medRxiv 2020. [Google Scholar] [CrossRef]
- Kim, S.; Yu, N.K.; Kaang, B.K. CTCF as a multifunctional protein in genome regulation and gene expression. Exp. Mol. Med. 2015, 47, e166. [Google Scholar] [CrossRef]
- Phillips, J.E.; Corces, V.G. CTCF: Master weaver of the genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef] [PubMed]
- Mehlotra, R.K. Chemokine receptor gene polymorphisms and COVID-19: Could knowledge gained from HIV/AIDS be important? Infect. Genet. Evol. 2020, 85, 104512. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.H.; Zimmerman, P.A.; Guignard, F.; Kleeberger, C.A.; Leitman, S.F.; Murphy, P.M. CCR5 promoter polymorphism and HIV-1 disease progression. Multicenter AIDS Cohort Study (MACS). Lancet 1998, 352, 866–870. [Google Scholar] [CrossRef]
- Kulkarni, S.; Lied, A.; Kulkarni, V.; Rucevic, M.; Martin, M.P.; Walker-Sperling, V.; Anderson, S.K.; Ewy, R.; Singh, S.; Nguyen, H.; et al. CCR5AS lncRNA variation differentially regulates CCR5, influencing HIV disease outcome. Nat. Immunol. 2019, 20, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Glass, W.G.; McDermott, D.H.; Lim, J.K.; Lekhong, S.; Yu, S.F.; Frank, W.A.; Pape, J.; Cheshier, R.C.; Murphy, P.M. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J. Exp. Med. 2006, 203, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Durrant, D.M.; Daniels, B.P.; Pasieka, T.; Dorsey, D.; Klein, R.S. CCR5 limits cortical viral loads during West Nile virus infection of the central nervous system. J. Neuroinflammation 2015, 12, 233. [Google Scholar] [CrossRef] [PubMed]
- Falcon, A.; Cuevas, M.T.; Rodriguez-Frandsen, A.; Reyes, N.; Pozo, F.; Moreno, S.; Ledesma, J.; Martinez-Alarcon, J.; Nieto, A.; Casas, I. CCR5 deficiency predisposes to fatal outcome in influenza virus infection. J. Gen. Virol. 2015, 96, 2074–2078. [Google Scholar] [CrossRef] [PubMed]
- Laing, A.G.; Lorenc, A.; Del Molino Del Barrio, I.; Das, A.; Fish, M.; Monin, L.; Munoz-Ruiz, M.; McKenzie, D.R.; Hayday, T.S.; Francos-Quijorna, I.; et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020, 26, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Sharif, F.A.; Ashour, M.J.; Badawi, N.T.; Al-Ashi, S.F. Polymorphism in Regulatory T-cell (Treg)-Related Genes Is Associated with Unexplained Recurrent Pregnancy Loss. Am. J. Clin. Exp. Med. 2016, 4, 63–67. [Google Scholar] [CrossRef][Green Version]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Initiative, C.-H.G. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 2020, 28, 715–718. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).