
Hijacked Immune Cells in the Tumor Microenvironment: Molecular Mechanisms of Immunosuppression and Cues to Improve T Cell-Based Immunotherapy of Solid Tumors


Abstract
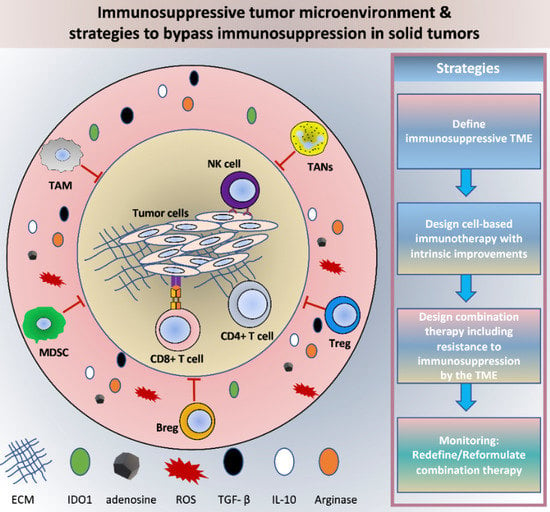
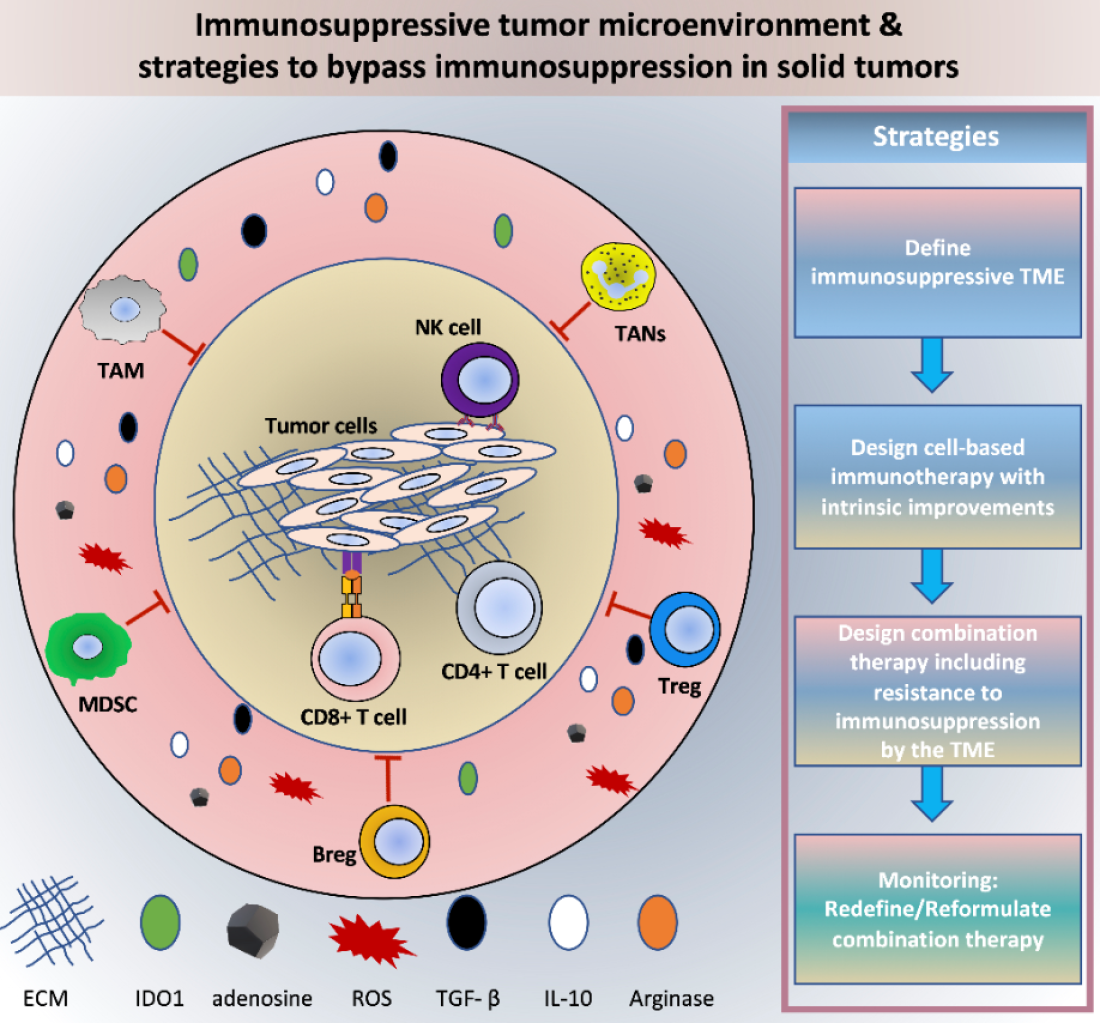
:
1. Composition and Heterogeneity of the Tumor Microenvironment
2. Tumor Immune Surveillance and Immunoediting
3. Immune Cell Constituents of the TME
3.1. Tumor-Associated Macrophages (TAMs)
3.2. Tumor-Associated Neutrophils (TANs)
3.3. Myeloid-Derived Suppressor Cells (MDSCs)
3.4. Natural Killer Cells (NK Cells)
3.5. B Cells
3.6. Dendritic Cells (DCs)
3.7. T Cells
3.7.1. CD8+ T Cells
3.7.2. CD4+ T Helper Cells
3.7.3. Regulatory T Cells
4. Molecular Constituents of an Immunosuppressive TME
4.1. Anti-Inflammatory Cytokines
4.2. Reactive Oxygen Species (ROS)
4.3. Metabolites
5. Immunotherapeutic Strategies against Solid Tumors
5.1. The Portfolio of T Cell-Based Immunotherapies
5.1.1. TIL Therapy
5.1.2. TCR Therapy
5.1.3. CAR T Cell Therapy
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Q.Q.; Zhang, H.L.; Zheng, J.N.; Zhang, L.J. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [Green Version]
- Krause, R.M.; Ehrlich, P.; Avery, O.T. Pathfinders in the search for immunity. Vaccine 1999, 17 (Suppl. 3), S64–S67. [Google Scholar] [CrossRef]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, P. Ueber den jetzigen Stand der Karzinomforschung. Ned. Tijdschr. Geneeskd. 1909, 5, 273–290. [Google Scholar]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Lin, K.; Li, X.; Yuan, X.; Xu, P.; Ni, P.; Xu, D. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front. Immunol. 2020, 11, 1731. [Google Scholar] [CrossRef] [PubMed]
- Bain, C.C.; Scott, C.L.; Uronen-Hansson, H.; Gudjonsson, S.; Jansson, O.; Grip, O.; Guilliams, M.; Malissen, B.; Agace, W.W.; Mowat, A.M. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal. Immunol. 2013, 6, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, D.; Zins, K.; Sioud, M.; Lucas, T.; Schafer, R.; Stanley, E.R.; Aharinejad, S. Stromal cell-derived CSF-1 blockade prolongs xenograft survival of CSF-1-negative neuroblastoma. Int. J. Cancer 2010, 126, 1339–1352. [Google Scholar] [CrossRef] [Green Version]
- Sousa, S.; Maatta, J. The role of tumour-associated macrophages in bone metastasis. J. Bone Oncol. 2016, 5, 135–138. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Valilou, S.F.; Soltani, A.; Ahmadi, S.; Abarghan, Y.J.; Rosengren, R.J.; Sahebkar, A. Tumor-associated macrophages: Role in cancer development and therapeutic implications. Cell Oncol. 2019, 42, 591–608. [Google Scholar] [CrossRef]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.H.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019, 35, 588–602.e510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front. Physiol. 2014, 5, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bercovici, N.; Guerin, M.V.; Trautmann, A.; Donnadieu, E. The Remarkable Plasticity of Macrophages: A Chance to Fight Cancer. Front. Immunol. 2019, 10, 1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wislez, M.; Rabbe, N.; Marchal, J.; Milleron, B.; Crestani, B.; Mayaud, C.; Antoine, M.; Soler, P.; Cadranel, J. Hepatocyte growth factor production by neutrophils infiltrating bronchioloalveolar subtype pulmonary adenocarcinoma: Role in tumor progression and death. Cancer Res. 2003, 63, 1405–1412. [Google Scholar] [PubMed]
- Schmidt, H.; Bastholt, L.; Geertsen, P.; Christensen, I.J.; Larsen, S.; Gehl, J.; von der Maase, H. Elevated neutrophil and monocyte counts in peripheral blood are associated with poor survival in patients with metastatic melanoma: A prognostic model. Br. J. Cancer 2005, 93, 273–278. [Google Scholar] [CrossRef]
- Jensen, H.K.; Donskov, F.; Marcussen, N.; Nordsmark, M.; Lundbeck, F.; von der Maase, H. Presence of intratumoral neutrophils is an independent prognostic factor in localized renal cell carcinoma. J. Clin. Oncol. 2009, 27, 4709–4717. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, X.; Li, R.; Yan, J.; Xiao, Y.; Li, W.; Shen, H. Neutrophil-to-Lymphocyte Ratio Is Independently Associated With Severe Psychopathology in Schizophrenia and Is Changed by Antipsychotic Administration: A Large-Scale Cross-Sectional Retrospective Study. Front. Psychiatry 2020, 11, 581061. [Google Scholar] [CrossRef]
- Gago-Dominguez, M.; Matabuena, M.; Redondo, C.M.; Patel, S.P.; Carracedo, A.; Ponte, S.M.; Martinez, M.E.; Castelao, J.E. Neutrophil to lymphocyte ratio and breast cancer risk: Analysis by subtype and potential interactions. Sci. Rep. 2020, 10, 13203. [Google Scholar] [CrossRef]
- He, G.; Zhang, H.; Zhou, J.; Wang, B.; Chen, Y.; Kong, Y.; Xie, X.; Wang, X.; Fei, R.; Wei, L.; et al. Peritumoural neutrophils negatively regulate adaptive immunity via the PD-L1/PD-1 signalling pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 141. [Google Scholar] [CrossRef] [Green Version]
- Amarnath, S.; Mangus, C.W.; Wang, J.C.; Wei, F.; He, A.; Kapoor, V.; Foley, J.E.; Massey, P.R.; Felizardo, T.C.; Riley, J.L.; et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci. Transl. Med. 2011, 3, 111ra120. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.Y. Searching for the real function of mTOR signaling in the regulation of PD-L1 expression. Transl. Oncol. 2020, 13, 100847. [Google Scholar] [CrossRef] [PubMed]
- Klemke, M.; Wabnitz, G.H.; Funke, F.; Funk, B.; Kirchgessner, H.; Samstag, Y. Oxidation of cofilin mediates T cell hyporesponsiveness under oxidative stress conditions. Immunity 2008, 29, 404–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wabnitz, G.H.; Goursot, C.; Jahraus, B.; Kirchgessner, H.; Hellwig, A.; Klemke, M.; Konstandin, M.H.; Samstag, Y. Mitochondrial translocation of oxidized cofilin induces caspase-independent necrotic-like programmed cell death of T cells. Cell Death Dis. 2010, 1, e58. [Google Scholar] [CrossRef] [Green Version]
- Feldmeyer, N.; Wabnitz, G.; Leicht, S.; Luckner-Minden, C.; Schiller, M.; Franz, T.; Conradi, R.; Kropf, P.; Muller, I.; Ho, A.D.; et al. Arginine deficiency leads to impaired cofilin dephosphorylation in activated human T lymphocytes. Int. Immunol. 2012, 24, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Kuttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361. [Google Scholar] [CrossRef] [Green Version]
- Teijeira, A.; Garasa, S.; Gato, M.; Alfaro, C.; Migueliz, I.; Cirella, A.; de Andrea, C.; Ochoa, M.C.; Otano, I.; Etxeberria, I.; et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity 2020, 52, 856–871.e858. [Google Scholar] [CrossRef]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.W.; Punnoose, E.A.; Januario, T.; Lawrence, D.A.; Pitti, R.M.; Lancaster, K.; Lee, D.; von Goetz, M.; Yee, S.F.; Totpal, K.; et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat. Med. 2007, 13, 1070–1077. [Google Scholar] [CrossRef]
- Koga, Y.; Matsuzaki, A.; Suminoe, A.; Hattori, H.; Hara, T. Neutrophil-derived TNF-related apoptosis-inducing ligand (TRAIL): A novel mechanism of antitumor effect by neutrophils. Cancer Res. 2004, 64, 1037–1043. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, R.W.; Frew, A.J.; Smyth, M.J. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer 2008, 8, 782–798. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Hsu, B.E.; Tabaries, S.; Johnson, R.M.; Andrzejewski, S.; Senecal, J.; Lehuede, C.; Annis, M.G.; Ma, E.H.; Vols, S.; Ramsay, L.; et al. Immature Low-Density Neutrophils Exhibit Metabolic Flexibility that Facilitates Breast Cancer Liver Metastasis. Cell Rep. 2019, 27, 3902–3915.e3906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagiv, J.Y.; Michaeli, J.; Assi, S.; Mishalian, I.; Kisos, H.; Levy, L.; Damti, P.; Lumbroso, D.; Polyansky, L.; Sionov, R.V.; et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015, 10, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Boettcher, S.; Manz, M.G. Regulation of Inflammation- and Infection-Driven Hematopoiesis. Trends Immunol. 2017, 38, 345–357. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef] [Green Version]
- Tcyganov, E.; Mastio, J.; Chen, E.; Gabrilovich, D.I. Plasticity of myeloid-derived suppressor cells in cancer. Curr. Opin. Immunol. 2018, 51, 76–82. [Google Scholar] [CrossRef]
- Li, C.; Liu, T.; Bazhin, A.V.; Yang, Y. The Sabotaging Role of Myeloid Cells in Anti-Angiogenic Therapy: Coordination of Angiogenesis and Immune Suppression by Hypoxia. J. Cell Physiol. 2017, 232, 2312–2322. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, C.; Liu, T.; Dai, X.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 2020, 11, 1371. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef] [PubMed]
- Kesarwani, P.; Murali, A.K.; Al-Khami, A.A.; Mehrotra, S. Redox regulation of T-cell function: From molecular mechanisms to significance in human health and disease. Antioxid. Redox Signal. 2013, 18, 1497–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.R.; Lee, W.; Cho, S.K.; Park, S.G. Characterization of Multiple Cytokine Combinations and TGF-beta on Differentiation and Functions of Myeloid-Derived Suppressor Cells. Int. J. Mol. Sci. 2018, 19, 869. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Ochoa, A.C. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: Mechanisms and therapeutic perspectives. Immunol. Rev. 2008, 222, 180–191. [Google Scholar] [CrossRef]
- Platten, M.; Wick, W.; Van den Eynde, B.J. Tryptophan catabolism in cancer: Beyond IDO and tryptophan depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef] [Green Version]
- Sitkovsky, M.; Lukashev, D.; Deaglio, S.; Dwyer, K.; Robson, S.C.; Ohta, A. Adenosine A2A receptor antagonists: Blockade of adenosinergic effects and T regulatory cells. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S457–S464. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Ballbach, M.; Dannert, A.; Singh, A.; Siegmund, D.M.; Handgretinger, R.; Piali, L.; Rieber, N.; Hartl, D. Expression of checkpoint molecules on myeloid-derived suppressor cells. Immunol. Lett. 2017, 192, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pinton, L.; Solito, S.; Damuzzo, V.; Francescato, S.; Pozzuoli, A.; Berizzi, A.; Mocellin, S.; Rossi, C.R.; Bronte, V.; Mandruzzato, S. Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget 2016, 7, 1168–1184. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Redd, P.S.; Lee, J.R.; Savage, N.; Liu, K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology 2016, 5, e1247135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgun, G.; Samur, M.K.; Cowens, K.B.; Paula, S.; Bianchi, G.; Anderson, J.E.; White, R.E.; Singh, A.; Ohguchi, H.; Suzuki, R.; et al. Lenalidomide Enhances Immune Checkpoint Blockade-Induced Immune Response in Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4607–4618. [Google Scholar] [CrossRef] [Green Version]
- Iwata, T.; Kondo, Y.; Kimura, O.; Morosawa, T.; Fujisaka, Y.; Umetsu, T.; Kogure, T.; Inoue, J.; Nakagome, Y.; Shimosegawa, T. PD-L1(+)MDSCs are increased in HCC patients and induced by soluble factor in the tumor microenvironment. Sci. Rep. 2016, 6, 39296. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Lanier, L.L. NK cell development, homeostasis and function: Parallels with CD8(+) T cells. Nat. Rev. Immunol. 2011, 11, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Freud, A.G.; Mundy-Bosse, B.L.; Yu, J.H.; Caligiuri, M.A. The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity 2017, 47, 820–833. [Google Scholar] [CrossRef] [Green Version]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell. Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Menard, C.; Blay, J.Y.; Borg, C.; Michiels, S.; Ghiringhelli, F.; Robert, C.; Nonn, C.; Chaput, N.; Taieb, J.; Delahaye, N.F.; et al. Natural Killer Cell IFN-gamma Levels Predict Long-term Survival with Imatinib Mesylate Therapy in Gastrointestinal Stromal Tumor-Bearing Patients. Cancer Res. 2009, 69, 3563–3569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semeraro, M.; Rusakiewicz, S.; Minard-Colin, V.; Delahaye, N.F.; Enot, D.; Vely, F.; Marabelle, A.; Papoular, B.; Piperoglou, C.; Ponzoni, M.; et al. Clinical impact of the NKp30/B7-H6 axis in high-risk neuroblastoma patients. Sci. Transl. Med. 2015, 7, 283ra255. [Google Scholar] [CrossRef]
- Villegas, F.R.; Coca, S.; Villarrubia, V.G.; Jimenez, R.; Chillon, M.J.; Jareno, J.; Zuil, M.; Callol, L. Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer 2002, 35, 23–28. [Google Scholar] [CrossRef]
- Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front. Immunol. 2019, 10, 3038. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, W.; Hu, B.; Wang, P.; Lv, X.; Chen, S.; Shao, Z. Prognostic Significance of Tumor-Infiltrating Natural Killer Cells in Solid Tumors: A Systematic Review and Meta-Analysis. Front. Immunol. 2020, 11, 1242. [Google Scholar] [CrossRef] [PubMed]
- Judge, S.J.; Murphy, W.J.; Canter, R.J. Characterizing the Dysfunctional NK Cell: Assessing the Clinical Relevance of Exhaustion, Anergy, and Senescence. Front. Cell Infect. Microbiol. 2020, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Russick, J.; Joubert, P.E.; Gillard-Bocquet, M.; Torset, C.; Meylan, M.; Petitprez, F.; Dragon-Durey, M.A.; Marmier, S.; Varthaman, A.; Josseaume, N.; et al. Natural killer cells in the human lung tumor microenvironment display immune inhibitory functions. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Carrega, P.; Morandi, B.; Costa, R.; Frumento, G.; Forte, G.; Altavilla, G.; Ratto, G.B.; Mingari, M.C.; Moretta, L.; Ferlazzo, G. Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56 bright CD16(-) cells and display an impaired capability to kill tumor cells. Cancer 2008, 112, 863–875. [Google Scholar] [CrossRef]
- Delahaye, N.F.; Rusakiewicz, S.; Martins, I.; Menard, C.; Roux, S.; Lyonnet, L.; Paul, P.; Sarabi, M.; Chaput, N.; Semeraro, M.; et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat. Med. 2011, 17, 700–707. [Google Scholar] [CrossRef]
- Stiff, A.; Trikha, P.; Mundy-Bosse, B.; McMichael, E.; Mace, T.A.; Benner, B.; Kendra, K.; Campbell, A.; Gautam, S.; Abood, D.; et al. Nitric Oxide Production by Myeloid-Derived Suppressor Cells Plays a Role in Impairing Fc Receptor-Mediated Natural Killer Cell Function. Clin. Cancer Res. 2018, 24, 1891–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingoni, A.; Vulpis, E.; Nardone, I.; Soriani, A.; Fionda, C.; Cippitelli, M.; Santoni, A. Targeting NKG2D and NKp30 Ligands Shedding to Improve NK Cell-Based Immunotherapy. Crit. Rev. Immunol. 2016, 36, 445–459. [Google Scholar] [CrossRef]
- Bassani, B.; Baci, D.; Gallazzi, M.; Poggi, A.; Bruno, A.; Mortara, L. Natural Killer Cells as Key Players of Tumor Progression and Angiogenesis: Old and Novel Tools to Divert Their Pro-Tumor Activities into Potent Anti-Tumor Effects. Cancers 2019, 11, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Kroeger, D.R.; Milne, K.; Nelson, B.H. Tumor-Infiltrating Plasma Cells Are Associated with Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer. Clin. Cancer Res. 2016, 22, 3005–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wouters, M.C.A.; Nelson, B.H. Prognostic Significance of Tumor-Infiltrating B Cells and Plasma Cells in Human Cancer. Clin. Cancer Res. 2018, 24, 6125–6135. [Google Scholar] [CrossRef] [Green Version]
- Edin, S.; Kaprio, T.; Hagstrom, J.; Larsson, P.; Mustonen, H.; Bockelman, C.; Strigard, K.; Gunnarsson, U.; Haglund, C.; Palmqvist, R. The Prognostic Importance of CD20(+) B lymphocytes in Colorectal Cancer and the Relation to Other Immune Cell subsets. Sci. Rep. 2019, 9, 19997. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Germain, C.; Gnjatic, S.; Dieu-Nosjean, M.C. Tertiary Lymphoid Structure-Associated B Cells are Key Players in Anti-Tumor Immunity. Front. Immunol. 2015, 6, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sautes-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, R.A.M.; Lorenzi, N.P.C.; Yokochi, K.; Rosa, M.B.S.D.; Benevides, L.; Margarido, P.F.R.; Baracat, E.C.; Carvalho, J.P.; Villa, L.L.; Lepique, A.P. B lymphocytes can be activated to act as antigen presenting cells to promote anti-tumor responses. PLoS ONE 2018, 13, e0199034. [Google Scholar] [CrossRef]
- Shi, J.Y.; Gao, Q.; Wang, Z.C.; Zhou, J.; Wang, X.Y.; Min, Z.H.; Shi, Y.H.; Shi, G.M.; Ding, Z.B.; Ke, A.W.; et al. Margin-Infiltrating CD20(+) BCells Display an Atypical Memory Phenotype and Correlate with Favorable Prognosis in Hepatocellular Carcinoma. Clin. Cancer Res. 2013, 19, 5994–6005. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, J.S.; Sahota, R.A.; Milne, K.; Kost, S.E.; Nesslinger, N.J.; Watson, P.H.; Nelson, B.H. CD20(+) Tumor-Infiltrating Lymphocytes Have an Atypical CD27(-) Memory Phenotype and Together with CD8(+) T Cells Promote Favorable Prognosis in Ovarian Cancer. Clin. Cancer Res. 2012, 18, 3281–3292. [Google Scholar] [CrossRef] [Green Version]
- Garaud, S.; Buisseret, L.; Solinas, C.; Gu-Trantien, C.; de Wind, A.; Van den Eynden, G.; Naveaux, C.; Lodewyckx, J.N.; Boisson, A.; Duvillier, H.; et al. Tumor infiltrating B-cells signal functional humoral immune responses in breast cancer. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Iglesia, M.D.; Vincent, B.G.; Parker, J.S.; Hoadley, K.A.; Carey, L.A.; Perou, C.M.; Serody, J.S. Prognostic B-cell signatures using mRNA-seq in patients with subtype-specific breast and ovarian cancer. Clin. Cancer Res. 2014, 20, 3818–3829. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, R.; Morello, S.; Forte, G.; Montinaro, A.; De Vita, G.; Luciano, A.; Palma, G.; Arra, C.; Maiolino, P.; Adcock, I.M.; et al. B Cells Contribute to the Antitumor Activity of CpG-Oligodeoxynucleotide in a Mouse Model of Metastatic Lung Carcinoma. Am. J. Respir. Crit. Care 2011, 183, 1369–1379. [Google Scholar] [CrossRef]
- Coronella, J.A.; Spier, C.; Welch, M.; Trevor, K.T.; Stopeck, A.T.; Villar, H.; Hersh, E.M. Antigen-driven oligoclonal expansion of tumor-infiltrating B cells in infiltrating ductal carcinoma of the breast. J. Immunol. 2002, 169, 1829–1836. [Google Scholar] [CrossRef] [Green Version]
- Bolotin, D.A.; Poslavsky, S.; Davydov, A.N.; Frenkel, F.E.; Fanchi, L.; Zolotareva, O.I.; Hemmers, S.; Putintseva, E.V.; Obraztsova, A.S.; Shugay, M.; et al. Antigen receptor repertoire profiling from RNA-seq data. Nat. Biotechnol. 2017, 35, 908–911. [Google Scholar] [CrossRef]
- Reuschenbach, M.; von Knebel Doeberitz, M.; Wentzensen, N. A systematic review of humoral immune responses against tumor antigens. Cancer Immunol. Immunother. 2009, 58, 1535–1544. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, B.; Li, X.; Zhou, D.; Li, Y.; Jia, S.; Qi, S.; Xu, A.; Zhao, X.; Wang, J.; et al. Identification and Validation of Novel Serum Autoantibody Biomarkers for Early Detection of Colorectal Cancer and Advanced Adenoma. Front. Oncol. 2020, 10, 1081. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, I.; Nagata, M.; Takiguchi, N.; Nabeya, Y.; Ikeda, A.; Yokoi, S.; Kuwajima, A.; Tagawa, M.; Matsushita, K.; Satoshi, Y.; et al. Panel of autoantibodies against multiple tumor-associated antigens for detecting gastric cancer. Cancer Sci. 2017, 108, 308–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.M.; Ling, Z.G.; Wang, C.M.; Wu, Y.B.; Kong, J.L. Serum tumor-associated autoantibodies as diagnostic biomarkers for lung cancer: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0182117. [Google Scholar] [CrossRef] [Green Version]
- Berntsson, J.; Nodin, B.; Eberhard, J.; Micke, P.; Jirstrom, K. Prognostic impact of tumour-infiltrating B cells and plasma cells in colorectal cancer. Int. J. Cancer 2016, 139, 1129–1139. [Google Scholar] [CrossRef]
- Germain, C.; Gnjatic, S.; Tamzalit, F.; Knockaert, S.; Remark, R.; Goc, J.; Lepelley, A.; Becht, E.; Katsahian, S.; Bizouard, G.; et al. Presence of B Cells in Tertiary Lymphoid Structures Is Associated with a Protective Immunity in Patients with Lung Cancer. Am. J. Resp. Crit. Care 2014, 189, 832–844. [Google Scholar] [CrossRef]
- Milne, K.; Kobel, M.; Kalloger, S.E.; Barnes, R.O.; Gao, D.X.; Gilks, C.B.; Watson, P.H.; Nelson, B.H. Systematic Analysis of Immune Infiltrates in High-Grade Serous Ovarian Cancer Reveals CD20, FoxP3 and TIA-1 as Positive Prognostic Factors. PLoS ONE 2009, 4, e6412. [Google Scholar] [CrossRef] [Green Version]
- Garg, K.; Maurer, M.; Griss, J.; Bruggen, M.C.; Wolf, I.H.; Wagner, C.; Willi, N.; Mertz, K.D.; Wagner, S.N. Tumor-associated B cells in cutaneous primary melanoma and improved clinical outcome. Hum. Pathol. 2016, 54, 157–164. [Google Scholar] [CrossRef]
- Ou, Z.Y.; Wang, Y.J.; Liu, L.F.; Li, L.; Yeh, S.Y.; Qi, L.; Chang, C.S. Tumor microenvironment B cells increase bladder cancer metastasis via modulation of the IL-8/androgen receptor (AR)/MMPs signals. Oncotarget 2015, 6, 26065–26078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Tai, Y.T.; Ho, M.; Xing, L.; Chauhan, D.; Gang, A.; Qiu, L.; Anderson, K.C. Regulatory B cell-myeloma cell interaction confers immunosuppression and promotes their survival in the bone marrow milieu. Blood Cancer J. 2017, 7, e547. [Google Scholar] [CrossRef] [Green Version]
- Olkhanud, P.B.; Damdinsuren, B.; Bodogai, M.; Gress, R.E.; Sen, R.; Wejksza, K.; Malchinkhuu, E.; Wersto, R.P.; Biragyn, A. Tumor-Evoked Regulatory B Cells Promote Breast Cancer Metastasis by Converting Resting CD4(+) T Cells to T-Regulatory Cells. Cancer Res. 2011, 71, 3505–3515. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Eliav, Y.; Shin, S.U.; Schreiber, T.H.; Podack, E.R.; Tadmor, T.; Rosenblatt, J.D. B lymphocyte inhibition of anti-tumor response depends on expansion of Treg but is independent of B-cell IL-10 secretion. Cancer Immunol. Immunother. 2013, 62, 87–99. [Google Scholar] [CrossRef]
- Lindner, S.; Dahlke, K.; Sontheimer, K.; Hagn, M.; Kaltenmeier, C.; Barth, T.F.; Beyer, T.; Reister, F.; Fabricius, D.; Lotfi, R.; et al. Interleukin 21-induced granzyme B-expressing B cells infiltrate tumors and regulate T cells. Cancer Res. 2013, 73, 2468–2479. [Google Scholar] [CrossRef] [Green Version]
- Shalapour, S.; Lin, X.J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; Vrbanac, A.F.; Li, W.H.; Perkins, A.; Matsutani, T.; et al. Inflammation-induced IgA(+) cells dismantle anti-liver cancer immunity. Nature 2017, 551, 340–345. [Google Scholar] [CrossRef]
- Sharonov, G.V.; Serebrovskaya, E.O.; Yuzhakova, D.V.; Britanova, O.V.; Chudakov, D.M. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol. 2020, 20, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, P.M.; Ardavin, C. Differentiation and function of mouse monocyte-derived dendritic cells in steady state and inflammation. Immunol. Rev. 2010, 234, 90–104. [Google Scholar] [CrossRef]
- Mildner, A.; Jung, S. Development and function of dendritic cell subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [Green Version]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [Green Version]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.S.; Angel, C.E.; Chen, C.J.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141(+) (BDCA-3)(+) dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef] [Green Version]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Sousa, C.R.E. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022. [Google Scholar] [CrossRef] [Green Version]
- Zong, J.; Keskinov, A.A.; Shurin, G.V.; Shurin, M.R. Tumor-derived factors modulating dendritic cell function. Cancer Immunol. Immunother. 2016, 65, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.Y.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Challier, J.; Bruniquel, D.; Sewell, A.K.; Laugel, B. Adenosine and cAMP signalling skew human dendritic cell differentiation towards a tolerogenic phenotype with defective CD8(+) T-cell priming capacity. Immunology 2013, 138, 402–410. [Google Scholar] [CrossRef]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Melief, C.J. Cancer immunotherapy by dendritic cells. Immunity 2008, 29, 372–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nouri-Shirazi, M.; Banchereau, J.; Bell, D.; Burkeholder, S.; Kraus, E.T.; Davoust, J.; Palucka, K.A. Dendritic cells capture killed tumor cells and present their antigens to elicit tumor-specific immune responses. J. Immunol. 2000, 165, 3797–3803. [Google Scholar] [CrossRef] [PubMed]
- Schoenberger, S.P.; Toes, R.E.M.; van der Voort, E.I.H.; Offringa, R.; Melief, C.J.M. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [Green Version]
- St. Paul, M.; Ohashi, P.S. The Roles of CD8(+) T Cell Subsets in Antitumor Immunity. Trends Cell Biol. 2020, 30, 695–704. [Google Scholar] [CrossRef]
- Masopust, D.; Schenkel, J.M. The integration of T cell migration, differentiation and function. Nat. Rev. Immunol. 2013, 13, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Vihervuori, H.; Autere, T.A.; Repo, H.; Kurki, S.; Kallio, L.; Lintunen, M.M.; Talvinen, K.; Kronqvist, P. Tumor-infiltrating lymphocytes and CD8(+) T cells predict survival of triple-negative breast cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 3105–3114. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.W.; Hu, P. Tumor-Infiltrating CD8 T Cells Predict Clinical Breast Cancer Outcomes in Young Women. Cancers 2020, 12, 1076. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piras, F.; Colombari, R.; Minerba, L.; Murtas, D.; Floris, C.; Maxia, C.; Corbu, A.; Perra, M.T.; Sirigu, P. The predictive value of CD8, CD4, CD68, and human leukocyte antigen-D-related cells in the prognosis of cutaneous malignant melanoma with vertical growth phase. Cancer 2005, 104, 1246–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maecker, H.T.; McCoy, J.P.; Nussenblatt, R. Standardizing immunophenotyping for the Human Immunology Project. Nat. Rev. Immunol. 2012, 12, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8(+) T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Ito, M.; Srirat, T.; Kondo, T.; Yoshimura, A. Memory T cell, exhaustion, and tumor immunity. Immunol. Med. 2020, 43, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e110. [Google Scholar] [CrossRef] [Green Version]
- Li, H.J.; van der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; van Akkooi, A.C.J.; van den Braber, M.; Rozeman, E.A.; Haanen, J.B.A.G.; Blank, C.U.; et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2019, 176, 775. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, W.; Jankovic, V.; Golubov, J.; Poon, P.; Oswald, E.M.; Gurer, C.; Wei, J.; Ramos, I.; Wu, Q.; et al. Combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8(+) T cell dysfunction and maintain memory phenotype. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.J.; Sun, Z.J.; Chen, L.G. Memory T cells: Strategies for optimizing tumor immunotherapy. Protein Cell 2020, 11, 549–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, M.; Kamphorst, A.O.; Im, S.J.; Kissick, H.T.; Pillai, R.N.; Ramalingam, S.S.; Araki, K.; Ahmed, R. CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu. Rev. Med. 2018, 69, 301–318. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Catakovic, K.; Klieser, E.; Neureiter, D.; Geisberger, R. T cell exhaustion: From pathophysiological basics to tumor immunotherapy. Cell Commun. Signal. 2017, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samstag, Y.; Eibert, S.M.; Klemke, M.; Wabnitz, G.H. Actin cytoskeletal dynamics in T lymphocyte activation and migration. J. Leukoc. Biol. 2003, 73, 30–48. [Google Scholar] [CrossRef] [Green Version]
- Melssen, M.; Slingluff, C.L., Jr. Vaccines targeting helper T cells for cancer immunotherapy. Curr. Opin. Immunol. 2017, 47, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.A.; Simpson, T.R.; Peggs, K.S.; Merghoub, T.; Vider, J.; Fan, X.; Blasberg, R.; Yagita, H.; Muranski, P.; Antony, P.A.; et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med. 2010, 207, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Juno, J.A.; van Bockel, D.; Kent, S.J.; Kelleher, A.D.; Zaunders, J.J.; Munier, C.M. Cytotoxic CD4 T Cells-Friend or Foe during Viral Infection? Front. Immunol. 2017, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Ahrends, T.; Busselaar, J.; Severson, T.M.; Babala, N.; de Vries, E.; Bovens, A.; Wessels, L.; van Leeuwen, F.; Borst, J. CD4(+) T cell help creates memory CD8(+) T cells with innate and help-independent recall capacities. Nat. Commun. 2019, 10, 5531. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.M.; Wilson, N.S.; Waithman, J.; Villadangos, J.A.; Carbone, F.R.; Heath, W.R.; Belz, G.T. Cognate CD4(+) T cell licensing of dendritic cells in CD8(+) T cell immunity. Nat. Immunol. 2004, 5, 1143–1148. [Google Scholar] [CrossRef]
- Ahrends, T.; Spanjaard, A.; Pilzecker, B.; Babala, N.; Bovens, A.; Xiao, Y.; Jacobs, H.; Borst, J. CD4(+) T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity 2017, 47, 848–861.e845. [Google Scholar] [CrossRef] [Green Version]
- Guy, T.V.; Terry, A.M.; Bolton, H.A.; Hancock, D.G.; Zhu, E.H.; Brink, R.; McGuire, H.M.; Shklovskaya, E.; Fazekas de St. Groth, B. Collaboration between tumor-specific CD4(+) T cells and B cells in anti-cancer immunity. Oncotarget 2016, 7, 30211–30229. [Google Scholar] [CrossRef] [Green Version]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Lower, M.; Diekmann, J.; Boegel, S.; Schrors, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef] [Green Version]
- Linnemann, C.; van Buuren, M.M.; Bies, L.; Verdegaal, E.M.; Schotte, R.; Calis, J.J.; Behjati, S.; Velds, A.; Hilkmann, H.; Atmioui, D.E.; et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 2015, 21, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Zander, R.; Schauder, D.; Xin, G.; Nguyen, C.; Wu, X.P.; Zajac, A.; Cui, W.G. CD4(+) T Cell Help Is Required for the Formation of a Cytolytic CD8(+) T Cell Subset that Protects against Chronic Infection and Cancer. Immunity 2019, 51, 1028–1042.e4. [Google Scholar] [CrossRef]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef]
- Sledzinska, A.; Vila de Mucha, M.; Bergerhoff, K.; Hotblack, A.; Demane, D.F.; Ghorani, E.; Akarca, A.U.; Marzolini, M.A.V.; Solomon, I.; Vargas, F.A.; et al. Regulatory T Cells Restrain Interleukin-2- and Blimp-1-Dependent Acquisition of Cytotoxic Function by CD4(+) T Cells. Immunity 2020, 52, 151–166.e156. [Google Scholar] [CrossRef] [PubMed]
- LaCasse, C.J.; Janikashvili, N.; Larmonier, C.B.; Alizadeh, D.; Hanke, N.; Kartchner, J.; Situ, E.; Centuori, S.; Har-Noy, M.; Bonnotte, B.; et al. Th-1 lymphocytes induce dendritic cell tumor killing activity by an IFN-gamma-dependent mechanism. J. Immunol. 2011, 187, 6310–6317. [Google Scholar] [CrossRef]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kuhnel, F.; Woller, N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef] [Green Version]
- Corthay, A.; Skovseth, D.K.; Lundin, K.U.; Rosjo, E.; Omholt, H.; Hofgaard, P.O.; Haraldsen, G.; Bogen, B. Primary antitumor immune response mediated by CD4+ T cells. Immunity 2005, 22, 371–383. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, S.L.; Bailey, D.; Zielinski, J.; Apte, A.; Musenge, F.; Karp, R.; Burke, S.; Garcon, F.; Mishra, A.; Gurumurthy, S.; et al. Intratumoral IL12 mRNA Therapy Promotes TH1 Transformation of the Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 6284–6298. [Google Scholar] [CrossRef]
- Mattes, J.; Hulett, M.; Xie, W.; Hogan, S.; Rothenberg, M.E.; Foster, P.; Parish, C. Immunotherapy of cytotoxic T cell-resistant tumors by T helper 2 cells: An eotaxin and STAT6-dependent process. J. Exp. Med. 2003, 197, 387–393. [Google Scholar] [CrossRef]
- Tepper, R.I.; Coffman, R.L.; Leder, P. An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science 1992, 257, 548–551. [Google Scholar] [CrossRef]
- Kim, H.J.; Cantor, H. CD4 T-cell subsets and tumor immunity: The helpful and the not-so-helpful. Cancer Immunol. Res. 2014, 2, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Asadzadeh, Z.; Mohammadi, H.; Safarzadeh, E.; Hemmatzadeh, M.; Mahdian-Shakib, A.; Jadidi-Niaragh, F.; Azizi, G.; Baradaran, B. The paradox of Th17 cell functions in tumor immunity. Cell Immunol. 2017, 322, 15–25. [Google Scholar] [CrossRef]
- Yan, C.; Richmond, A. Th9 and Th17 cells: The controversial twins in cancer immunity. J. Clin. Invest. 2020, 130, 3409–3411. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Livergood, R.S.; Peng, G. The role and regulation of human Th17 cells in tumor immunity. Am. J. Pathol. 2013, 182, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Janikashvili, N.; Chikovani, T.; Audia, S.; Bonnotte, B.; Larmonier, N. T Lymphocyte Plasticity in Autoimmunity and Cancer. Biomed. Res. Int. 2015, 2015, 540750. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Jahraus, B.; Balta, E.; Ziegler, J.D.; Hubner, K.; Blank, N.; Niesler, B.; Wabnitz, G.H.; Samstag, Y. Sulforaphane Inhibits Inflammatory Responses of Primary Human T-Cells by Increasing ROS and Depleting Glutathione. Front. Immunol. 2018, 9, 2584. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Bruchard, M.; Vegran, F.; Ghiringhelli, F. Regulation of T cell antitumor immune response by tumor induced metabolic stress. Cell Stress 2018, 3, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T Cells and Human Disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, M.; Takahashi, T.; Sakaguchi, N.; Kuniyasu, Y.; Shimizu, J.; Otsuka, F.; Sakaguchi, S. Thymus and autoimmunity: Production of CD25(+)CD4(+) naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J. Immunol. 1999, 162, 5317–5326. [Google Scholar] [PubMed]
- Kanamori, M.; Nakatsukasa, H.; Okada, M.; Lu, Q.; Yoshimura, A. Induced Regulatory T Cells: Their Development, Stability, and Applications. Trends Immunol. 2016, 37, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Paluskievicz, C.M.; Cao, X.; Abdi, R.; Zheng, P.; Liu, Y.; Bromberg, J.S. T Regulatory Cells and Priming the Suppressive Tumor Microenvironment. Front. Immunol. 2019, 10, 2453. [Google Scholar] [CrossRef] [PubMed]
- Plitas, G.; Rudensky, A.Y. Regulatory T Cells in Cancer. Annu. Rev. Cancer Biol. 2020, 4, 459–477. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [Green Version]
- Liang, B.T.; Workman, C.; Lee, J.; Chew, C.; Dale, B.M.; Colonna, L.; Flores, M.; Li, N.Y.; Schweighoffer, E.; Greenberg, S.; et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J. Immunol. 2008, 180, 5916–5926. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.; Verhagen, J.; Blaser, K.; Akdis, M.; Akdis, C.A. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: The role of T regulatory cells. Immunology 2006, 117, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular mechanisms of treg-mediated T cell suppression. Front. Immunol. 2012, 3, 51. [Google Scholar] [CrossRef] [Green Version]
- Pandiyan, P.; Zheng, L.; Ishihara, S.; Reed, J.; Lenardo, M.J. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 2007, 8, 1353–1362. [Google Scholar] [CrossRef]
- Klein, M.; Bopp, T. Cyclic AMP Represents a Crucial Component of Treg Cell-Mediated Immune Regulation. Front. Immunol. 2016, 7, 315. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Hasko, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, C.; Hossain, F.; Rodriguez, P.C.; Sierra, R.A.; Pannuti, A.; Osborne, B.A.; Minter, L.M.; Miele, L.; Morello, S. Adenosine A2A Receptor Stimulation Inhibits TCR-Induced Notch1 Activation in CD8+T-Cells. Front. Immunol. 2019, 10, 162. [Google Scholar] [CrossRef]
- Samstag, Y.; John, I.; Wabnitz, G.H. Cofilin: A redox sensitive mediator of actin dynamics during T-cell activation and migration. Immunol. Rev. 2013, 256, 30–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munder, M.; Engelhardt, M.; Knies, D.; Medenhoff, S.; Wabnitz, G.; Luckner-Minden, C.; Feldmeyer, N.; Voss, R.H.; Kropf, P.; Muller, I.; et al. Cytotoxicity of tumor antigen specific human T cells is unimpaired by arginine depletion. PLoS ONE 2013, 8, e63521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gostner, J.M.; Becker, K.; Fuchs, D.; Sucher, R. Redox regulation of the immune response. Redox Rep. 2013, 18, 88–94. [Google Scholar] [CrossRef]
- Wabnitz, G.H.; Balta, E.; Schindler, S.; Kirchgessner, H.; Jahraus, B.; Meuer, S.; Samstag, Y. The pro-oxidative drug WF-10 inhibits serial killing by primary human cytotoxic T-cells. Cell Death Discov. 2016, 2, 16057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llopiz, D.; Ruiz, M.; Infante, S.; Villanueva, L.; Silva, L.; Hervas-Stubbs, S.; Alignani, D.; Guruceaga, E.; Lasarte, J.J.; Sarobe, P. IL-10 expression defines an immunosuppressive dendritic cell population induced by antitumor therapeutic vaccination. Oncotarget 2017, 8, 2659–2671. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Perez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castanon, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Chen, Q.; Ames, B.N. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc. Natl. Acad. Sci. USA 1994, 91, 4130–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, J.; Liu, Z.; Dong, C.; Luan, Y.; Zhang, A.; Moore, C.; Fu, K.; Peng, J.; Wang, Y.; Ren, Z.; et al. Targeting Tumors with IL-10 Prevents Dendritic Cell-Mediated CD8(+) T Cell Apoptosis. Cancer Cell 2019, 35, 901–915.e904. [Google Scholar] [CrossRef] [PubMed]
- Ni, G.; Zhang, L.; Yang, X.; Li, H.; Ma, B.; Walton, S.; Wu, X.; Yuan, J.; Wang, T.; Liu, X. Targeting interleukin-10 signalling for cancer immunotherapy, a promising and complicated task. Hum. Vaccin. Immunother. 2020, 16, 2328–2332. [Google Scholar] [CrossRef]
- Fiaschi, T.; Chiarugi, P. Oxidative stress, tumor microenvironment, and metabolic reprogramming: A diabolic liaison. Int. J. Cell Biol. 2012, 2012, 762825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers 2019, 11, 1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, I.S.; DeNicola, G.M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 2020, 30, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 2009, 21, 894–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.; Sotgia, F.; Lisanti, M.P. Tumor microenvironment and metabolic synergy in breast cancers: Critical importance of mitochondrial fuels and function. Semin. Oncol. 2014, 41, 195–216. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, C.; Kovacs, K.; Polgar, Z.; Regdon, Z.; Szabo, E.; Robaszkiewicz, A.; Forman, H.J.; Martner, A.; Virag, L. Redox control of cancer cell destruction. Redox. Biol. 2018, 16, 59–74. [Google Scholar] [CrossRef]
- Ozben, T. Oxidative stress and apoptosis: Impact on cancer therapy. J. Pharm. Sci. 2007, 96, 2181–2196. [Google Scholar] [CrossRef] [PubMed]
- Watson, J. Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol. 2013, 3, 120144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef] [Green Version]
- Samaranayake, G.J.; Troccoli, C.I.; Huynh, M.; Lyles, R.D.Z.; Kage, K.; Win, A.; Lakshmanan, V.; Kwon, D.; Ban, Y.; Chen, S.X.; et al. Thioredoxin-1 protects against androgen receptor-induced redox vulnerability in castration-resistant prostate cancer. Nat. Commun. 2017, 8, 1204. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Balta, E.; Hardt, R.; Liang, J.; Kirchgessner, H.; Orlik, C.; Jahraus, B.; Hillmer, S.; Meuer, S.; Hubner, K.; Wabnitz, G.H.; et al. Spatial oxidation of L-plastin downmodulates actin-based functions of tumor cells. Nat. Commun. 2019, 10, 4073. [Google Scholar] [CrossRef] [Green Version]
- Balta, E.; Kramer, J.; Samstag, Y. Redox Regulation of the Actin Cytoskeleton in Cell Migration and Adhesion: On the Way to a Spatiotemporal View. Front. Cell Dev. Biol. 2020, 8, 618261. [Google Scholar] [CrossRef]
- Thoren, F.B.; Betten, A.; Romero, A.I.; Hellstrand, K. Cutting edge: Antioxidative properties of myeloid dendritic cells: Protection of T cells and NK cells from oxygen radical-induced inactivation and apoptosis. J. Immunol. 2007, 179, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Munder, M.; Schneider, H.; Luckner, C.; Giese, T.; Langhans, C.D.; Fuentes, J.M.; Kropf, P.; Mueller, I.; Kolb, A.; Modolell, M.; et al. Suppression of T-cell functions by human granulocyte arginase. Blood 2006, 108, 1627–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid Cell-Derived Arginase in Cancer Immune Response. Front. Immunol. 2020, 11, 938. [Google Scholar] [CrossRef]
- Meireson, A.; Devos, M.; Brochez, L. IDO Expression in Cancer: Different Compartment, Different Functionality? Front. Immunol. 2020, 11, 531491. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, J.; Shen, H.; Wang, J. The prognostic value of IDO expression in solid tumors: A systematic review and meta-analysis. BMC Cancer 2020, 20, 471. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. 2014, 20, 5290–5301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agliardi, G.; Patel, A.; Romiti, A.; Kalber, T.; Roberts, T.; Franz-Demane, D.; Lythgoe, M.; Anderson, J.; Quezada, S.; Pule, M.; et al. CAR T Cells Combined With PD1 Blockade Promote Long-Term Efficacy in a Mouse Model for High Grade Gliomas. Mol. Ther. 2017, 25, 154–155. [Google Scholar]
- Kakarla, S.; Gottschalk, S. CAR T cells for solid tumors: Armed and ready to go? Cancer J. 2014, 20, 151–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentjens, R.J.; Latouche, J.B.; Santos, E.; Marti, F.; Gong, M.C.; Lyddane, C.; King, P.D.; Larson, S.; Weiss, M.; Riviere, I.; et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat. Med. 2003, 9, 279–286. [Google Scholar] [CrossRef]
- Spiess, P.J.; Yang, J.C.; Rosenberg, S.A. In vivo antitumor activity of tumor-infiltrating lymphocytes expanded in recombinant interleukin-2. J. Natl. Cancer Inst. 1987, 79, 1067–1075. [Google Scholar] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.L.; Dudley, M.E.; Citrin, D.E.; Somerville, R.P.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Randomized, Prospective Evaluation Comparing Intensity of Lymphodepletion Before Adoptive Transfer of Tumor-Infiltrating Lymphocytes for Patients With Metastatic Melanoma. J. Clin. Oncol. 2016, 34, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J. Adoptive cellular therapies: The current landscape. Virchows Arch. 2019, 474, 449–461. [Google Scholar] [CrossRef] [Green Version]
- Lo Presti, V.; Buitenwerf, F.; van Til, N.P.; Nierkens, S. Gene Augmentation and Editing to Improve TCR Engineered T Cell Therapy against Solid Tumors. Vaccines 2020, 8, 733. [Google Scholar] [CrossRef]
- Porter, C.E.; Shaw, A.R.; Jung, Y.; Yip, T.; Castro, P.D.; Sandulache, V.C.; Sikora, A.; Gottschalk, S.; Ittman, M.M.; Brenner, M.K.; et al. Oncolytic Adenovirus Armed with BiTE, Cytokine, and Checkpoint Inhibitor Enables CAR T Cells to Control the Growth of Heterogeneous Tumors. Mol. Ther. 2020, 28, 1251–1262. [Google Scholar] [CrossRef]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor Regression in Patients With Metastatic Synovial Cell Sarcoma and Melanoma Using Genetically Engineered Lymphocytes Reactive With NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [Green Version]
- Geyer, M.B.; Brentjens, R.J. Review: Current clinical applications of chimeric antigen receptor (CAR) modified T cells. Cytotherapy 2016. [Google Scholar] [CrossRef] [Green Version]
- Knochelmann, H.M.; Smith, A.S.; Dwyer, C.J.; Wyatt, M.M.; Mehrotra, S.; Paulos, C.M. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front. Immunol. 2018, 9, 1740. [Google Scholar] [CrossRef] [Green Version]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.C.; Kapoor, V.; Predina, J.; Powell, D.J., Jr.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [Green Version]
- Zou, F.; Lu, L.; Liu, J.; Xia, B.; Zhang, W.; Hu, Q.; Liu, W.; Zhang, Y.; Lin, Y.; Jing, S.; et al. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat. Commun. 2019, 10, 4109. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Invest. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegram, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Curran, K.J.; Giralt, S.A.; Barker, J.N.; Brentjens, R.J. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia 2015, 29, 415–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Zhou, Y.; Li, Z.; Farooq, M.A.; Ajmal, I.; Zhang, H.; Zhang, L.; Tao, L.; Yao, J.; Du, B.; et al. Co-Expression of IL-7 Improves NKG2D-Based CAR T Cell Therapy on Prostate Cancer by Enhancing the Expansion and Inhibiting the Apoptosis and Exhaustion. Cancers 2020, 12, 1969. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Morello, A.; Tano, Z.; Adusumilli, P.S. CAR T-cell intrinsic PD-1 checkpoint blockade: A two-in-one approach for solid tumor immunotherapy. Oncoimmunology 2017, 6, e1273302. [Google Scholar] [CrossRef] [Green Version]
- Wing, A.; Fajardo, C.A.; Posey, A.D.; Shaw, C.; Da, T.; Young, R.M.; Alemany, R.; June, C.H.; Guedan, S. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus-Driven Production of a Bispecific T-cell Engager. Cancer Immunol. Res. 2018, 6, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.S.; Lotze, M.T.; Zhu, Z.; Storkus, W.J.; Song, X.T. Bi- and Tri-Specific T Cell Engager-Armed Oncolytic Viruses: Next-Generation Cancer Immunotherapy. Biomedicines 2020, 8, 204. [Google Scholar] [CrossRef]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef] [Green Version]
- Rashida Gnanaprakasam, J.N.; Wu, R.; Wang, R. Metabolic Reprogramming in Modulating T Cell Reactive Oxygen Species Generation and Antioxidant Capacity. Front. Immunol. 2018, 9, 1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; De Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxid Med. Cell Longev. 2016, 2016, 3907147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Mayer, A.T.; Li, R. Integrated imaging and molecular analysis to decipher tumor microenvironment in the era of immunotherapy. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]


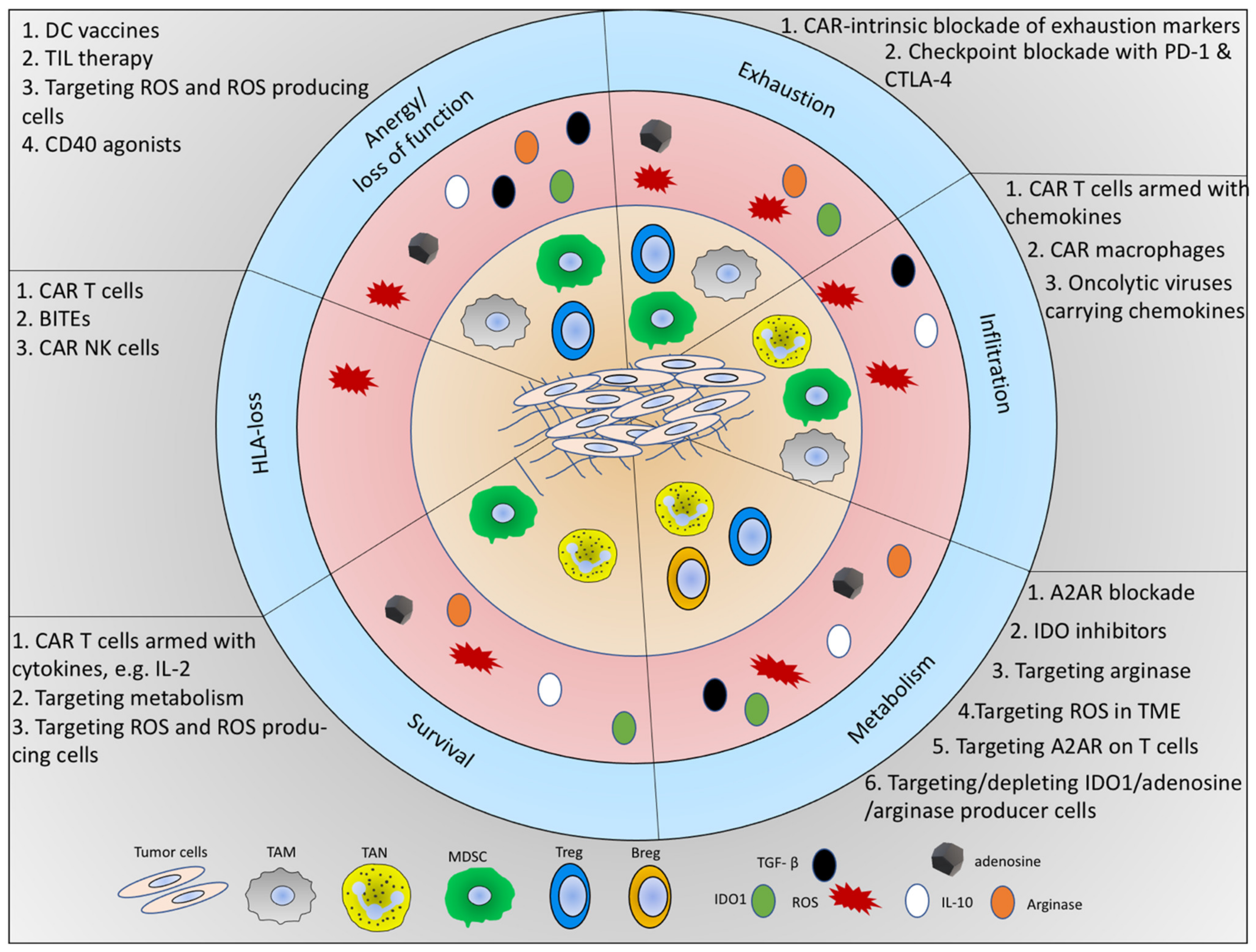

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T Cell (Memory) Subset | Surface Markers | Antitumor Functions | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CD45RA | CD45RO | CD28 | CD27 | CD57 | CD62L | CD127 | CCR7 (CD197) | Proliferation Potential | Effector Functions | |
| Naïve (TNA) | + | − | + | + | − | + | + | + | +/− | − |
| Central memor (TCM) | − | + | + | + | − | + | + | + | +/− | + |
| Effector memory (TEM) | − | + | − | +/− | + | − | +/− | − | − | + |
| Resident memory (TRM) | − | + | +/− | + | − | − | + | − | ? | + |
| Memory stem cells (TSCM) | + | + | + | + | − | + | + | + | + | + |
| Effector memory RA cells (TEMRA) | + | + | − | − | + | − | − | − | − | + |
| Terminally differentiated effector cells (TEF) | + | − | − | − | + | − | − | − | − | ++ |
| Exhaustion Marker Expressed on T Cell | Ligand | Cells Expressing the Ligand | Influence on Antitumor Functions of T Cells |
|---|---|---|---|
| PD-1 | PD-L1 | Tumor cells, DCs, TAMs, Bregs | Inhibitory |
| CTLA-4 | CD80/CD86 | DCs, B cells, TAMs, Treg | Inhibitory |
| TIM3 | Galectin 9 | APC | Both inhibitory and activating, but mostly inhibitory |
| LAG3 | MHC-I or MHC-II–Peptide complex | APC, tumor cells | Both inhibitory and activating, but mostly inhibitory |
| TIGIT | CD155 | APC | Inhibitory |
| Vista | ? | APC | Largely remains to be elucidated |
| CD96 | CD112 | APC, tumor cells | Largely remains to be elucidated |
| BTLA-4 | HVEM | T cells, Tregs, APCs, tumor cells | Largely remains to be elucidated |
| CD160 | HVEM | T cells, Tregs, APCs, tumor cells | Largely remains to be elucidated |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balta, E.; Wabnitz, G.H.; Samstag, Y. Hijacked Immune Cells in the Tumor Microenvironment: Molecular Mechanisms of Immunosuppression and Cues to Improve T Cell-Based Immunotherapy of Solid Tumors. Int. J. Mol. Sci. 2021, 22, 5736. https://doi.org/10.3390/ijms22115736
Balta E, Wabnitz GH, Samstag Y. Hijacked Immune Cells in the Tumor Microenvironment: Molecular Mechanisms of Immunosuppression and Cues to Improve T Cell-Based Immunotherapy of Solid Tumors. International Journal of Molecular Sciences. 2021; 22(11):5736. https://doi.org/10.3390/ijms22115736
Chicago/Turabian StyleBalta, Emre, Guido H. Wabnitz, and Yvonne Samstag. 2021. "Hijacked Immune Cells in the Tumor Microenvironment: Molecular Mechanisms of Immunosuppression and Cues to Improve T Cell-Based Immunotherapy of Solid Tumors" International Journal of Molecular Sciences 22, no. 11: 5736. https://doi.org/10.3390/ijms22115736