
Characterization of Three Complete Mitogenomes of Flatidae (Hemiptera: Fulgoroidea) and Compositional Heterogeneity Analysis in the Planthoppers’ Mitochondrial Phylogenomics



Abstract
:1. Introduction
2. Results and Discussion
2.1. Mitogenome Organization and Base Composition
2.2. Proteins-coding Genes and Codon Usage
2.3. Transfer and Ribosomal RNA Genes
2.4. Control Region
2.5. Heterogeneous Sequence Divergence within Planthopper Mitogenomes
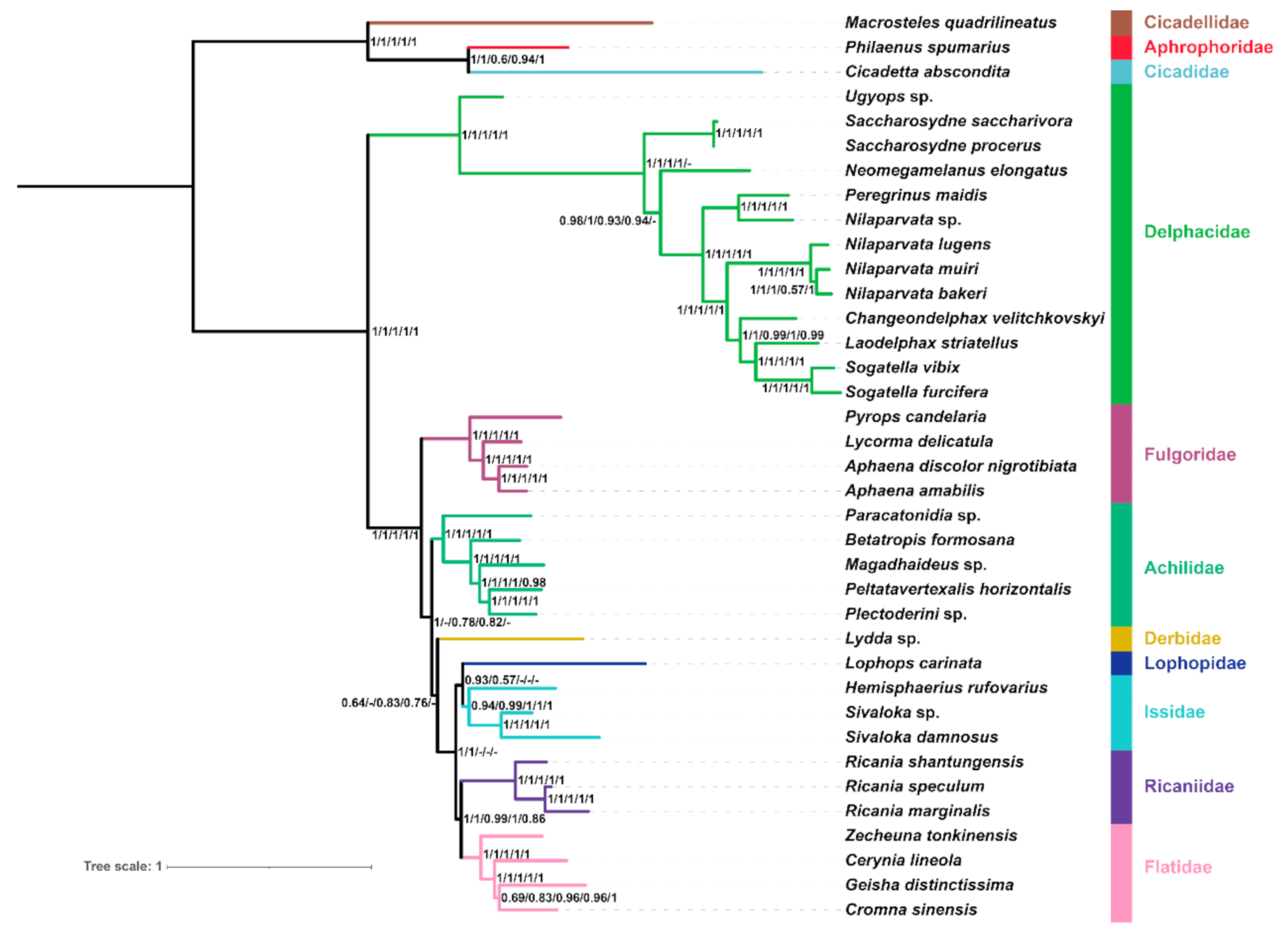
2.6. Phylogenetic Reconstruction Using Site-homogenous Models
2.7. Phylogenetic Reconstruction under Site-heterogeneous Mixture Model
2.8. Comparative Analysis of Site-homogenous and Site-heterogeneous Mixture Models
3. Materials and Methods
3.1. Sample Collection
3.2. DNA Extraction, Mitogenome Sequencing, Analysis, and Annotation
3.3. Mitogenome Sequence Alignment and Analyses of Sequence Heterogeneity
3.4. Phylogenetic Analyses Based on Site-homogeneous and Site-heterogeneous Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nick, L. Power, Sex, Suicide: Mitochondria and the Meaning of Life; Oxford University Press: Oxford, UK, 2005. [Google Scholar]
- Wolstenholme, D.R. Animal Mitochondrial DNA: Structure and Evolution. In International Review of Cytology; Wolstenholme, D.R., Jeon, K.W., Eds.; Academic Press: Cambridge, MA, USA, 1992; Volume 141, pp. 173–216. [Google Scholar]
- Bourguignon, T.; Lo, N.; Sobotnik, J.; Ho, S.Y.; Iqbal, N.; Coissac, E.; Lee, M.; Jendryka, M.M.; Sillam-Dusses, D.; Krizkova, B.; et al. Mitochondrial Phylogenomics Resolves the Global Spread of Higher Termites, Ecosystem Engineers of the Tropics. Mol. Biol. Evol. 2017, 34, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Hasegawa, H.; Cooley, J.R.; Simon, C.; Yoshimura, J.; Cai, W.; Sota, T.; Li, H. Mitochondrial Genomics Reveals Shared Phylogeographic Patterns and Demographic History among Three Periodical Cicada Species Groups. Mol. Biol. Evol. 2019, 36, 1187–1200. [Google Scholar] [CrossRef]
- Du, Z.Y.; Wu, Y.F.; Chen, Z.; Cao, L.M.; Ishikawa, T.; Kamitani, S.; Sota, T.; Song, F.; Tian, L.; Cai, W.Z.; et al. Global phylogeography and invasion history of the spotted lanternfly revealed by mitochondrial phylogenomics. Evol. Appl. 2020, 14, 915–930. [Google Scholar] [CrossRef] [PubMed]
- Bourgoin, T. FLOW (Fulgoromorpha Lists on The Web): A world knowledge base dedicated to Fulgoromorpha. Available online: https://www.hemiptera-databases.org/flow/ (accessed on 18 March 2021).
- Skinner, R.K.; Dietrich, C.H.; Walden, K.K.O.; Gordon, E.; Sweet, A.D.; Podsiadlowski, L.; Petersen, M.; Simon, C.; Takiya, D.M.; Johnson, K.P. Phylogenomics of Auchenorrhyncha (Insecta: Hemiptera) using transcriptomes: Examining controversial relationships via degeneracy coding and interrogation of gene conflict. Syst. Entomol. 2019, 45, 85–113. [Google Scholar] [CrossRef]
- Song, N.; Liang, A.P. A preliminary molecular phylogeny of planthoppers (Hemiptera: Fulgoroidea) based on nuclear and mitochondrial DNA sequences. PLoS ONE 2013, 8, e58400. [Google Scholar] [CrossRef] [Green Version]
- Urban, J.M.; Cryan, J.R. Evolution of the planthoppers (Insecta: Hemiptera: Fulgoroidea). Mol. Phylogenet. Evol. 2007, 42, 556–572. [Google Scholar] [CrossRef]
- Huang, Y.X.; Ren, F.J.; Bartlett, C.R.; Wei, Y.S.; Qin, D.Z. Contribution to the mitogenome diversity in Delphacinae: Phylogenetic and ecological implications. Genomics 2020, 112, 1363–1370. [Google Scholar] [CrossRef]
- Song, N.; Liang, A. The complete mitochondrial genome sequence of Geisha distinctissima (Hemiptera: Flatidae) and comparison with other hemipteran insects. Acta Biochim. Biophys. Sin. 2009, 41, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Huang, Y.; Bartlett, C.R.; Zhou, F.; Meng, R.; Qin, D. Characterization of the complete mitochondrial genomes of two species of the genus Aphaena Guerin-Meneville (Hemiptera: Fulgoridae) and its phylogenetic implications. Int. J. Biol. Macromol. 2019, 141, 29–40. [Google Scholar] [CrossRef]
- Xu, S.Y.; Long, J.K.; Chen, X.S. Comparative analysis of the complete mitochondrial genomes of five Achilidae species (Hemiptera: Fulgoroidea) and other Fulgoroidea reveals conserved mitochondrial genome organization. PeerJ 2019, 7, e6659. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-J.; Yang, L.; Chang, Z.-M.; Zhang, Y.-J.; Chen, X.-S. The complete mitochondrial genome of Hemisphaeriusrufovarius Walker, 1858. Mitochondrial DNA Part B 2020, 5, 2619–2620. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.W.; O’Brien, L.B. Survey of Planthopper Pests of Economically Important Plants (Homoptera: Fulgoroidea). Proceedings of 2nd International Workshop on Leafhoppers and Planthoppers of Economic Importance, Brigham Young University, Provo, UT, USA, 28 July–1 August 1986; Wilson, M.R., Nault, L.R., Eds.; CAB International Institute of Entomology: Oxfordshire, UK, 1986. [Google Scholar]
- Chou, I.; Lu, J.; Huang, J.; Wang, S. Economic Insect Fauna of China. Fasc. 36. Homoptera: Fulgoroidea; Science Press: Beijing, China, 1985; p. 152. [Google Scholar]
- Liu, Y.; Song, F.; Jiang, P.; Wilson, J.J.; Cai, W.; Li, H. Compositional heterogeneity in true bug mitochondrial phylogenomics. Mol. Phylogenet. Evol. 2018, 118, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Li, H.; Jiang, P.; Zhou, X.; Liu, J.; Sun, C.; Vogler, A.P.; Cai, W. Capturing the Phylogeny of Holometabola with Mitochondrial Genome Data and Bayesian Site-Heterogeneous Mixture Models. Genome Biol. Evol. 2016, 8, 1411–1426. [Google Scholar] [CrossRef]
- Song, N.; Zhang, H.; Zhao, T. Insights into the phylogeny of Hemiptera from increased mitogenomic taxon sampling. Mol. Phylogenet. Evol. 2019, 137, 236–249. [Google Scholar] [CrossRef]
- Cameron, S.L.; Barker, S.C.; Whiting, M.F. Mitochondrial genomics and the new insect order Mantophasmatodea. Mol. Phylogenet. Evol. 2006, 38, 274–279. [Google Scholar] [CrossRef]
- Lartillot, N.; Philippe, H. A Bayesian Mixture Model for Across-Site Heterogeneities in the Amino-Acid Replacement Process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef]
- Lartillot, N.; Lepage, T.; Blanquart, S. PhyloBayes 3: A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 2009, 25, 2286–2288. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.J.; Zhu, W.C.; Rong, X.; Zhang, Y.K.; Ding, X.L.; Liu, J.; Chen, D.S.; Du, Y.; Hong, X.Y. The complete mitochondrial genomes of two rice planthoppers, Nilaparvata lugens and Laodelphax striatellus: Conserved genome rearrangement in Delphacidae and discovery of new characteristics of atp8 and tRNA genes. BMC Genom. 2013, 14, 417. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.-J.; Shi, M.; Chen, X.-X.; Sharkey, M.J.; Van Achterberg, C.; Ye, G.-Y.; He, J.-H. New Views on Strand Asymmetry in Insect Mitochondrial Genomes. PLoS ONE 2010, 5, e12708. [Google Scholar] [CrossRef] [Green Version]
- Garey, J.R.; Wolstenholme, D.R. Platyhelminth mitochondrial DNA: Evidence for early evolutionary origin of a tRNAserAGN that contains a dihydrouridine arm replacement loop, and of serine-specifying AGA and AGG codons. J. Mol. Evol. 1989, 28, 374–387. [Google Scholar] [CrossRef]
- Song, N.; Liang, A.P.; Ma, C. The complete mitochondrial genome sequence of the planthopper, Sivaloka damnosus. J. Insect Sci. 2010, 10, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgoin, T. Female genitalia in Hemiptera Fulgoromorpha, morphological and phylogenetic data. Ann. Soc. Entomol. France 1993, 29, 225–244. [Google Scholar]
- Yeh, W.-B.; Yang, C.T.; Hui, C.F. A Molecular Phylogeny of Planthoppers (Hemiptera: Fulgoroidea) Inferred from Mitochondrial 16S rDNA Sequences. Zool. Stud. 2005, 44, 519–535. [Google Scholar]
- Li, H.; Leavengood, J.M.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P.; Liu, J.; Zhou, X.; Cai, W. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. R. Soc. B 2017, 284, 1223. [Google Scholar] [CrossRef] [PubMed]
- Medler, J.T. New concepts in classification of the Flatidae. Mitt. Schweiz. Entomol. Ges. 1984, 57, 433–434. [Google Scholar]
- Jermiin, L.S.; Ho, S.Y.W.; Ababneh, F.; Robinson, J.; Larkum, A.W.D. The Biasing Effect of Compositional Heterogeneity on Phylogenetic Estimates May be Underestimated. Syst. Biol. 2004, 53, 638–643. [Google Scholar] [CrossRef] [Green Version]
- Bourgoin, T. In A new interpretation of the homologies of the Hemiptera male genitalia illustrated by the Tettigometridae. In Proceedings of the 6th Auchenorrhyncha Meeting, Turin, Italy, 7–11 September 1987; pp. 113–120. [Google Scholar]
- Simon, A. FASTQC. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 18 June 2020).
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CG View Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kück, P.; Meid, S.A.; Groß, C.; Wägele, J.W.; Misof, B. AliGROOVE—Visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Bioinform. 2014, 15, 294. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.; Pfeiffer, W.T.; Schwartz, T. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; Institute of Electrical and Electronics Engineers (IEEE): New Orleans, LA, USA, 2010; pp. 1–8. [Google Scholar]
- Minh, B.Q.; Nguyen, M.A.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Yang, X.; Bennett, G. The complete mitochondrial genome of Macrosteles quadrilineatus (Hemiptera: Cicadellidae). Mitochondrial DNA B Resour. 2017, 2, 173–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.B.; Beckenbach, A.T. Insect mitochondrial genomics: The complete mitochondrial genome sequence of the meadow spittlebug Philaenus spumarius (Hemiptera: Auchenorrhyncha: Cercopoidae). Genome 2005, 48, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Liang, A.P. The Complete Mitochondrial Genome of Ugyops sp. (Hemiptera: Delphacidae). J. Insect Sci. 2018, 18, 25. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.X.; Qin, D.Z. The complete mitochondrial genome sequence of the corn planthopper, Peregrinus maidis (Hemiptera: Fulgoroidea). Mitochondrial DNA B Resour. 2017, 2, 783–784. [Google Scholar] [CrossRef]
- Huang, Y.X.; Qin, D.Z. Sequencing and analysis of the complete mitochondrial genome of Changeondelphax velitchkovskyi (Hemiptera: Fulgoroidea). Mitochondrial DNA B Resour. 2018, 3, 90–91. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.; Peng, X.; Jing, S.; Liu, B.; Zhu, L.; He, G. Intraspecific and Interspecific Variations in the Mitochondrial Genomes of Nilaparvata (Hemiptera: Delphacidae). J. Econ. Entomol. 2015, 108, 2021–2029. [Google Scholar] [CrossRef]
- Zhang, K.J.; Zhu, W.C.; Rong, X.; Liu, J.; Ding, X.L.; Hong, X.Y. The complete mitochondrial genome sequence of Sogatella furcifera (Horvath) and a comparative mitogenomic analysis of three predominant rice planthoppers. Gene 2014, 533, 100–109. [Google Scholar] [CrossRef]
- Sun, J.T.; Duan, X.Z.; Hoffmann, A.A.; Liu, Y.; Garvin, M.R.; Chen, L.; Hu, G.; Zhou, J.C.; Huang, H.J.; Xue, X.F.; et al. Mitochondrial variation in small brown planthoppers linked to multiple traits and probably reflecting a complex evolutionary trajectory. Mol. Ecol. 2019, 28, 3306–3323. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Cai, W.; Li, H. Deep-level phylogeny of Cicadomorpha inferred from mitochondrial genomes sequenced by NGS. Sci. Rep. 2017, 7, 10429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, N.; Liang, A.P.; Bu, C.P. A molecular phylogeny of Hemiptera inferred from mitochondrial genome sequences. PLoS ONE 2012, 7, e48778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, J.; Li, M.; Dong, P.; Cui, Y.; Xie, Q.; Bu, W. Phylogenetic analysis of the true water bugs (Insecta: Hemiptera: Heteroptera: Nepomorpha): Evidence from mitochondrial genomes. BMC Evol. Biol. 2009, 9, 134. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.X.; Guan, D.L.; Niu, Y.; Sang, L.Q.; Zhang, X.X.; Xu, S.Q. Characterization of the complete mitochondrial genome of the Asian planthopper Ricania speculum (Hemiptera: Fulgoroidea: Ricannidae). Conserv. Genet. Resour. 2016, 8, 463–466. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Superfamily | Family | Species | GenBank Number | References |
|---|---|---|---|---|
| Outgroups | ||||
| Membracoidea | Cicadellidae | Macrosteles quadrilineatus | NC_034781 | [51] |
| Cercopoidea | Aphrophoridae | Philaenus spumarius | NC_005944 | [52] |
| Cicadoidea | Cicadidae | Cicadetta abscondita | MW123088 | Unpublished |
| Ingroup | ||||
| Fulgoroidea | Delphacidae | Ugyops sp. | MH352481 | [53] |
| Sogatella vibix | NC_042180 | Unpublished | ||
| Peregrinus maidis | NC_037182 | [54] | ||
| Changeondelphax velitchkovskyi | NC_037181 | [55] | ||
| Nilaparvata bakeri | NC_033388 | Unpublished | ||
| Nilaparvata muiri | NC_024627 | [56] | ||
| Sogatella furcifera | NC_021417 | [57] | ||
| Laodelphax striatellus | MK292897 | [58] | ||
| Nilaparvata sp. | KY039125 | [59] | ||
| Nilaparvata lugens | JN563995 | [56] | ||
| Neomegamelanus elongatus | MK251068 | [20] | ||
| Saccharosydne procerus | NC_042179 | Unpublished | ||
| Saccharosydne saccharivora | MK251072 | [20] | ||
| Fulgoridae | Pyrops candelaria | FJ006724 | [60] | |
| Aphaena amabilis | NC_045075 | [13] | ||
| Aphaena discolor | MN025523 | [13] | ||
| Lycorma delicatula | NC_012835 | [61] | ||
| Achilidae | Paracatonidia sp. | MH324931 | [14] | |
| Betatropis formosana | MH324927 | [14] | ||
| Magadhaideus sp. | MH324928 | [14] | ||
| Plectoderini sp. | MH324930 | [14] | ||
| Peltatavertexalis horizontalis | MH324929 | [14] | ||
| Derbidae | Lydda sp. | KY039126 | [59] | |
| Lophopidae | Lophops carinata | MT990448 | Unpublished | |
| Issidae | Hemisphaerius rufovarius | MT210096 | [15] | |
| Sivaloka damnosus | FJ360694 | [27] | ||
| Sivaloka sp. | KY039137 | [59] | ||
| Ricaniidae | Ricania shantungensis | MT898421 | Unpublished | |
| Ricania marginalis | NC_019597 | [60] | ||
| Ricania speculum | NC_031369 | [62] | ||
| Flatidae | Cerynia lineola | MW872011 | Present study | |
| Cromna sinensis | MW872012 | Present study | ||
| Geisha distinctissima | NC_012617 | [12] | ||
| Zecheuna tonkinensis | MW872013 | Present study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ai, D.; Peng, L.; Qin, D.; Zhang, Y. Characterization of Three Complete Mitogenomes of Flatidae (Hemiptera: Fulgoroidea) and Compositional Heterogeneity Analysis in the Planthoppers’ Mitochondrial Phylogenomics. Int. J. Mol. Sci. 2021, 22, 5586. https://doi.org/10.3390/ijms22115586
Ai D, Peng L, Qin D, Zhang Y. Characterization of Three Complete Mitogenomes of Flatidae (Hemiptera: Fulgoroidea) and Compositional Heterogeneity Analysis in the Planthoppers’ Mitochondrial Phylogenomics. International Journal of Molecular Sciences. 2021; 22(11):5586. https://doi.org/10.3390/ijms22115586
Chicago/Turabian StyleAi, Deqiang, Lingfei Peng, Daozheng Qin, and Yalin Zhang. 2021. "Characterization of Three Complete Mitogenomes of Flatidae (Hemiptera: Fulgoroidea) and Compositional Heterogeneity Analysis in the Planthoppers’ Mitochondrial Phylogenomics" International Journal of Molecular Sciences 22, no. 11: 5586. https://doi.org/10.3390/ijms22115586