
Repression of MUC1 Promotes Expansion and Suppressive Function of Myeloid-Derived Suppressor Cells in Pancreatic and Breast Cancer Murine Models


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
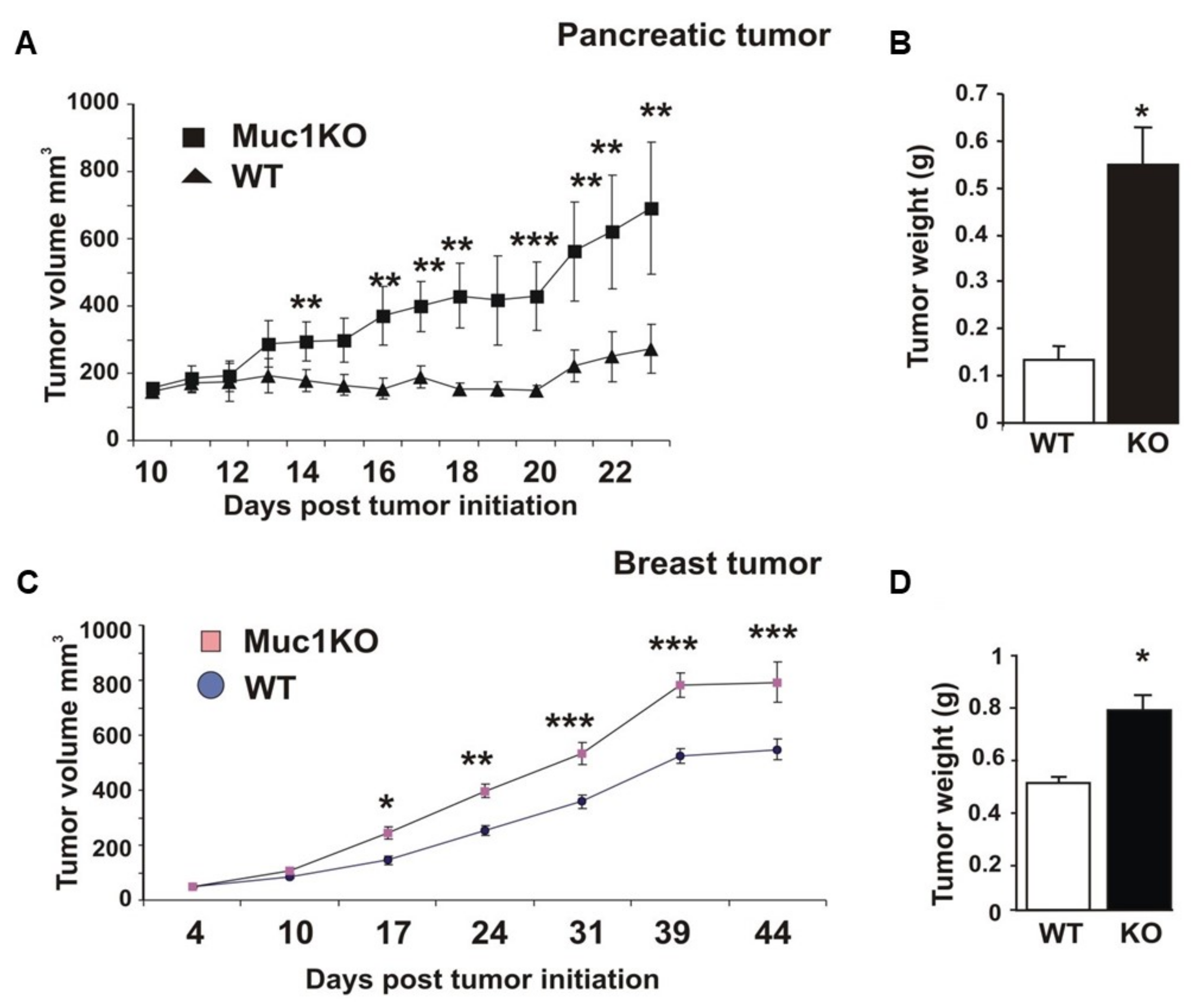
2.1. MUC1KO Mice Are More Susceptible to Tumor Growth
2.2. Increased Expansion and Migration of MDSCs to the Spleen of MUC1KO Mice and Higher Levels of TGF-β in Their Serum Compared to That of WT Mice
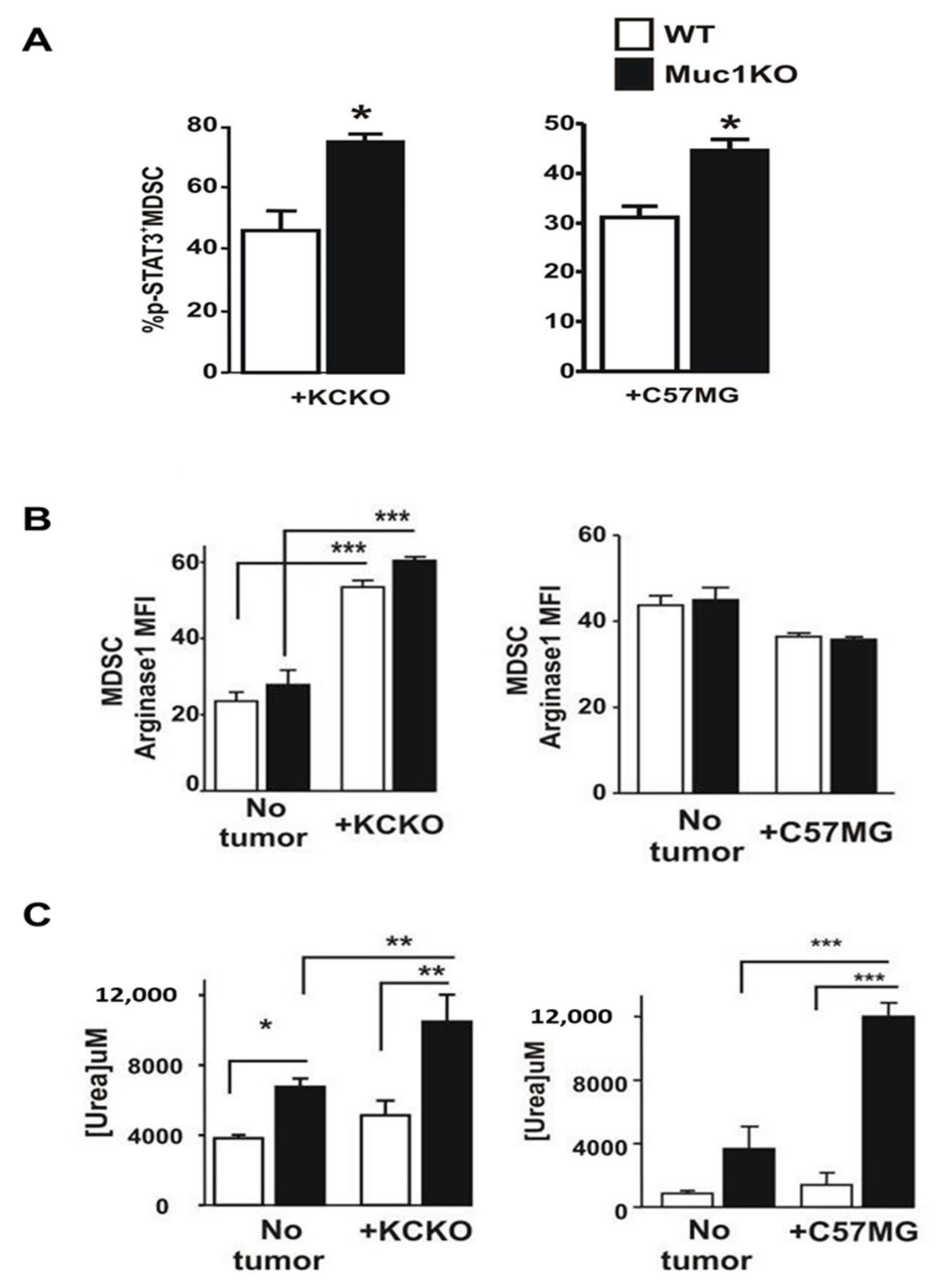
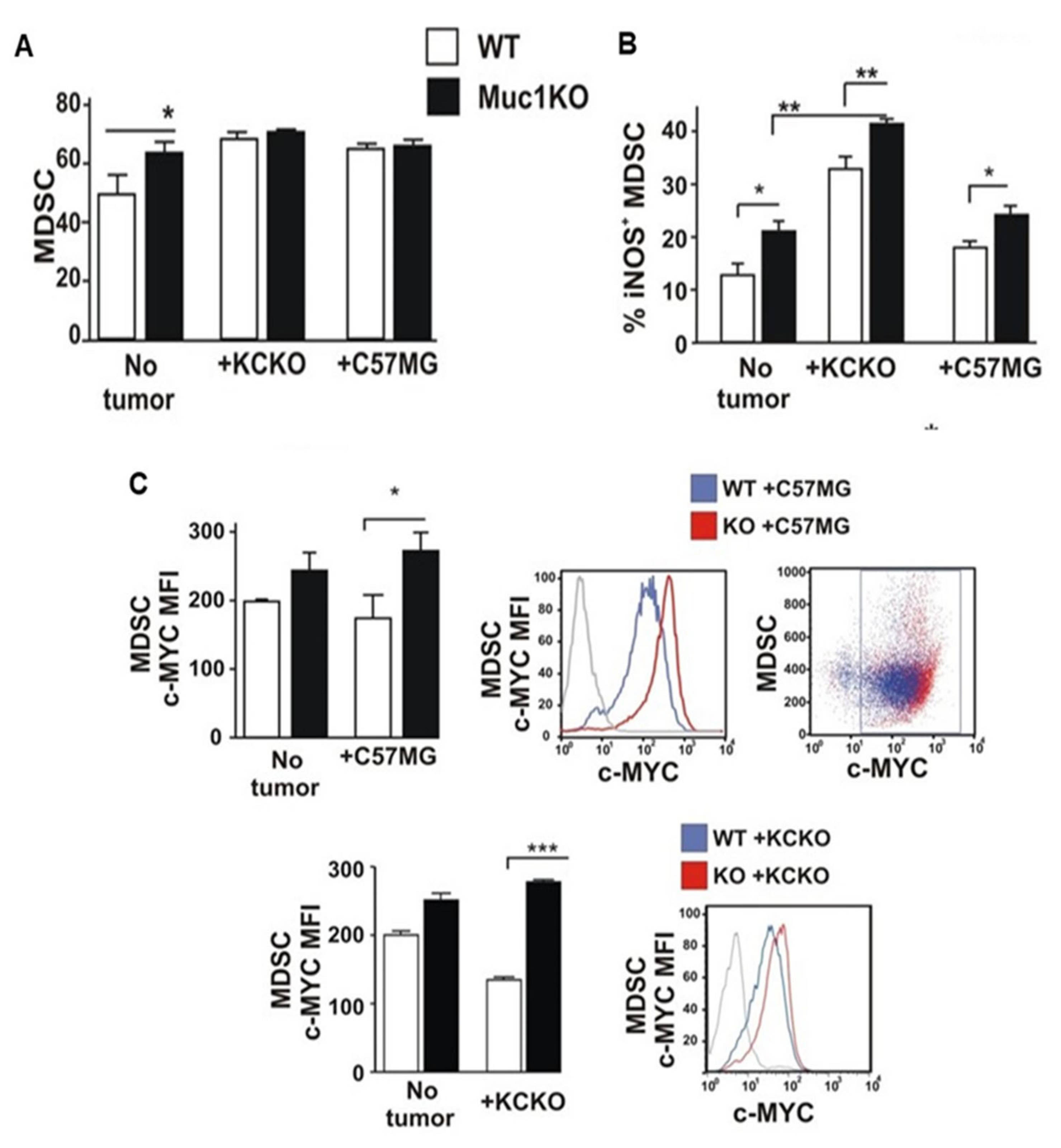
2.3. Spleen-Derived MDSCs from MUC1KO Mice Have Higher Immunosuppressive Phenotype
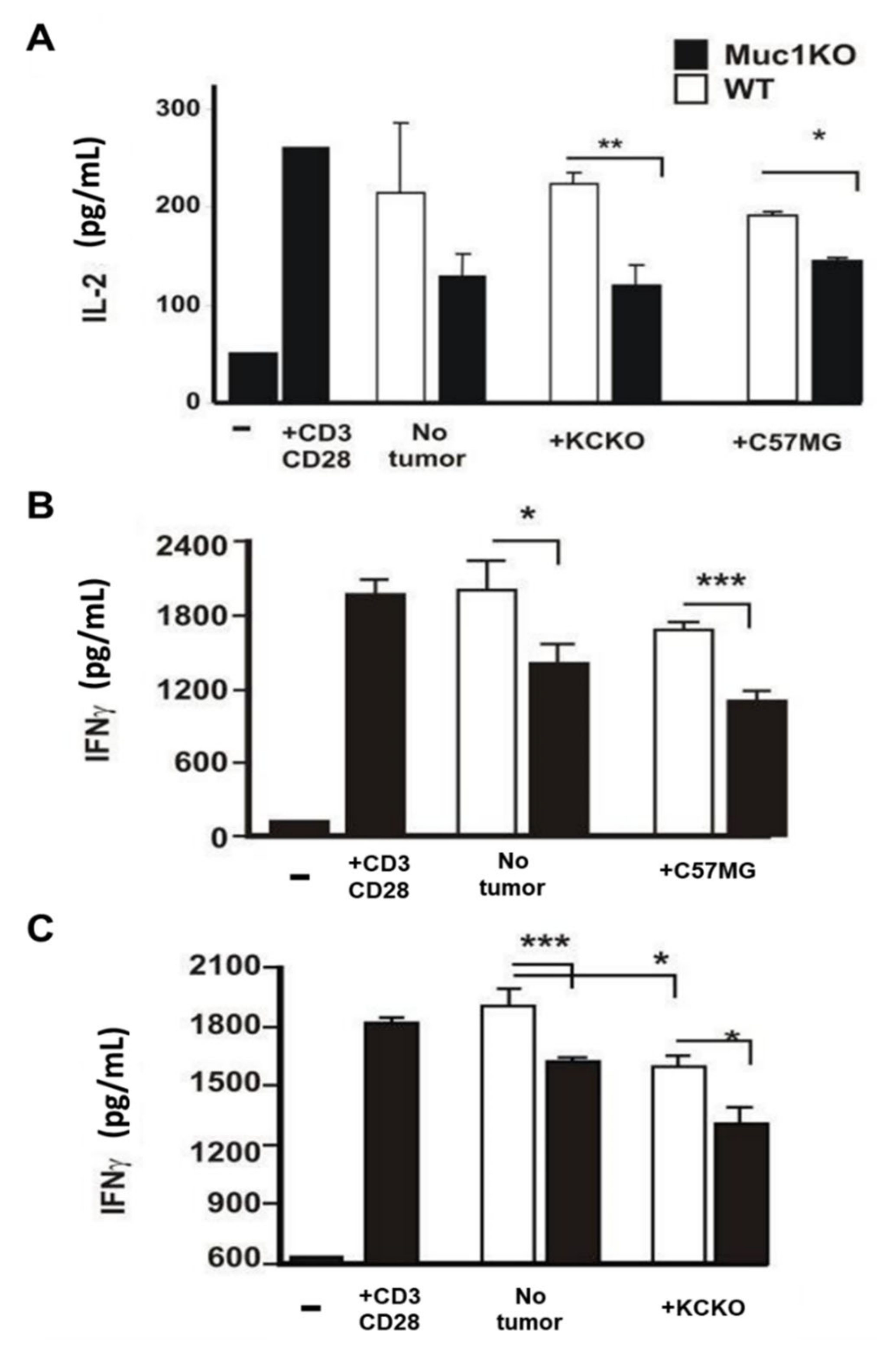
2.4. MDSCs from the Spleen of MUC1KO Tumor Bearing Mice Lead to Increased Suppression of Cytotoxic T Cells Compared to WT MDSCs
2.5. BM-MDSCS from MUC1KO Mice Have Different Rates of Expansion and Proliferation Compared to That from WT Mice
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Mouse Models
4.3. Flow Cytometry
4.4. Antibodies
4.5. Arginase Assay
4.6. ELISA
4.7. MDSC Suppression Assay
4.8. T Cell Proliferation Assay
4.9. Immunohistochemistry
4.10. Proteomics
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Parker, K.H.; Beury, D.W.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells: Critical Cells Driving Immune Suppression in the Tumor Microenvironment. In Advances in Cancer Research; Elsevier: Cambridge, MA, USA, 2015; Volume 128, pp. 95–139. [Google Scholar]
- Meirow, Y.; Kanterman, J.; Baniyash, M. Paving the road to tumor development and spreading: Myeloid-derived suppressor cells are ruling the fate. Front. Immunol. 2015, 6, 523. [Google Scholar] [CrossRef] [Green Version]
- Solito, S.; Marigo, I.; Pinton, L.; Damuzzo, V.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann. N. Y. Acad. Sci. 2014, 1319, 47–65. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condamine, T.; Mastio, J.; Gabrilovich, D.I. Transcriptional regulation of myeloid-derived suppressor cells. J. Leukoc. Biol. 2015, 98, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Filipazzi, P.; Huber, V.; Rivoltini, L. Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol. Immunother. 2012, 61, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Poschke, I.; Kiessling, R. On the armament and appearances of human myeloid-derived suppressor cells. Clin. Immunol. 2012, 144, 250–268. [Google Scholar] [CrossRef] [PubMed]
- Serafini, P.; Borrello, I.; Bronte, V. Myeloid suppressor cells in cancer: Recruitment, phenotype, properties, and mechanisms of immune suppression. Semin. Cancer Biol. 2006, 16, 53–65. [Google Scholar] [CrossRef]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef]
- Baniyash, M. Myeloid-derived suppressor cells as intruders and targets: Clinical implications in cancer therapy. Cancer Immunol. Immunother. 2016, 65, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: More mechanisms for inhibiting antitumor immunity. Cancer Immunol. Immunother. 2010, 59, 1593–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raber, P.; Ochoa, A.C.; Rodríguez, P.C. Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: Mechanisms of T cell suppression and therapeutic perspectives. Immunol. Investig. 2012, 41, 614–634. [Google Scholar] [CrossRef] [PubMed]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Woo, S.-R.; Zha, Y.; Spaapen, R.; Zheng, Y.; Corrales, L.; Spranger, S. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 268–276. [Google Scholar] [CrossRef]
- Tartour, E.; Pere, H.; Maillere, B.; Terme, M.; Merillon, N.; Taieb, J.; Sandoval, F.; Quintin-Colonna, F.; Lacerda, K.; Karadimou, A. Angiogenesis and immunity: A bidirectional link potentially relevant for the monitoring of antiangiogenic therapy and the development of novel therapeutic combination with immunotherapy. Cancer Metastasis. Rev. 2011, 30, 83–95. [Google Scholar] [CrossRef]
- Binsfeld, M.; Muller, J.; Lamour, V.; De Veirman, K.; De Raeve, H.; Bellahcène, A.; Van Valckenborgh, E.; Baron, F.; Beguin, Y.; Caers, J. Granulocytic myeloid-derived suppressor cells promote angiogenesis in the context of multiple myeloma. Oncotarget 2016, 7, 37931. [Google Scholar] [CrossRef] [Green Version]
- Qu, P.; Yan, C.; Du, H. Matrix metalloproteinase 12 overexpression in myeloid lineage cells plays a key role in modulating myelopoiesis, immune suppression, and lung tumorigenesis. Blood J. Am. Soc. Hematol. 2011, 117, 4476–4489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, P.-Y.; Ma, G.; Weber, K.J.; Ozao-Choy, J.; Wang, G.; Yin, B.; Divino, C.M.; Chen, S.-H. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010, 70, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gendler, S.J.; Spicer, A.P. Epithelial mucin genes. Annu. Rev. Physiol. 1995, 57, 607–634. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.F.; Zhao, H.L.; Phillips, J.; Greenburg, G. The epithelial mucin, MUC1, is expressed on resting T lymphocytes and can function as a negative regulator of T cell activation. Cell. Immunol. 2000, 201, 83–88. [Google Scholar] [CrossRef]
- Lau, S.K.; Weiss, L.M.; Chu, P.G. Differential expression of MUC1, MUC2, and MUC5AC in carcinomas of various sites. Am. J. Clin. Pathol. 2004, 122, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Brockhausen, I.; Yang, J.M.; Burchell, J.; Whitehouse, C.; Taylor-Papadimitriou, J. Mechanisms underlying aberrant glycosylation of MUC1 mucin in breast cancer cells. Eur. J. Biochem. 1995, 233, 607–617. [Google Scholar] [CrossRef]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [Green Version]
- Umansky, V.; Sevko, A. Melanoma-Induced Immunosuppression and its Neutralization. In Proceedings of Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2012; pp. 319–326. [Google Scholar]
- Kanterman, J.; Sade-Feldman, M.; Baniyash, M. New insights into chronic inflammation-induced immunosuppression. Semin. Cancer Biol. 2012, 22, 307–318. [Google Scholar] [CrossRef]
- Umansky, V.; Sevko, A.; Gebhardt, C.; Utikal, J. Myeloid-derived suppressor cells in malignant melanoma. JDDG J. Der. Dtsch. Dermatol. Ges. 2014, 12, 1021–1027. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Kanterman, J.; Ish-Shalom, E.; Elnekave, M.; Horwitz, E.; Baniyash, M. Tumor necrosis factor-α blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation. Immunity 2013, 38, 541–554. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.; Roy, L.D.; Grover, P.; Rao, S.; Mukherjee, P. Mucin 1 regulates Cox-2 gene in pancreatic cancer. Pancreas 2015, 44, 909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poh, T.W.; Bradley, J.M.; Mukherjee, P.; Gendler, S.J. Lack of Muc1-regulated beta-catenin stability results in aberrant expansion of CD11b+Gr1+ myeloid-derived suppressor cells from the bone marrow. Cancer Res. 2009, 69, 3554–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidiyoor, A.; Schettini, J.; Besmer, D.M.; Rego, S.L.; Nath, S.; Curry, J.M.; Roy, L.D.; Dreau, D.; Mukherjee, P. Pancreatic Cancer Cells Isolated from Muc1-Null Tumors Favor the Generation of a Mature Less Suppressive MDSC Population. Front. Immunol. 2014, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Bose, M.; Mukherjee, P. A Novel Antibody Blocks Anti-Apoptotic Activity of MUC1 in Pancreatic Cancer Cell Lines; AACR: Atlanta, GA, USA, 2019. [Google Scholar]
- Yazdanifar, M.; Zhou, R.; Grover, P.; Williams, C.; Bose, M.; Moore, L.J.; Wu, S.-T.; Maher, J.; Dreau, D.; Mukherjee, P. Overcoming Immunological Resistance Enhances the Efficacy of a Novel Anti-tMUC1-CAR T Cell Treatment against Pancreatic Ductal Adenocarcinoma. Cells 2019, 8, 1070. [Google Scholar] [CrossRef] [Green Version]
- Kruger, W.; Kroger, N.; Zander, A.R. MUC1 expression in hemopoietic tissues. J. Hematother. Stem. Cell Res. 2000, 9, 409–410. [Google Scholar] [CrossRef]
- Dent, G.A.; Civalier, C.J.; Brecher, M.E.; Bentley, S.A. MUC1 expression in hematopoietic tissues. Am. J. Clin. Pathol. 1999, 111, 741–747. [Google Scholar] [CrossRef] [Green Version]
- Alshetaiwi, H.; Pervolarakis, N.; McIntyre, L.L.; Ma, D.; Nguyen, Q.; Rath, J.A.; Nee, K.; Hernandez, G.; Evans, K.; Torosian, L. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Thyagarajan, A.; Alshehri, M.S.A.; Miller, K.L.; Sherwin, C.M.; Travers, J.B.; Sahu, R.P. Myeloid-derived suppressor cells and pancreatic cancer: Implications in novel therapeutic approaches. Cancers 2019, 11, 1627. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaraj, S.; Youn, J.-I.; Gabrilovich, D.I. Reciprocal relationship between myeloid-derived suppressor cells and T cells. J. Immunol. 2013, 191, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The role of myeloid-derived suppressor cells (MDSC) in cancer progression. Vaccines 2016, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Nefedova, Y.; Huang, M.; Kusmartsev, S.; Bhattacharya, R.; Cheng, P.; Salup, R.; Jove, R.; Gabrilovich, D. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J. Immunol. 2004, 172, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Panni, R.Z.; Sanford, D.E.; Belt, B.A.; Mitchem, J.B.; Worley, L.A.; Goetz, B.D.; Mukherjee, P.; Wang-Gillam, A.; Link, D.C.; Denardo, D.G.; et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol. Immunother. 2014, 63, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Morris, S.M., Jr. Arginine metabolism: Nitric oxide and beyond. Biochem. J. 1998, 336, 1–17. [Google Scholar] [CrossRef]
- Pyzer, A.R.; Stroopinsky, D.; Rajabi, H.; Washington, A.; Tagde, A.; Coll, M.; Fung, J.; Bryant, M.P.; Cole, L.; Palmer, K. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2017, 129, 1791–1801. [Google Scholar] [CrossRef] [Green Version]
- Rowse, G.J.; Tempero, R.M.; VanLith, M.L.; Hollingsworth, M.A.; Gendler, S.J. Tolerance and immunity to MUC1 in a human MUC1 transgenic murine model. Cancer Res. 1998, 58, 315–321. [Google Scholar]
- Nagaraj, S.; Gabrilovich, D.I. Myeloid-derived suppressor cells. Adv. Exp. Med. Biol. 2007, 601, 213–223. [Google Scholar]
- Gendler, S.J. MUC1, the renaissance molecule. J. Mammary Gland Biol. Neoplasia 2001, 6, 339–353. [Google Scholar] [CrossRef]
- Khaled, Y.S.; Ammori, B.J.; Elkord, E. Myeloid-derived suppressor cells in cancer: Recent progress and prospects. Immunol. Cell Biol. 2013, 91, 493–502. [Google Scholar] [CrossRef]
- Schlecker, E.; Stojanovic, A.; Eisen, C.; Quack, C.; Falk, C.S.; Umansky, V.; Cerwenka, A. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J. Immunol. 2012, 189, 5602–5611. [Google Scholar] [CrossRef] [Green Version]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouillez, A.; Rajabi, H.; Pitroda, S.; Jin, C.; Alam, M.; Kharbanda, A.; Tagde, A.; Wong, K.K.; Kufe, D. Inhibition of MUC1-C Suppresses MYC Expression and Attenuates Malignant Growth in KRAS Mutant Lung Adenocarcinomas. Cancer Res. 2016, 76, 1538–1548. [Google Scholar] [CrossRef] [Green Version]
- Tagde, A.; Rajabi, H.; Bouillez, A.; Alam, M.; Gali, R.; Bailey, S.; Tai, Y.T.; Hideshima, T.; Anderson, K.; Avigan, D.; et al. MUC1-C drives MYC in multiple myeloma. Blood 2016, 127, 2587–2597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Nguyen-Jackson, H.; Panopoulos, A.D.; Li, H.S.; Murray, P.J.; Watowich, S.S. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood 2010, 116, 2462–2471. [Google Scholar] [CrossRef] [Green Version]
- Safarzadeh, E.; Orangi, M.; Mohammadi, H.; Babaie, F.; Baradaran, B. Myeloid-derived suppressor cells: Important contributors to tumor progression and metastasis. J. Cell Physiol. 2018, 233, 3024–3036. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Schrum, A.G.; Cho, H.I.; Celis, E.; Gabrilovich, D.I. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J. Immunol. 2010, 184, 3106–3116. [Google Scholar] [CrossRef]
- Bose, M.; Mukherjee, P. Microbe–MUC1 Crosstalk in Cancer-Associated Infections. Trends Mol. Med. 2020, 26, 324–336. [Google Scholar] [CrossRef]
- Curry, J.M.; Thompson, K.J.; Rao, S.G.; Besmer, D.M.; Murphy, A.M.; Grdzelishvili, V.Z.; Ahrens, W.A.; McKillop, I.H.; Sindram, D.; Iannitti, D.A.; et al. The use of a novel MUC1 antibody to identify cancer stem cells and circulating MUC1 in mice and patients with pancreatic cancer. J. Surg. Oncol. 2013, 107, 713–722. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.T.; Williams, C.D.; Grover, P.A.; Moore, L.J.; Mukherjee, P. Early detection of pancreatic cancer in mouse models using a novel antibody, TAB004. PLoS ONE 2018, 13, e0193260. [Google Scholar] [CrossRef] [Green Version]
- Besmer, D.M.; Curry, J.M.; Roy, L.D.; Tinder, T.L.; Sahraei, M.; Schettini, J.; Hwang, S.I.; Lee, Y.Y.; Gendler, S.J.; Mukherjee, P. Pancreatic ductal adenocarcinoma mice lacking mucin 1 have a profound defect in tumor growth and metastasis. Cancer Res. 2011, 71, 4432–4442. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, H.; Julius, M.A.; Giarre, M.; Zheng, Z.; Brown, A.M.; Kitajewski, J. Transformation by Wnt family proteins correlates with regulation of beta-catenin. Cell Growth Differ. 1997, 8, 1349–1358. [Google Scholar] [PubMed]
- Spicer, A.P.; Rowse, G.J.; Lidner, T.K.; Gendler, S.J. Delayed mammary tumor progression in Muc-1 null mice. J. Biol. Chem. 1995, 270, 30093–30101. [Google Scholar] [CrossRef] [Green Version]
- Corraliza, I.; Campo, M.; Soler, G.; Modolell, M. Determination of arginase activity in macrophages: A micromethod. J. Immunol. Methods 1994, 174, 231–235. [Google Scholar] [CrossRef]
- Kropf, P.; Fuentes, J.M.; Fähnrich, E.; Arpa, L.; Herath, S.; Weber, V.; Soler, G.; Celada, A.; Modolell, M.; Müller, I. Arginase and polyamine synthesis are key factors in the regulation of experimental leishmaniasis in vivo. FASEB J. 2005, 19, 1000–1002. [Google Scholar] [CrossRef]
- Ribechini, E.; Leenen, P.J.; Lutz, M.B. Gr-1 antibody induces STAT signaling, macrophage marker expression and abrogation of myeloid-derived suppressor cell activity in BM cells. Eur. J. Immunol. 2009, 39, 3538–3551. [Google Scholar] [CrossRef]
- Mukherjee, P.; Ginardi, A.R.; Madsen, C.S.; Tinder, T.L.; Jacobs, F.; Parker, J.; Agrawal, B.; Longenecker, B.M.; Gendler, S.J. MUC1-specific CTLs are non-functional within a pancreatic tumor microenvironment. Glycoconj. J. 2001, 18, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.D.; Sahraei, M.; Subramani, D.B.; Besmer, D.; Nath, S.; Tinder, T.L.; Bajaj, E.; Shanmugam, K.; Lee, Y.Y.; Hwang, S.I. MUC1 enhances invasiveness of pancreatic cancer cells by inducing epithelial to mesenchymal transition. Oncogene 2011, 30, 1449–1459. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahraei, M.; Bose, M.; Sanders, J.A.; De, C.; DasRoy, L.; Nath, S.; Brouwer, C.R.; Mukherjee, P. Repression of MUC1 Promotes Expansion and Suppressive Function of Myeloid-Derived Suppressor Cells in Pancreatic and Breast Cancer Murine Models. Int. J. Mol. Sci. 2021, 22, 5587. https://doi.org/10.3390/ijms22115587
Sahraei M, Bose M, Sanders JA, De C, DasRoy L, Nath S, Brouwer CR, Mukherjee P. Repression of MUC1 Promotes Expansion and Suppressive Function of Myeloid-Derived Suppressor Cells in Pancreatic and Breast Cancer Murine Models. International Journal of Molecular Sciences. 2021; 22(11):5587. https://doi.org/10.3390/ijms22115587
Chicago/Turabian StyleSahraei, Mahnaz, Mukulika Bose, J. Alexa Sanders, Chandrav De, Lopamudra DasRoy, Sritama Nath, Cory R. Brouwer, and Pinku Mukherjee. 2021. "Repression of MUC1 Promotes Expansion and Suppressive Function of Myeloid-Derived Suppressor Cells in Pancreatic and Breast Cancer Murine Models" International Journal of Molecular Sciences 22, no. 11: 5587. https://doi.org/10.3390/ijms22115587
APA StyleSahraei, M., Bose, M., Sanders, J. A., De, C., DasRoy, L., Nath, S., Brouwer, C. R., & Mukherjee, P. (2021). Repression of MUC1 Promotes Expansion and Suppressive Function of Myeloid-Derived Suppressor Cells in Pancreatic and Breast Cancer Murine Models. International Journal of Molecular Sciences, 22(11), 5587. https://doi.org/10.3390/ijms22115587